
Design, Synthesis, and Evaluation of Doxifluridine Derivatives as Nitroreductase-Responsive Anticancer Prodrugs


Abstract
1. Introduction
2. Results and Discussion
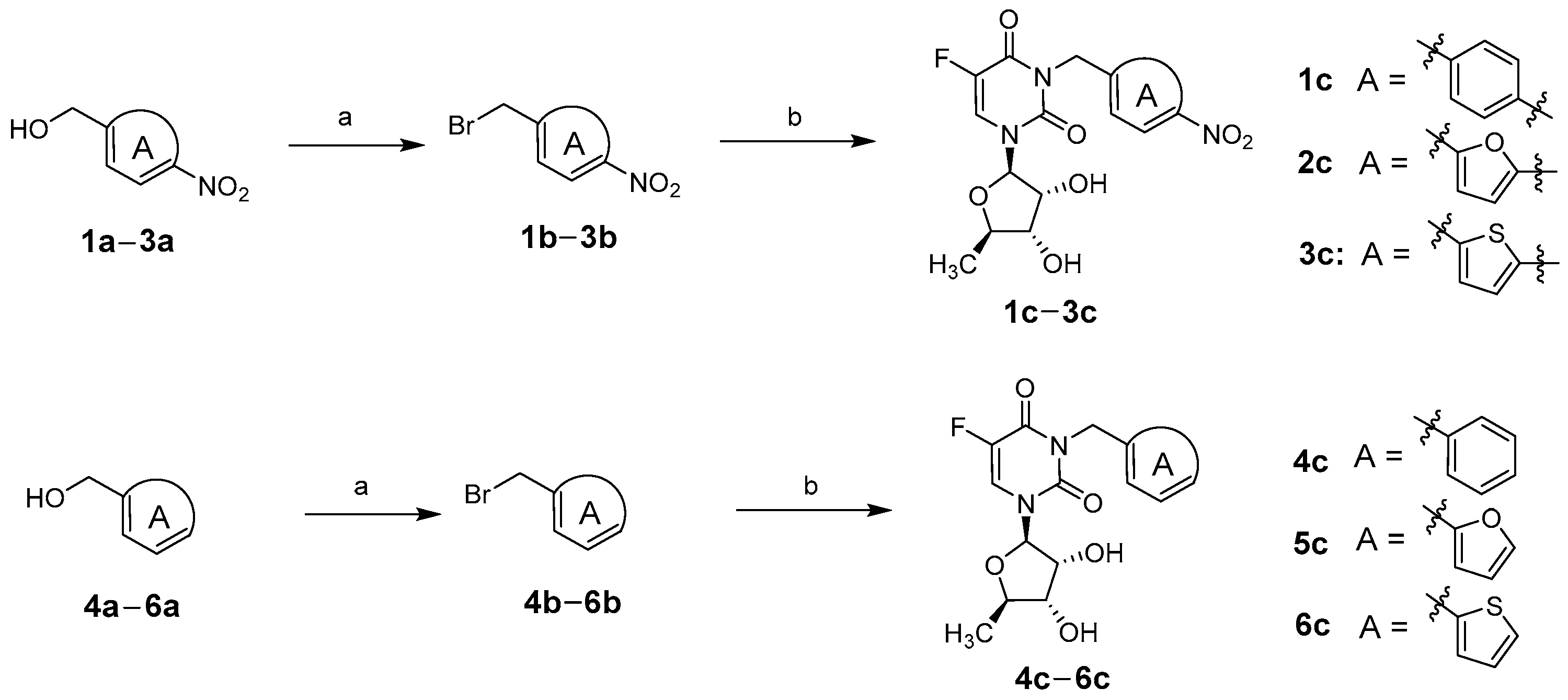
2.1. Chemistry
2.2. Biological Evaluation
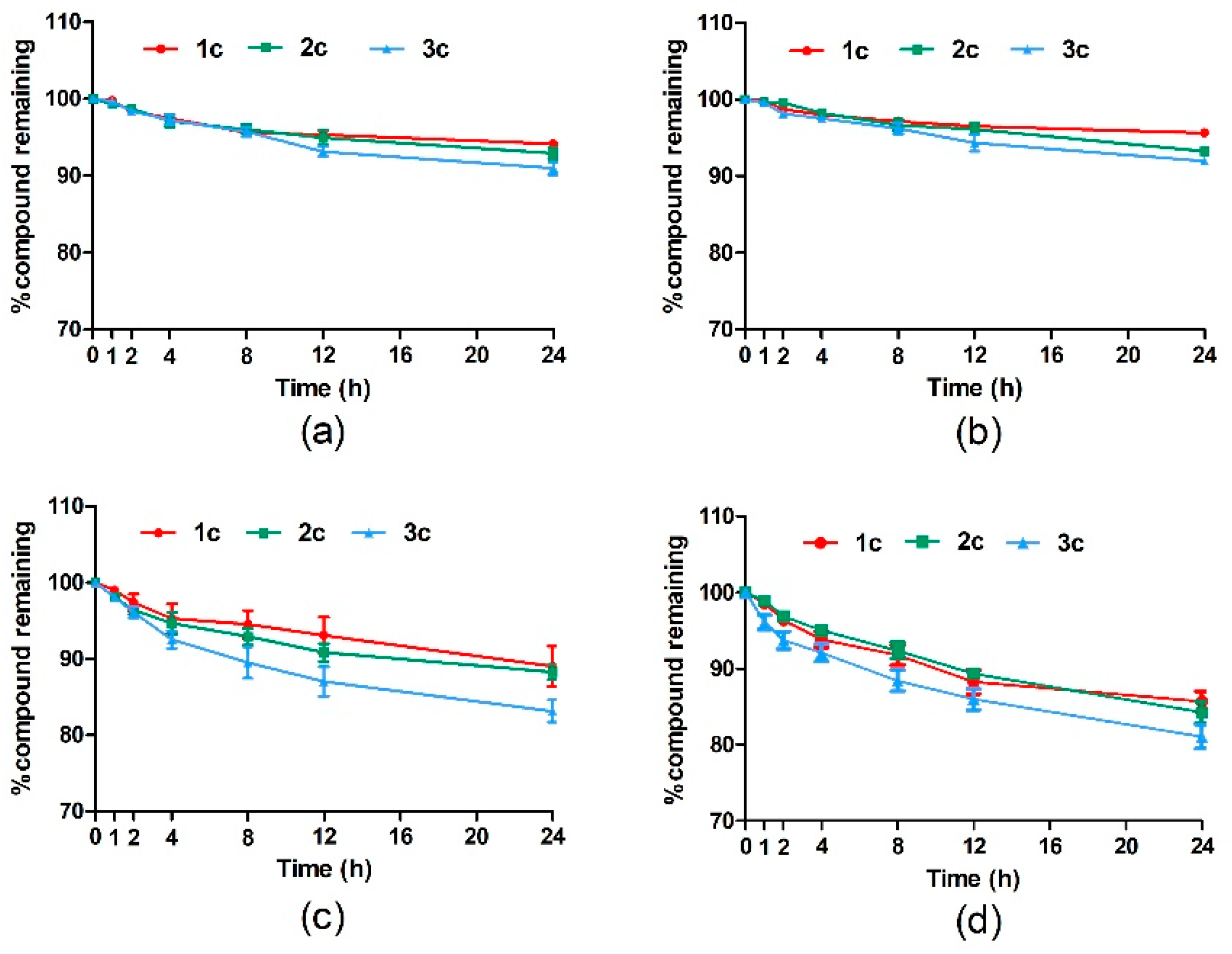
2.2.1. Stability Evaluation of Target Compounds in Phosphate-Buffered Saline Versus Plasma
2.2.2. In Vitro Release Evaluation of Target Compounds
2.2.3. In Vitro Cytotoxicity Evaluation
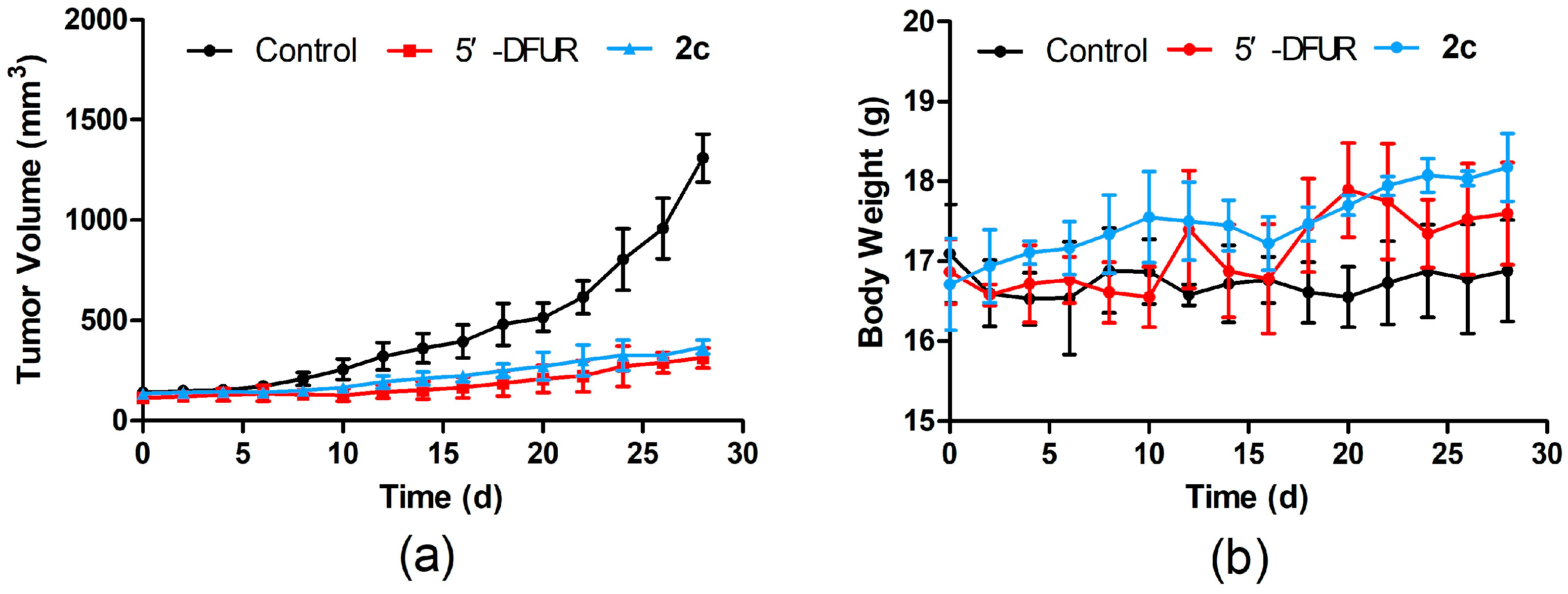
2.2.4. In Vivo Antitumor Activity Evaluation
3. Materials and Methods
3.1. Chemistry
3.1.1. General Synthetic Procedure for Bromide Intermediates 1b–6b
3.1.2. General Synthetic Procedure for Target Compounds 1c–6c
3.2. Biological Evaluation
3.2.1. Stability Evaluation of Target Compounds in PBS Versus Plasma
3.2.2. In Vitro Release Evaluation of Target Compounds
3.2.3. In Vitro Cytotoxicity Evaluation
3.2.4. In Vivo Antitumor Activity Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Der Kraak, L.; Goel, G.; Ramanan, K.; Kaltenmeier, C.; Zhang, L.; Normolle, D.P.; Freeman, G.J.; Tang, D.; Nason, K.S.; Davison, J.M.; et al. 5-Fluorouracil upregulates cell surface B7-H1 (PD-L1) expression in gastrointestinal cancers. J. Immunother. Cancer 2016, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Sadowska, A.; Sawicka, D.; Godlewska, K.; Guzińska-Ustymowicz, K.; Zapora, E.; Sokołowska, E.; Car, H. Beneficial Proapoptotic Effect of Heterobasidion Annosum Extract in Colorectal Cancer Xenograft Mouse Model. Molecules 2023, 28, 1352. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Yoshida, Y.; Yamada, T.; Aisu, N.; Yoshimatsu, G.; Yoshimura, F.; Hasegawa, S. Current Status of Therapeutic Drug Monitoring of 5-Fluorouracil Prodrugs. Anticancer Res. 2020, 40, 4655–4661. [Google Scholar] [CrossRef]
- Cassidy, J.; Saltz, L.; Twelves, C.; Van Cutsem, E.; Hoff, P.; Kang, Y.; Saini, J.P.; Gilberg, F.; Cunningham, D. Efficacy of capecitabine versus 5-fluorouracil in colorectal and gastric cancers: A meta-analysis of individual data from 6171 patients. Ann. Oncol. 2011, 22, 2604–2609. [Google Scholar] [CrossRef]
- Armstrong, R.D.; Cadman, E. 5′-Deoxy-5-fluorouridine selective toxicity for human tumor cells compared to human bone marrow. Cancer Res. 1983, 43, 2525–2528. [Google Scholar]
- Kim, D.W.; Kim, H.-J.; Kim, D.-E.; Roh, J.-K. Doxifluridine-induced neurotoxicity with normal dihydropyrimidine dehydrogenase activity. Neurology 2004, 62, 2136–2137. [Google Scholar] [CrossRef]
- Au, J.L.S.; Walker, J.S.; Rustum, Y.M. Pharmacokinetic studies of 5-fluorouracil and 5′-deoxy-5-fluorouridine in rats. J. Pharmacol. Exp. Ther. 1983, 227, 174–180. [Google Scholar]
- Tarar, A.; Alyami, E.M.; Peng, C.-A. Mesenchymal stem cells anchored with thymidine phosphorylase for doxifluridine-mediated cancer therapy. RSC Adv. 2021, 11, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Si, S.; Xu, J.; Xue, P.; Li, K. Construction of synergistic pH/H2O2-responsive prodrug for prolonging blood circulation and accelerating cellular internalization. Bioorg. Chem. 2022, 119, 105510. [Google Scholar] [CrossRef]
- Xue, F.; Lin, X.; Cai, Z.; Liu, X.; Ma, Y.; Wu, M. Doxifluridine-based pharmacosomes delivering miR-122 as tumor microenvironments-activated nanoplatforms for synergistic treatment of hepatocellular carcinoma. Colloid. Surf. B 2021, 197, 111367. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Danquah, M.K.; Zhang, X.A.; Mahato, R.I. Extravasation of polymeric nanomedicines across tumor vasculature. Adv. Drug. Deliver. Rev. 2011, 63, 623–639. [Google Scholar] [CrossRef]
- Katayama, Y.; Uchino, J.; Chihara, Y.; Tamiya, N.; Kaneko, Y.; Yamada, T.; Takayama, K. Tumor Neovascularization and Developments in Therapeutics. Cancers 2019, 11, 316. [Google Scholar] [CrossRef]
- He, Z.; Chen, K.; An, Y.; He, J.; Zhang, X.; Tang, L.; Sun, F.; Jiang, K. BSA modification of bacterial surface: A promising anti-cancer therapeutic strategy. BMC Microbiol. 2023, 23, 105. [Google Scholar] [CrossRef]
- Ngabire, D.; Kim, G.-D. Autophagy and inflammatory response in the tumor microenvironment. Int. J. Mol. Sci. 2017, 18, 2016. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Li, J.; Entenberg, D.; Xue, A.; Wang, W.; Condeelis, J. Direct visualization of the phenotype of hypoxic tumor cells at single cell resolution in vivo using a new hypoxia probe. Intravital 2016, 5, e1187803. [Google Scholar] [CrossRef]
- Wu, Q.; You, L.; Nepovimova, E.; Heger, Z.; Wu, W.; Kuca, K.; Adam, V. Hypoxia-inducible factors: Master regulators of hypoxic tumor immune escape. J. Hematol. Oncol. 2022, 15, 77. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ohh, M. Oxygen-mediated endocytosis in cancer. J. Cell Mol. Med. 2010, 14, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Abe, F.; Matsuda, Y.; Kitadate, A.; Takahashi, N.; Tagawa, H. Hypoxia-inducible hexokinase-2 enhances anti-apoptotic function via activating autophagy in multiple myeloma. Cancer Sci. 2020, 111, 4088–4101. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Wu, F.; Zeng, Y.; Liu, B.; Peng, R.; Zheng, J. An azoreductase activatable, endonuclease-gated nanodevice for spatiotemporal amplification imaging of microRNA-21 in hypoxic tumor cells. Chem. Commun. 2023, 59, 7411–7414. [Google Scholar] [CrossRef]
- Yang, Y.; Zhai, H.; Yuan, J.; Wang, K.; Zhang, H. Recent Advances in the Fluorescent Probes for Flavinase Activity: Design and Applications. Chem-Asian J. 2022, 17, e202200043. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.-L.; Guo, L.; Chen, L.-L.; Li, H.; Yang, Y.-S.; Jiang, A.-Q.; Zhu, H.-L. Recent progress in the design principles, sensing mechanisms, and applications of small-molecule probes for nitroreductases. Coordin. Chem. Rev. 2020, 421, 213460. [Google Scholar] [CrossRef]
- Ross, C.L.; Lawer, A.; Sircombe, K.J.; Pletzer, D.; Gamble, A.B.; Hook, S. Site-Specific Antimicrobial Activity of a Dual-Responsive Ciprofloxacin Prodrug. J. Med. Chem. 2024, 67, 9599–9612. [Google Scholar] [CrossRef]
- Güngör, T.; Tokay, E.; Güven Gülhan, Ü.; Hacıoğlu, N.; Çelik, A.; Köçkar, F.; Ay, M. Prodrugs for nitroreductase based cancer therapy-4: Towards prostate cancer targeting: Synthesis of N-heterocyclic nitro prodrugs, Ssap-NtrB enzymatic activation and anticancer evaluation. Bioorg. Chem. 2020, 105, 104450. [Google Scholar] [CrossRef]
- McKeage, M.J.; Gu, Y.; Wilson, W.R.; Hill, A.; Amies, K.; Melink, T.J.; Jameson, M.B. A phase I trial of PR-104, a pre-prodrug of the bioreductive prodrug PR-104A, given weekly to solid tumour patients. BMC Cancer 2011, 11, 432. [Google Scholar] [CrossRef]
- Shi, S.; Du, Y.; Zou, Y.; Niu, J.; Cai, Z.; Wang, X.; Qiu, F.; Ding, Y.; Yang, G.; Wu, Y.; et al. Rational Design for Nitroreductase (NTR)-Responsive Proteolysis Targeting Chimeras (PROTACs) Selectively Targeting Tumor Tissues. J. Med.Chem. 2022, 65, 5057–5071. [Google Scholar] [CrossRef]
- Parkinson, G.N.; Skelly, J.V.; Neidle, S. Crystal Structure of FMN-Dependent Nitroreductase from Escherichia coli B: A Prodrug-Activating Enzyme. J. Med. Chem. 2000, 43, 3624–3631. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Wen, S.; Zhang, Q.; Zhu, Q.; Yu, J.; Lu, W. Synthesis and Biological Evaluation of Paclitaxel and Camptothecin Prodrugs on the Basis of 2-Nitroimidazole. ACS Med. Chem. Lett. 2017, 8, 762–765. [Google Scholar] [CrossRef]
- Jin, C.; Zhang, Q.; Lu, W. Synthesis and biological evaluation of hypoxia-activated prodrugs of SN-38. Eur. J. Med. Chem. 2017, 132, 135–141. [Google Scholar] [CrossRef]
- Liang, D.; Wu, X.; Hasinoff, B.B.; Herbert, D.E.; Tranmer, G.K. Evaluation of Nitrobenzyl Derivatives of Camptothecin as Anti-Cancer Agents and Potential Hypoxia Targeting Prodrugs. Molecules 2018, 23, 2041. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.-X.; Jiao, H.; Kaizerman, J.; Stanton, T.; Evans, J.W.; Lan, L.; Lorente, G.; Banica, M.; Jung, D.; Wang, J.; et al. Potent and Highly Selective Hypoxia-Activated Achiral Phosphoramidate Mustards as Anticancer Drugs. J. Med. Chem. 2008, 51, 2412–2420. [Google Scholar] [CrossRef]
- Ji, G.; Guo, Q.; Xue, Q.; Kong, R.; Wang, S.; Lei, K.; Liu, R.; Wang, X. Novel GPR120 Agonists with Improved Pharmacokinetic Profiles for the Treatment of Type 2 Diabetes. Molecules 2021, 26, 6907. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MCF-7 | HT29 | ||||
|---|---|---|---|---|---|---|
| IC50 (µM) | IC50 (NTR-Free)/ IC50 (NTR) | IC50 (µM) | IC50 (NTR-Free)/ IC50 (NTR) | |||
| NTR-Free | NTR | NTR-Free | NTR | |||
| 5′-DFUR | 83.61 | 86.73 | 0.96 | 50.37 | 48.11 | 1.05 |
| 1c | 469.85 | 106.27 | 4.42 | 583.74 | 78.96 | 7.39 |
| 2c | 400.36 | 80.26 | 4.99 | 436.86 | 57.36 | 7.62 |
| 3c | 435.26 | 96.35 | 4.52 | 456.93 | 63.28 | 7.22 |
| 4c | 495.38 | 536.54 | 0.92 | 621.08 | 614.27 | 1.01 |
| 5c | 412.35 | 423.24 | 0.97 | 459.36 | 421.63 | 1.09 |
| 6c | 467.83 | 472.61 | 0.99 | 473.62 | 465.28 | 1.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Dong, T.; Li, X.; Xu, C.; Chen, F.; Wang, S.; Wang, X. Design, Synthesis, and Evaluation of Doxifluridine Derivatives as Nitroreductase-Responsive Anticancer Prodrugs. Molecules 2024, 29, 5077. https://doi.org/10.3390/molecules29215077
Zhang X, Dong T, Li X, Xu C, Chen F, Wang S, Wang X. Design, Synthesis, and Evaluation of Doxifluridine Derivatives as Nitroreductase-Responsive Anticancer Prodrugs. Molecules. 2024; 29(21):5077. https://doi.org/10.3390/molecules29215077
Chicago/Turabian StyleZhang, Xinmeng, Taimin Dong, Xu Li, Changjie Xu, Fanghui Chen, Shiben Wang, and Xuekun Wang. 2024. "Design, Synthesis, and Evaluation of Doxifluridine Derivatives as Nitroreductase-Responsive Anticancer Prodrugs" Molecules 29, no. 21: 5077. https://doi.org/10.3390/molecules29215077
APA StyleZhang, X., Dong, T., Li, X., Xu, C., Chen, F., Wang, S., & Wang, X. (2024). Design, Synthesis, and Evaluation of Doxifluridine Derivatives as Nitroreductase-Responsive Anticancer Prodrugs. Molecules, 29(21), 5077. https://doi.org/10.3390/molecules29215077