
Kratom Alkaloids: A Blood–Brain Barrier Specific Membrane Permeability Assay-Guided Isolation and Cyclodextrin Complexation Study

 , , , ,
, , , ,  ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Extraction Studies
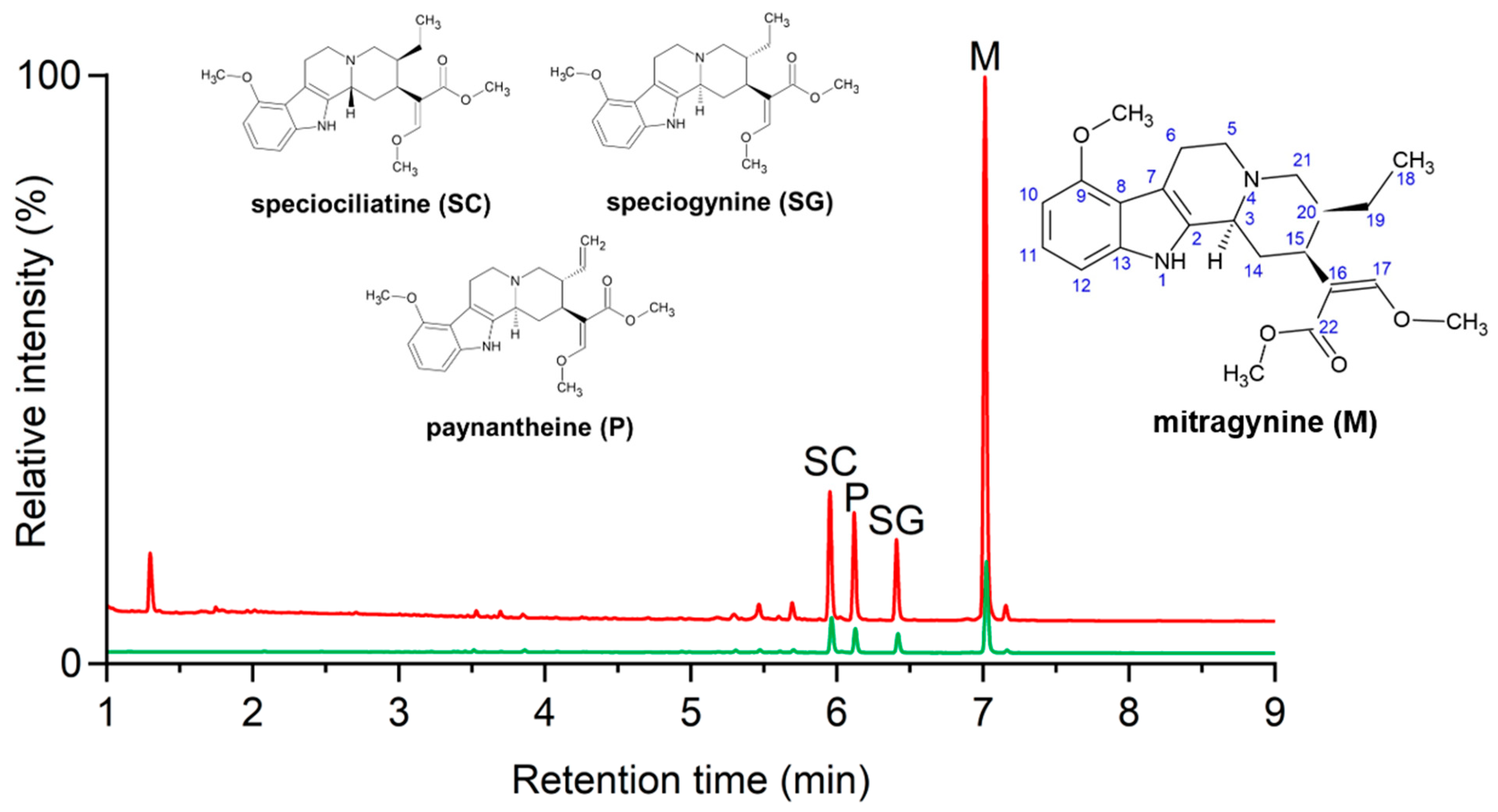
2.2. PAMPA-BBB-Guided Isolation Studies
2.3. Determination of the Alkaloid–CD Complex Stabilities by ACE
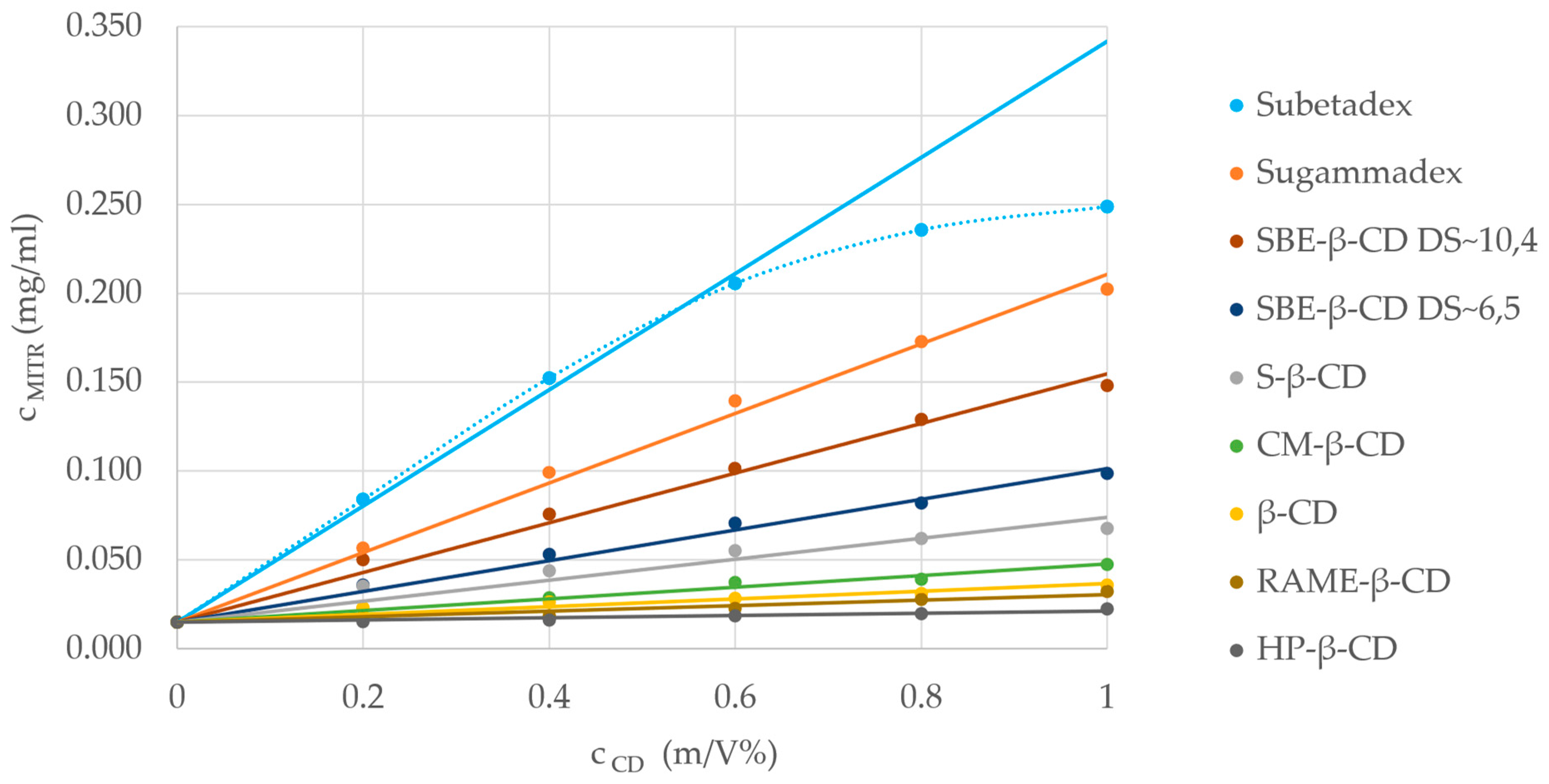
2.4. Phase-Solubility Studies
3. Materials and Methods
3.1. Materials
3.2. Extraction Procedures
3.2.1. Laboratory-Scale Extraction Methods
3.2.2. Pilot-Scale Supercritical Fluid Extraction
3.3. UHPLC-UV Method
3.4. Parallel Artificial Membrane Permeability Assay
3.5. Isolation Procedure
3.6. NMR
3.7. Affinity Capillary Electrophoresis
3.8. Phase-Solubility Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takayama, H. Chemistry and pharmacology of analgesic indole alkaloids from the rubiaceous plant, Mitragyna speciosa. Chem. Pharm. Bull. 2004, 52, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Cinosi, E.; Martinotti, G.; Simonato, P.; Singh, D.; Demetrovics, Z.; Roman-Urrestarazu, A.; Bersani, F.S.; Vicknasingam, B.; Piazzon, G.; Li, J.H.; et al. Following “the Roots” of Kratom (Mitragyna speciosa): The Evolution of an Enhancer from a Traditional Use to Increase Work and Productivity in Southeast Asia to a Recreational Psychoactive Drug in Western Countries. Biomed Res. Int. 2015, 2015, 968786. [Google Scholar] [CrossRef] [PubMed]
- Boyer, E.W.; Babu, K.M.; Adkins, J.E.; Mccurdy, C.R.; Halpern, J.H. Self-treatment of opioid withdrawal using kratom (Mitragynia speciosa korth). Wiley Online Libr. 2008, 103, 1048–1050. [Google Scholar] [CrossRef]
- Váradi, A.; Marrone, G.F.; Palmer, T.C.; Narayan, A.; Szabó, M.R.; Le Rouzic, V.; Grinnell, S.G.; Subrath, J.J.; Warner, E.; Kalra, S.; et al. Mitragynine/Corynantheidine Pseudoindoxyls as Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit β-Arrestin-2. J. Med. Chem. 2016, 59, 8381–8397. [Google Scholar] [CrossRef] [PubMed]
- Kruegel, A.C.; Gassaway, M.M.; Kapoor, A.; Váradi, A.; Majumdar, S.; Filizola, M.; Javitch, J.A.; Sames, D. Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J. Am. Chem. Soc. 2016, 138, 6754–6764. [Google Scholar] [CrossRef]
- Karunakaran, T.; Ngew, K.Z.; Zailan, A.A.D.; Mian Jong, V.Y.; Abu Bakar, M.H. The Chemical and Pharmacological Properties of Mitragynine and Its Diastereomers: An Insight Review. Front. Pharmacol. 2022, 13, 805986. [Google Scholar] [CrossRef]
- Hiranita, T.; Obeng, S.; Sharma, A.; Wilkerson, J.L.; McCurdy, C.R.; McMahon, L.R. In vitro and in vivo pharmacology of kratom. Adv. Pharmacol. 2022, 93, 35–76. [Google Scholar] [CrossRef]
- Herman, T.F.; Cascella, M.; Muzio, M.R. Mu Receptors; StatPearls Publishing: Tampa, FL, USA, 2024. [Google Scholar]
- Ramanathan, S.; Parthasarathy, S.; Murugaiyah, V.; Magosso, E.; Tan, S.C.; Mansor, S.M. Understanding the Physicochemical Properties of Mitragynine, a Principal Alkaloid of Mitragyna speciosa, for Preclinical Evaluation. Molecules 2015, 20, 4915. [Google Scholar] [CrossRef]
- Manda, V.K.; Avula, B.; Ali, Z.; Khan, I.A.; Walker, L.A.; Khan, S.I. Evaluation of in vitro absorption, distribution, metabolism, and excretion (ADME) properties of mitragynine, 7-hydroxymitragynine, and mitraphylline. Planta Med. 2014, 80, 568–576. [Google Scholar] [CrossRef]
- Ya, K.; Tangamornsuksan, W.; Scholfield, C.N.; Methaneethorn, J.; Lohitnavy, M. Pharmacokinetics of mitragynine, a major analgesic alkaloid in kratom (Mitragyna speciosa): A systematic review. Asian J. Psychiatr. 2019, 43, 73–82. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Ramanathan, S.; Ismail, S.; Adenan, M.I.; Mansor, S.M.; Murugaiyah, V. Determination of mitragynine in plasma with solid-phase extraction and rapid HPLC-UV analysis, and its application to a pharmacokinetic study in rat. Anal. Bioanal. Chem. 2010, 397, 2023–2030. [Google Scholar] [CrossRef] [PubMed]
- Várnai, B.; Zsila, F.; Szakács, Z.; Garádi, Z.; Malanga, M.; Béni, S. Sulfobutylation of Beta-Cyclodextrin Enhances the Complex Formation with Mitragynine: An NMR and Chiroptical Study. Int. J. Mol. Sci. 2022, 23, 3844. [Google Scholar] [CrossRef] [PubMed]
- Olabi, M.; Stein, M.; Wätzig, H. Affinity capillary electrophoresis for studying interactions in life sciences. Methods 2018, 146, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Dohárszky, A.; Kalydi, E.; Völgyi, G.; Béni, S.; Fejős, I. Cyclodextrin-Enabled Enantioselective Complexation Study of Cathinone Analogs. Molecules 2024, 29, 876. [Google Scholar] [CrossRef]
- Citti, C.; Laganà, A.; Capriotti, A.L.; Montone, C.M.; Cannazza, G. Kratom: The analytical challenge of an emerging herbal drug. J. Chromatogr. A 2023, 1703, 464094. [Google Scholar] [CrossRef]
- ICH Q2(R2) Validation of Analytical Procedures—Scientific Guideline | European Medicines Agency. Available online: https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures-scientific-guideline (accessed on 17 June 2024).
- Harizal, S.N.; Mansor, S.M.; Hasnan, J.; Tharakan, J.K.J.; Abdullah, J. Acute toxicity study of the standardized methanolic extract of Mitragyna speciosa Korth in rodent. J. Ethnopharmacol. 2010, 131, 404–409. [Google Scholar] [CrossRef]
- Haziqah, N.; Razak, Z.A.; Basyaruddin, M.; Rahman, A.; Ashari, S.E. Optimization of Extraction Yield and Phytochemical Characterization of Crude Methanolic Extract and Its Fractions of Mitragyna speciosa Leaves. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Kikura-Hanajiri, R.; Kawamura, M.; Maruyama, T.; Kitajima, M.; Takayama, H.; Goda, Y. Simultaneous analysis of mitragynine, 7-hydroxymitragynine, and other alkaloids in the psychotropic plant “kratom” (Mitragyna speciosa) by LC-ESI-MS. Forensic Toxicol. 2009, 27, 67–74. [Google Scholar] [CrossRef]
- Orio, L.; Alexandru, L.; Cravotto, G.; Mantegna, S.; Barge, A. UAE, MAE, SFE-CO2 and classical methods for the extraction of Mitragyna speciosa leaves. Ultrason. Sonochem. 2012, 19, 591–595. [Google Scholar] [CrossRef]
- Tohar, N.; Shilpi, J.A.; Sivasothy, Y.; Ahmad, S.; Awang, K. Chemical constituents and nitric oxide inhibitory activity of supercritical carbon dioxide extracts from Mitragyna speciosa leaves. Arab. J. Chem. 2019, 12, 350–359. [Google Scholar] [CrossRef]
- Kong, W.M.; Chik, Z.; Mohamed, Z.; Alshawsh, M.A. Physicochemical Characterization of Mitragyna speciosa Alkaloid Extract and Mitragynine using In Vitro High Throughput Assays. Comb. Chem. High Throughput Screen. 2017, 20, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Könczöl, Á.; Müller, J.; Földes, E.; Béni, Z.; Végh, K.; Kéry, Á.; Balogh, G.T. Applicability of a blood-brain barrier specific artificial membrane permeability assay at the early stage of natural product-based CNS drug discovery. J. Nat. Prod. 2013, 76, 655–663. [Google Scholar] [CrossRef]
- Flores-Bocanegra, L.; Raja, H.A.; Graf, T.N.; Augustinović, M.; Wallace, E.D.; Hematian, S.; Kellogg, J.J.; Todd, D.A.; Cech, N.B.; Oberlies, N.H. The Chemistry of Kratom [Mitragyna speciosa]: Updated Characterization Data and Methods to Elucidate Indole and Oxindole Alkaloids. J. Nat. Prod. 2020, 83, 2165–2177. [Google Scholar] [CrossRef] [PubMed]
- Garba, S.A.; Shaari, K.; Abdul Manap, M.R.; Lee, S.Y.; Abdulazeez, I.; Mohd Faudzi, S.M. Quantitative analysis of selected alkaloids of Mitragyna speciosa using 1H quantitative nuclear magnetic resonance spectroscopy. Magn. Reson. Chem. 2024, 62, 803–813. [Google Scholar] [CrossRef]
- Kong, W.M.; Mohamed, Z.; Alshawsh, M.A.; Chik, Z. Evaluation of pharmacokinetics and blood-brain barrier permeability of mitragynine using in vivo microdialysis technique. J. Pharm. Biomed. Anal. 2017, 143, 43–47. [Google Scholar] [CrossRef]
- Posch, T.N.; Müller, A.; Schulz, W.; Pütz, M.; Huhn, C. Implementation of a design of experiments to study the influence of the background electrolyte on separation and detection in non-aqueous capillary electrophoresis-mass spectrometry. Electrophoresis 2012, 33, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Vági, E.; Balázs, M.; Komoczi, A.; Mihalovits, M.; Székely, E. Fractionation of phytocannabinoids from industrial hemp residues with high-pressure technologies. J. Supercrit. Fluids 2020, 164, 104898. [Google Scholar] [CrossRef]
- Avdeef, A. Permeability—PAMPA. In Absorption and Drug Development; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012; pp. 319–498. [Google Scholar]
- Dubský, P.; Ördögová, M.; Malý, M.; Riesová, M. CEval: All-in-one software for data processing and statistical evaluations in affinity capillary electrophoresis. J. Chromatogr. A 2016, 1445, 158–165. [Google Scholar] [CrossRef]
- Østergaard, J.; Jensen, H.; Holm, R. Affinity capillary electrophoresis method for investigation of bile salts complexation with sulfobutyl ether-β-cyclodextrin. J. Sep. Sci. 2012, 35, 2764–2772. [Google Scholar] [CrossRef]
- Beneš, M.; Zusková, I.; Svobodová, J.; Gaš, B. Determination of stability constants of complexes of neutral analytes with charged cyclodextrins by affinity capillary electrophoresis. Electrophoresis 2012, 33, 1032–1039. [Google Scholar] [CrossRef]
- Higuchi, T.; Connors, K.A. Phase Solubility Techniques: Advances in Analytical Chemistry and Instrumentation. Adv. Anal. Chem. Instrum. 1965, 4, 117–212. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | logPe PAMPA-BBB (n = 9) |
|---|---|
| Mitragynine | −4.42 ± 0.61 |
| Paynantheine | −4.61 ± 0.31 |
| Speciociliatine | −4.58 ± 0.85 |
| Speciogynine | −4.77 ± 0.42 |
| Cyclodextrin | Mitragynine | Paynantheine | Speciociliatine | Speciogynine | |
|---|---|---|---|---|---|
| Native CDs | α-CD | 25 ± 2 | 20 ± 2 | 22 ± 3 | 23 ± 5 |
| β-CD | 145 ± 20 | 145 ± 15 | 35 ± 3 | 65 ± 9 | |
| γ-CD | 55 ± 5 | 140 ± 5 | 15 ± 1 | 45 ± 4 | |
| Negatively charged CD derivatives | Succ-β-CD | 375± 20 | 360 ± 30 | 470 ± 35 | 840 ± 80 |
| Phos-β-CD | 290 ± 30 | 335 ± 30 | 215 ± 15 | 420 ± 30 | |
| SBE-β-CD DS~4 | 550 ± 35 | 830 ± 50 | 670 ± 60 | 610 ± 60 | |
| SBE-β-CD DS~6.5 | 1530 ± 55 | 2000 ± 75 | 1700 ± 75 | 1800 ± 100 | |
| SBE-β-CD DS~10.4 | 4900 ± 420 | 5300 ± 280 | 5530 ± 435 | 9200 ± 890 | |
| S-β-CD | 1650 ± 30 | 1420 ± 145 | 2410 ± 185 | 970 ± 80 | |
| HS-β-CD | 1250 ± 105 | 575 ± 35 | 850 ± 30 | 645 ± 30 | |
| HDAS-β-CD | 4100 ± 180 | 1720 ± 125 | 3000 ± 275 | 2350 ± 115 | |
| HDMS-β-CD | 175 ± 7 | 135 ± 14 | 105 ± 12 | 30 ± 3 | |
| Subetadex | 1800 ± 80 | 3600 ± 300 | 2700 ± 100 | n.d. | |
| Sugammadex | 2050 ± 125 | 2750 ± 175 | 700 ± 45 | n.d. |
| Cyclodextrin | Kstab | CE | R2 | |
|---|---|---|---|---|
| Native CD | β-CD | 55 | 0.0021 | 0.9463 |
| Neutral CD derivatives | RAME-β-CD | 60 | 0.0023 | 0.9598 |
| HP-β-CD | 30 | 0.0011 | 0.9334 | |
| Negatively charged CD derivatives | CM-β-CD | 300 | 0.0113 | 0.9849 |
| Subetadex | 1730 | 0.0650 | 0.9964 | |
| Sugammadex | 1150 | 0.0431 | 0.9953 | |
| SBE-β-CD DS~6.5 | 480 | 0.0181 | 0.9933 | |
| SBE-β-CD DS~10.4 | 1020 | 0.0382 | 0.9938 | |
| S-β-CD | 340 | 0.0127 | 0.9531 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dohárszky, A.; Vági, E.M.; Könczöl, Á.; Simon, A.; Várnagy, E.; Muratov, M.; Steiger, K.I.; Várnai, B.; Béni, S.; Riethmüller, E.; et al. Kratom Alkaloids: A Blood–Brain Barrier Specific Membrane Permeability Assay-Guided Isolation and Cyclodextrin Complexation Study. Molecules 2024, 29, 5302. https://doi.org/10.3390/molecules29225302
Dohárszky A, Vági EM, Könczöl Á, Simon A, Várnagy E, Muratov M, Steiger KI, Várnai B, Béni S, Riethmüller E, et al. Kratom Alkaloids: A Blood–Brain Barrier Specific Membrane Permeability Assay-Guided Isolation and Cyclodextrin Complexation Study. Molecules. 2024; 29(22):5302. https://doi.org/10.3390/molecules29225302
Chicago/Turabian StyleDohárszky, András, Erika Mária Vági, Árpád Könczöl, Alexandra Simon, Erzsébet Várnagy, Miras Muratov, Kristóf István Steiger, Bianka Várnai, Szabolcs Béni, Eszter Riethmüller, and et al. 2024. "Kratom Alkaloids: A Blood–Brain Barrier Specific Membrane Permeability Assay-Guided Isolation and Cyclodextrin Complexation Study" Molecules 29, no. 22: 5302. https://doi.org/10.3390/molecules29225302
APA StyleDohárszky, A., Vági, E. M., Könczöl, Á., Simon, A., Várnagy, E., Muratov, M., Steiger, K. I., Várnai, B., Béni, S., Riethmüller, E., & Fejős, I. (2024). Kratom Alkaloids: A Blood–Brain Barrier Specific Membrane Permeability Assay-Guided Isolation and Cyclodextrin Complexation Study. Molecules, 29(22), 5302. https://doi.org/10.3390/molecules29225302