
The Role of Peptides in Combatting HIV Infection: Applications and Insights


Abstract
1. Human Immunodeficiency Virus (HIV): Structure and Prevalence of Infection
2. Anti-HIV Agents
3. Peptide-Based Anti-HIV Agents
3.1. Peptide-Based Entry Inhibitors
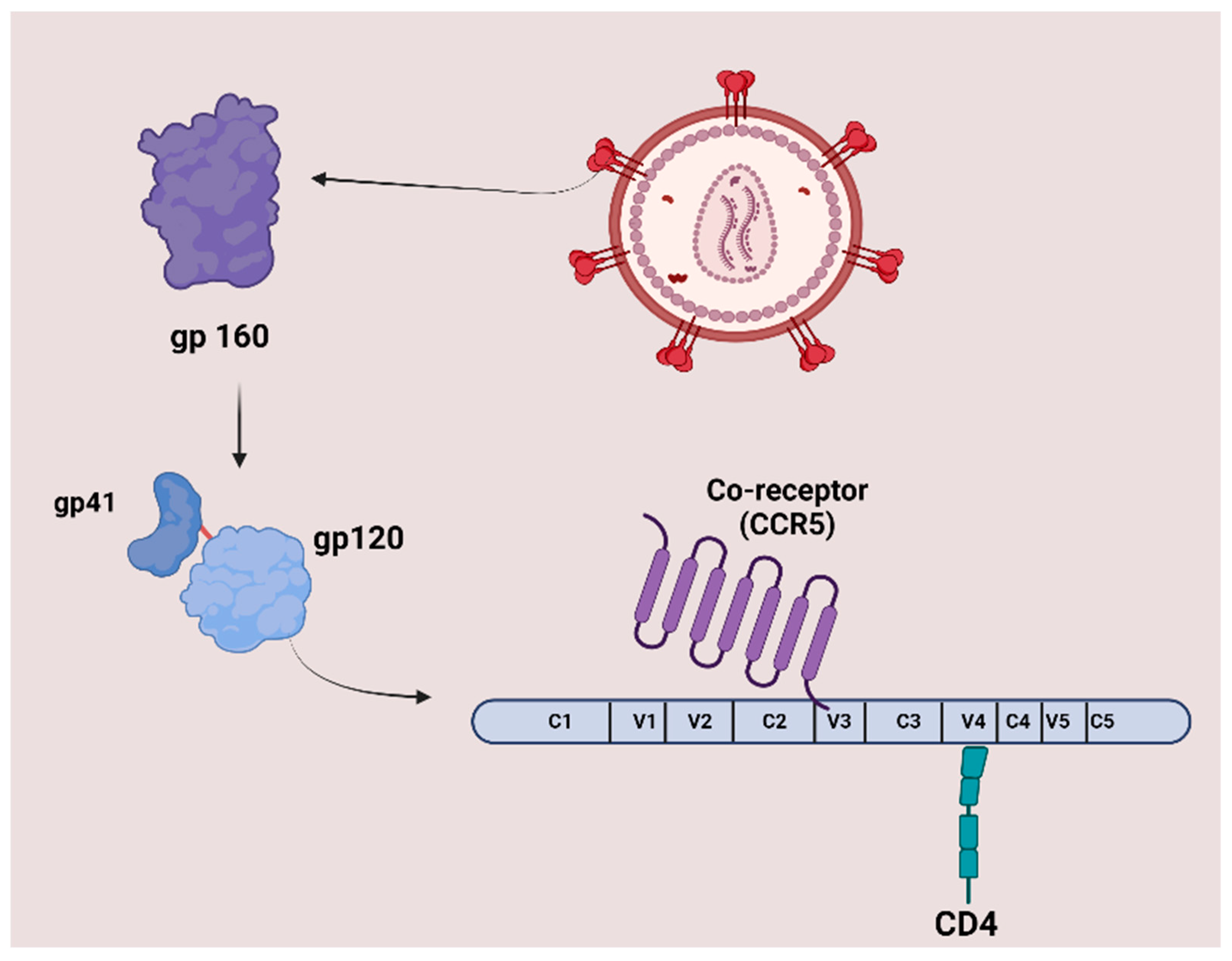
3.1.1. Peptide-Based HIV Entry Inhibitors Targeting gp120 or gp41
Inhibitors Targeting gp41 N-Heptad Repeat (NHR) Interactions
Inhibitors Targeting gp41 Heptad Repeat (CHR) Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitors Targeting gp120 CD4 Binding Site Interactions | ||
| CD4M33 | TpaNLHFCQLRCKSLGLLGKCAGSBipCACV-NH2 Tpa = Thiopropionic acid; Bip = biphenylalanine. | [43] |
| M48U1 M48U2 M48U3 | 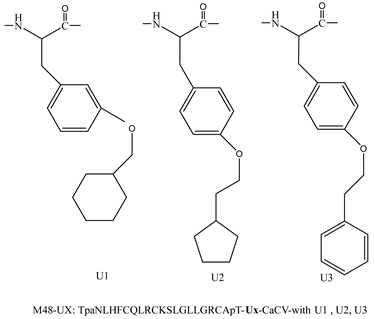 | [44,45] |
| Inhibitors Targeting gp41 N-heptad Repeat (NHR) Interactions | ||
| SJ-2176 | Amino acid residues 637–666: EWDREINNYTSLIHSLIEESQNQQEKNEQEGGC | [46,47] |
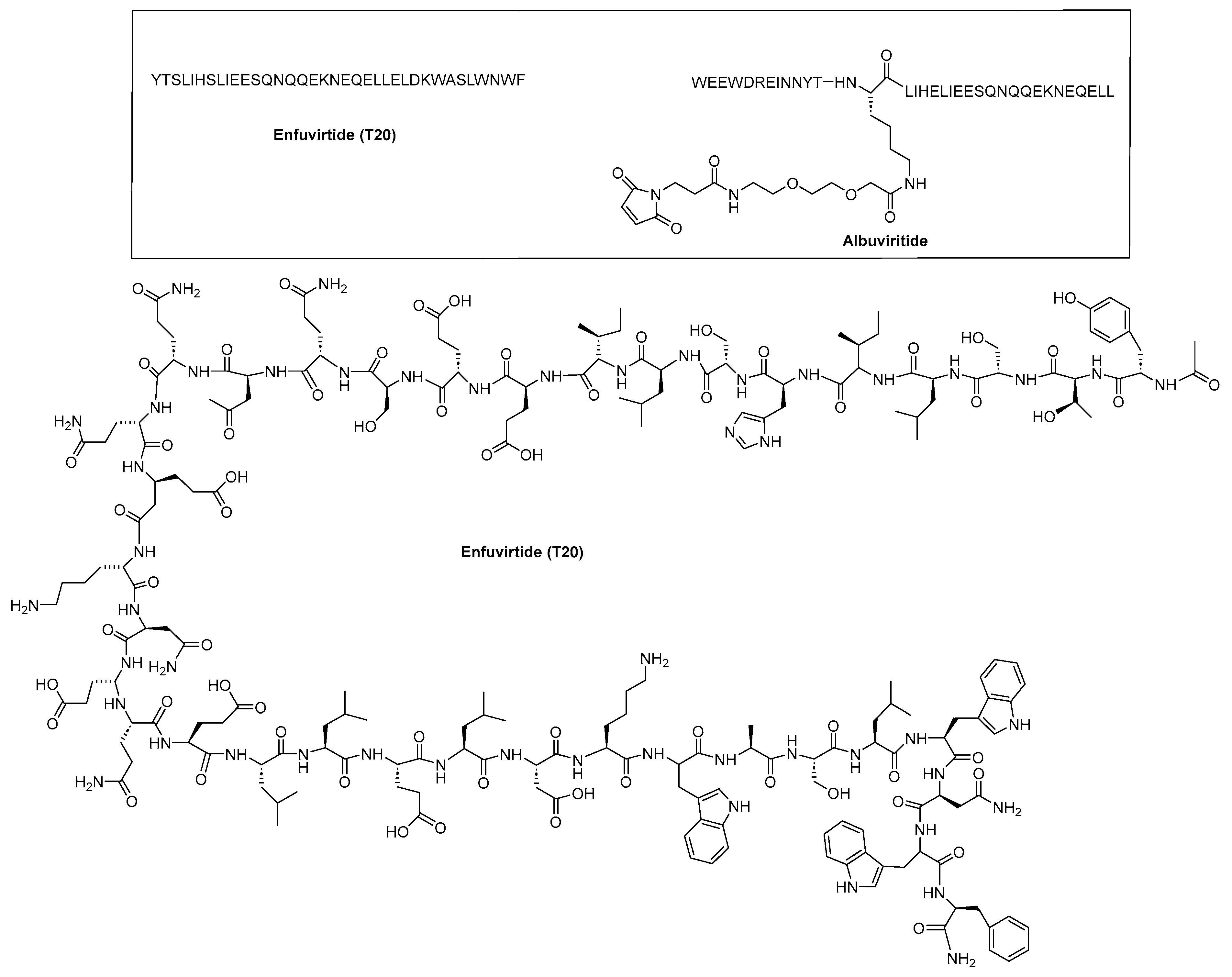
| T20 (ENF) | YTSLIHSLIEESQNQQEKNEQELLEDKWASLWNWF | [48] |
| C34 | WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL | [31,49] |
| PEG2kC34 and PEG5kC34 | PEGylated variants of C34 | [31] |
| C34-cholesterol (C34-Chol) | C34-GlySerGly-Cys(Chol) | [50] |
| CP34 RE | CP34-Arg-Glu | [35] |
| SFT | 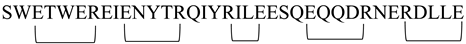 | [51] |
| SC29EK |  | [52] |
| T1249 | WQEWEQKITALLEQAQIQQEKNEYELQKLDKWASL WEWF | [53] |
| T1144 | TTWEAWDRAIAEYAARIEALLRALQEQQEKNEAALREL | [54] |
| FB006M (ABT) | 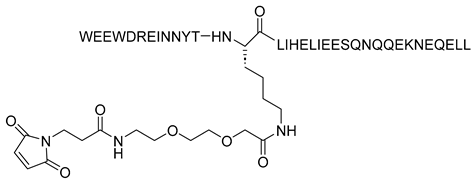 | [29,55] |
| CP32M | 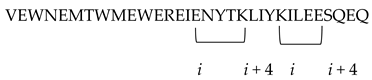 i to i + 4 position of the helical conformation. | [56] |
| HP23 | EMTWEEWEKKIEEYTKKIEEILK | [57] |
| AP3 | KKISEEQKKIQEEIKKILEESKKILEEIKKDWEEWIM | [58] |
| HP23-E6-IDL (626–656) | EMTWEEWEKKIEEYTKKIEEILKKSQNQQIDL IDL = Ile-Asp-Leu) | [59] |
| YIK-C16 | EMTWEEWEKIEEYIKKIEEILKKSQNQQIDLGSG-PEG4-K(Palm) | [60] |
| MT-WQ-IDL (626–656) | MTWEEWDKKIEEYTKKIEELIKKSQNQQIDL | [61] |
| LP-11 | EMTWEEWEKKIEEYTKKIEEILK-PEG8-K(C16) | [62] |
| LP-19 | EMTWEEWEKKVEELEKKIEELLK-PEG8-K(C16) | [32] |
| LP40 | YTSLIHSLIEESQNQQEKNEQELLELDK(C16) | [62] |
| LP46 | WQEWEQKI-------TALLEQAQIQQEKNEYELQKLDK(C16) | [63] |
| LP52 |  | [63] |
| LP-83 | WEQKIEELLKKAEEQQKKNEEELKKLEKC(Chol) | [33] |
| LP-86 | LEANIEELLKKAEEQQKKNEEELKKLEKC(Chol) | [33] |
| Hcs6ERE | IEELI^A AQ^QQRK NEEALRE L | [34] |
| N36 | SGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARIL | [35] |
| Inhibitors Targeting gp41 Heptad Repeat (CHR) Interactions | ||
| T21/DP107 | Ac-NNLLRAIEAQQHLLQLTVWGIKQLQARILAVERYLKDQ-NH2 | [36,37] |
| IZN17 | Ac-IKKEIEAIKKEQEAIKKKIEAIEK...............LLQLTVWGIKQLQARL-NH2 | [38] |
| C14linkmid | 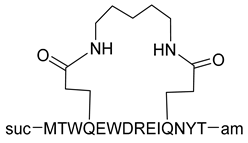 | [39] |
| C14Aib |  | [39] |
| DP-178 | YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF | [40] |
| CHR-derived α/β-peptide sequences | AcTTWEAWDRAIAEYAARIEALIRAAQEQQEKNEAALREL-NH2 AcTTWEXWDZAIAEYAXRIEXLIZAAQEQQEKNEXALZEL-NH2 | [42] |
| Inhibitors Containing a Pocket-Binding Domain | ||
| P35A4 | QEESIKKWEEWSKKIEELIKKSEELIKKIEEQIKK | [64] |
| PP24C | QEESIKKWEEWSKKIEELIKKIEEQIKK-PEG24(NH2-(PEG)24-CH2CH2COOH)-C(Chol) | [64] |
| PIE-12 | 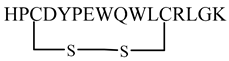 | [65] |
| Inhibitors Targeting Coreceptor CXCR4 or CCR5 Binding Interactions | ||
| pV2α-Tys | Lys-Val-Gln-Lys-Glu-Tyr(SO3H)-Ala-Leu-Phe-Tyr(SO3H)-Glu-Leu-Asp-Ile-Val-Pro-Ile-Asp | [66] |
| pCCR5-Tys | Met-Asp-Tyr-Gln-Val-Ser-Ser-Pro-Ile-Tyr(SO3H)-Asp-Ile-Asn-Tyr(SO3H)-Tyr-Thr-Ser-Glu-Pro-Ser-Gln-Lys | [66] |
| Cyclic disulfide peptides (L and D) | 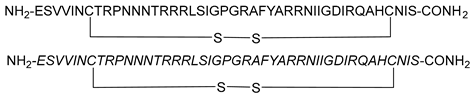 | [67] |
| EPI-X4 JM#173-C | d-I-LRWSRKC | [68] |
| Trifunctional construct | SAv-VIR-102C9-EPI-X4 | [68] |
| Irreversible Env Inactivators | ||
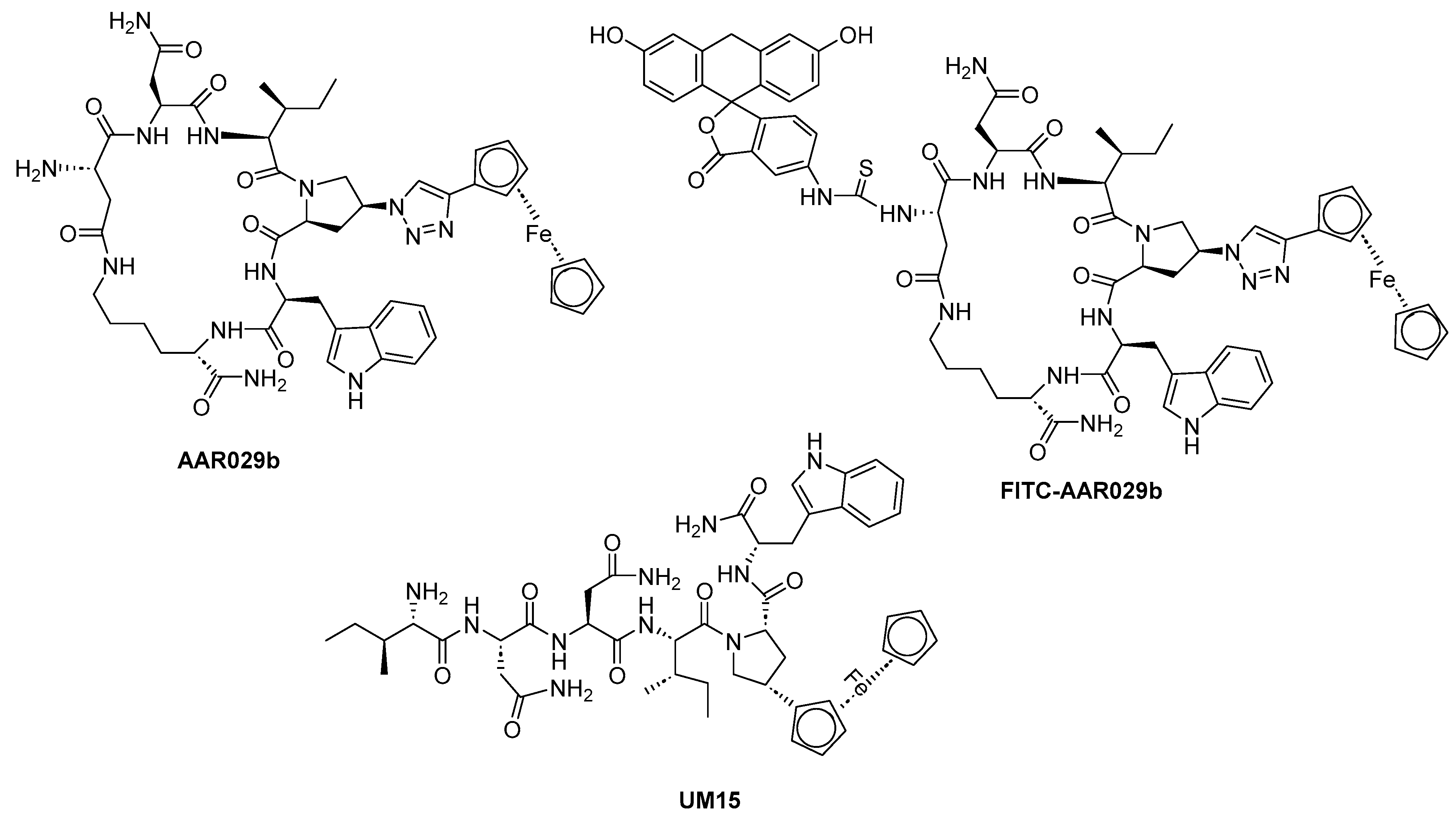
| Macrocyclic peptide triazole AAR029b | 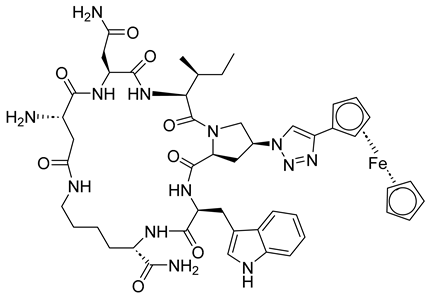 | [69] |
| FITC-AAR029b | 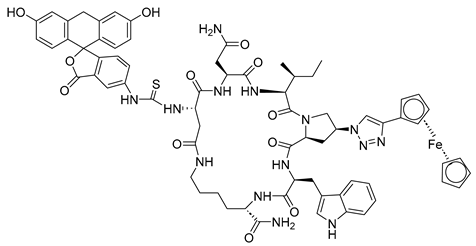 | [69] |
| UM15 | 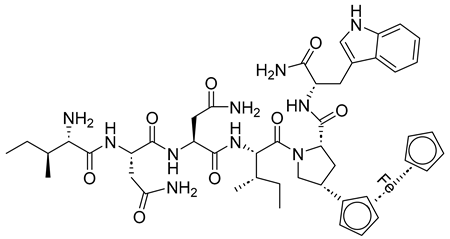 | [69] |
| Inhibitors Targeting gp41 Fusion Peptide (FP) Interactions | ||
| VIR-576 VIR-353 | (LEAIPCSIPPEFLFGKPFVF) × 2 LEAIPCSIPPCFLFNKPFVF | [70,71] |
| E1P47 E1P47-1 E1P47-2 | WILEYLWKVPFDFWRGVI ILEYLWKVPFDFWRGVIS LEYLWKVPFDFWRGVISL | [72] |
| Transmembrane Protein Sequence-Derived Anti-HIV Peptides | ||
| P3 | WQEWEQQVRYFLEANISQRLEQAQIQQEKNMYELQKLNSWDVFGNWF | [73] |
Inhibitors Containing a Pocket-Binding Domain
Inhibitors Targeting Coreceptor CXCR4 or CCR5 Binding Interactions
Irreversible Env Inactivators
Inhibitors Targeting gp41 Fusion Peptide (FP) Interactions
Transmembrane Protein Sequence-Derived Anti-HIV Peptides
3.2. Peptide-Based Vaccines
3.3. Peptides Targeting RNA
3.4. Peptide-Based Capsid Inhibitors
3.5. Defensins
3.6. Plant-Derived Anti-HIV Peptides
4. Conclusions
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Irvin, J.D.; Uckun, F.M. Pokeweed antiviral protein: Ribosome inactivation and therapeutic applications. Pharmacol. Ther. 1992, 55, 279–302. [Google Scholar] [CrossRef] [PubMed]
- Wilbourne, M.; Zhang, P. Visualizing HIV-1 Capsid and Its Interactions with Antivirals and Host Factors. Viruses 2021, 13, 246. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.G.; Summers, M.F. Structural biology of HIV. J. Mol. Biol. 1999, 285, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Fanales-Belasio, E.; Raimondo, M.; Suligoi, B.; Buttò, S. HIV virology and pathogenetic mechanisms of infection: A brief overview. Ann. Ist. Super. Sanita 2010, 46, 5–14. [Google Scholar] [CrossRef]
- Ennifar, E.; Paillart, J.C.; Bernacchi, S.; Walter, P.; Pale, P.; Decout, J.L.; Marquet, R.; Dumas, P. A structure-based approach for targeting the HIV-1 genomic RNA dimerization initiation site. Biochimie 2007, 89, 1195–1203. [Google Scholar] [CrossRef]
- Lai, Y.-T. Small Molecule HIV-1 Attachment Inhibitors: Discovery, Mode of Action and Structural Basis of Inhibition. Viruses 2021, 13, 843. [Google Scholar] [CrossRef]
- Kerman, K.; Kraatz, H.B. Electrochemical probing of HIV enzymes using ferrocene-conjugated peptides on surfaces. Analyst 2009, 134, 2400–2404. [Google Scholar] [CrossRef]
- Sever, B.; Otsuka, M.; Fujita, M.; Ciftci, H. A Review of FDA-Approved Anti-HIV-1 Drugs, Anti-Gag Compounds, and Potential Strategies for HIV-1 Eradication. Int. J. Mol. Sci. 2024, 25, 3659. [Google Scholar] [CrossRef]
- Trezza, C.; Ford, S.L.; Spreen, W.; Pan, R.; Piscitelli, S. Formulation and pharmacology of long-acting cabotegravir. Curr. Opin. HIV AIDS 2015, 10, 239–245. [Google Scholar] [CrossRef]
- Menéndez-Arias, L.; Delgado, R. Update and latest advances in antiretroviral therapy. Trends Pharmacol. Sci. 2022, 43, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zhou, R.; Huang, Y. Role of inflammasomes in HIV-1 infection and treatment. Trends Mol. Med. 2022, 28, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Carter, E.P.; G. Ang, C.; M. Chaiken, I. Peptide Triazole Inhibitors of HIV-1: Hijackers of Env Metastability. Curr. Protein Pept. Sci. 2023, 24, 59–77. [Google Scholar] [CrossRef]
- Soler, Y.; Rodriguez, M.; Austin, D.; Gineste, C.; Gelber, C.; El-Hage, N. SERPIN-Derived Small Peptide (SP16) as a Potential Therapeutic Agent against HIV-Induced Inflammatory Molecules and Viral Replication in Cells of the Central Nervous System. Cells 2023, 12, 632. [Google Scholar] [CrossRef] [PubMed]
- Huther, A.; Dietrich, U. The emergence of peptides as therapeutic drugs for the inhibition of HIV-1. AIDS Rev. 2007, 9, 208–217. [Google Scholar]
- Anderson, J.; Schiffer, C.; Lee, S.K.; Swanstrom, R. Viral protease inhibitors. Handb. Exp. Pharmacol. 2009, 189, 85–110. [Google Scholar] [PubMed]
- Abdel-Rahman, H.M.; Al-karamany, G.S.; El-Koussi, N.A.; Youssef, A.F.; Kiso, Y. HIV protease inhibitors: Peptidomimetic drugs and future perspectives. Curr. Med. Chem. 2002, 9, 1905–1922. [Google Scholar] [CrossRef]
- Li, G.R.; He, L.Y.; Liu, X.Y.; Liu, A.P.; Huang, Y.B.; Qiu, C.; Zhang, X.Y.; Xu, J.Q.; Yang, W.; Chen, Y.X. Rational design of peptides with anti-HCV/HIV activities and enhanced specificity. Chem. Biol. Drug Des. 2011, 78, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pozniak, A.; Wildfire, A.; Stanfield-Oakley, S.A.; Mosier, S.M.; Ratcliffe, D.; Workman, J.; Joall, A.; Myers, R.; Smit, E.; et al. Emergence and evolution of enfuvirtide resistance following long-term therapy involves heptad repeat 2 mutations within gp41. Antimicrob. Agents Chemother. 2005, 49, 1113–1119. [Google Scholar] [CrossRef]
- Caillat, C.; Guilligay, D.; Torralba, J.; Friedrich, N.; Nieva, J.L.; Trkola, A.; Chipot, C.J.; Dehez, F.L.; Weissenhorn, W. Structure of HIV-1 gp41 with its membrane anchors targeted by neutralizing antibodies. Elife 2021, 10, e65005. [Google Scholar] [CrossRef]
- Alkhatib, G. The biology of CCR5 and CXCR4. Curr. Opin. HIV AIDS 2009, 4, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Arrildt, K.T.; Joseph, S.B.; Swanstrom, R. The HIV-1 env protein: A coat of many colors. Curr. HIV/AIDS Rep. 2012, 9, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Wang, Q.; Xu, W.; Lu, L.; Jiang, S. Development of Protein- and Peptide-Based HIV Entry Inhibitors Targeting gp120 or gp41. Viruses 2019, 11, 705. [Google Scholar] [CrossRef]
- Pu, J.; Wang, Q.; Jiang, S. Peptide-Based HIV Entry Inhibitors. Adv. Exp. Med. Biol. 2022, 1366, 15–26. [Google Scholar] [PubMed]
- Chen, R.Y.; Kilby, J.M.; Saag, M.S. Enfuvirtide. Expert. Opin. Investig. Drugs 2002, 11, 1837–1843. [Google Scholar] [CrossRef]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; DeMasi, R.; Bolognesi, D. Enfuvirtide: The first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 2004, 3, 215–225. [Google Scholar] [CrossRef]
- Cheng, S.; Chang, X.; Wang, Y.; Gao, G.F.; Shao, Y.; Ma, L.; Li, X. Glycosylated enfuvirtide: A long-lasting glycopeptide with potent anti-HIV activity. J. Med. Chem. 2015, 58, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Wang, Y.; Zhang, Z.; Lv, X.; Gao, G.F.; Shao, Y.; Ma, L.; Li, X. Enfuvirtide-PEG conjugate: A potent HIV fusion inhibitor with improved pharmacokinetic properties. Eur. J. Med. Chem. 2016, 121, 232–237. [Google Scholar] [CrossRef]
- Chen, L.; Dai, H.; Wang, J.; Wang, M.; Ma, Y.; Ma, F.; Li, C.; Bai, L.; Du, L.; Tang, H. Comparing albuvirtide plus ritonavir-boosted lopinavir regimen to two nucleotide reverse transcriptase inhibitors plus ritonavir-boosted lopinavir in HIV-infected individuals who failed initial treatment: A retrospective comparative cohort study. Ann. Transl. Med. 2022, 10, 1125. [Google Scholar] [CrossRef]
- Su, B.; Yao, C.; Zhao, Q.X.; Cai, W.P.; Wang, M.; Lu, H.Z.; Chen, Y.Y.; Liu, L.; Wang, H.; He, Y.; et al. Efficacy and safety of the long-acting fusion inhibitor albuvirtide in antiretroviral-experienced adults with human immunodeficiency virus-1: Interim analysis of the randomized, controlled, phase 3, non-inferiority TALENT study. Chin. Med. J. 2020, 133, 2919–2927. [Google Scholar] [CrossRef]
- Wang, C.; Cheng, S.; Zhang, Y.; Ding, Y.; Chong, H.; Xing, H.; Jiang, S.; Li, X.; Ma, L. Long-Acting HIV-1 Fusion Inhibitory Peptides and their Mechanisms of Action. Viruses 2019, 11, 811. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, C.; Zhang, Y.; Chong, H.; Hu, X.; Li, D.; Xing, H.; He, Y.; Shao, Y.; Hong, K.; et al. Broad-spectrum anti-HIV activity and high drug resistance barrier of lipopeptide HIV fusion inhibitor LP-19. Front. Immunol. 2023, 14, 1199938. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chong, H.; Yu, D.; Guo, Y.; Zhou, Y.; He, Y. Design and Characterization of Cholesterylated Peptide HIV-1/2 Fusion Inhibitors with Extremely Potent and Long-Lasting Antiviral Activity. J. Virol. 2019, 93, 10-1128. [Google Scholar] [CrossRef]
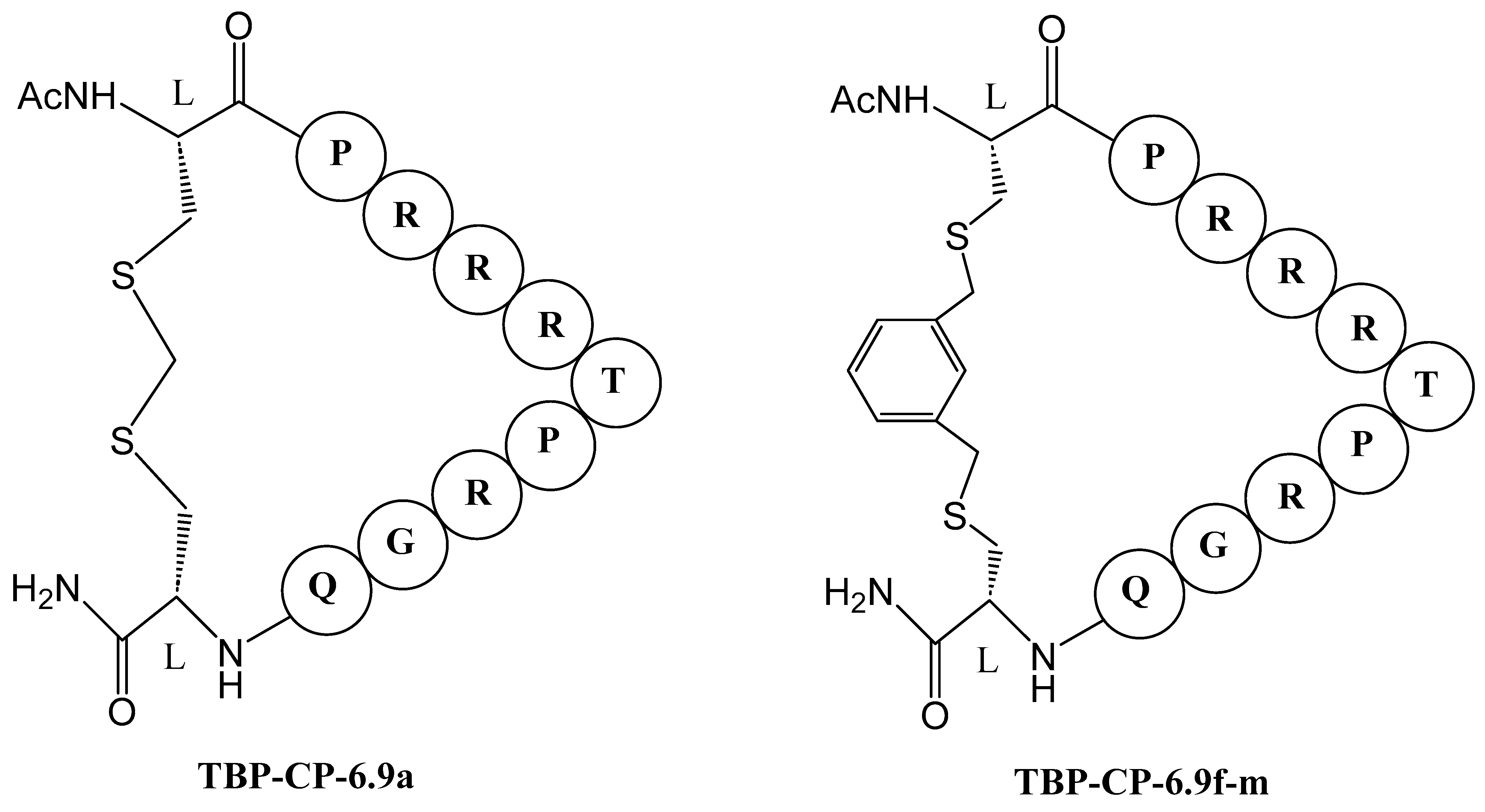
- Meng, G.; Pu, J.; Li, Y.; Han, A.; Tian, Y.; Xu, W.; Zhang, T.; Li, X.; Lu, L.; Wang, C.; et al. Design and Biological Evaluation of m-Xylene Thioether-Stapled Short Helical Peptides Targeting the HIV-1 gp41 Hexameric Coiled-Coil Fusion Complex. J. Med. Chem. 2019, 62, 8773–8783. [Google Scholar] [CrossRef] [PubMed]
- Mzoughi, O.; Teixido, M.; Planès, R.; Serrero, M.; Hamimed, I.; Zurita, E.; Moreno, M.; Granados, G.; Lakhdar-Ghazal, F.; BenMohamed, L.; et al. Trimeric heptad repeat synthetic peptides HR1 and HR2 efficiently inhibit HIV-1 entry. Biosci. Rep. 2019, 39, BSR20192196. [Google Scholar] [CrossRef]
- Su, S.B.; Gao, J.; Gong, W.; Dunlop, N.M.; Murphy, P.M.; Oppenheim, J.J.; Wang, J.M. T21/DP107, A synthetic leucine zipper-like domain of the HIV-1 envelope gp41, attracts and activates human phagocytes by using G-protein-coupled formyl peptide receptors. J. Immunol. 1999, 162, 5924–5930. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Chong, H.; Yao, X.; Su, Y.; Cui, S.; He, Y. Identification and characterization of a subpocket on the N-trimer of HIV-1 Gp41: Implication for viral entry and drug target. Aids 2015, 29, 1015–1024. [Google Scholar] [CrossRef]
- Eckert, D.M.; Kim, P.S. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc. Natl. Acad. Sci. USA 2001, 98, 11187–11192. [Google Scholar] [CrossRef]
- Sia, S.K.; Carr, P.A.; Cochran, A.G.; Malashkevich, V.N.; Kim, P.S. Short constrained peptides that inhibit HIV-1 entry. Proc. Natl. Acad. Sci. USA 2002, 99, 14664–14669. [Google Scholar] [CrossRef]
- Wild, C.T.; Shugars, D.C.; Greenwell, T.K.; McDanal, C.B.; Matthews, T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 9770–9774. [Google Scholar] [CrossRef]
- Werner, H.M.; Horne, W.S. Folding and function in α/β-peptides: Targets and therapeutic applications. Curr. Opin. Chem. Biol. 2015, 28, 75–82. [Google Scholar] [CrossRef]
- Horne, W.S.; Johnson, L.M.; Ketas, T.J.; Klasse, P.J.; Lu, M.; Moore, J.P.; Gellman, S.H. Structural and biological mimicry of protein surface recognition by α/β-peptide foldamers. Proc. Natl. Acad. Sci. USA 2009, 106, 14751–14756. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Stricher, F.; Missé, D.; Sironi, F.; Pugnière, M.; Barthe, P.; Prado-Gotor, R.; Freulon, I.; Magne, X.; Roumestand, C.; et al. Rational design of a CD4 mimic that inhibits HIV-1 entry and exposes cryptic neutralization epitopes. Nat. Biotechnol. 2003, 21, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Van Herrewege, Y.; Morellato, L.; Descours, A.; Aerts, L.; Michiels, J.; Heyndrickx, L.; Martin, L.; Vanham, G. CD4 mimetic miniproteins: Potent anti-HIV compounds with promising activity as microbicides. J. Antimicrob. Chemother. 2008, 61, 818–826. [Google Scholar] [CrossRef]
- Dereuddre-Bosquet, N.; Morellato-Castillo, L.; Brouwers, J.; Augustijns, P.; Bouchemal, K.; Ponchel, G.; Ramos, O.H.; Herrera, C.; Stefanidou, M.; Shattock, R.; et al. MiniCD4 microbicide prevents HIV infection of human mucosal explants and vaginal transmission of SHIV(162P3) in cynomolgus macaques. PLoS Pathog. 2012, 8, e1003071. [Google Scholar] [CrossRef]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. HIV-1 inhibition by a peptide. Nature 1993, 365, 113. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. Inhibition of HIV-1 infection by a fusion domain binding peptide from the HIV-1 envelope glycoprotein GP41. Biochem. Biophys. Res. Commun. 1993, 195, 533–538. [Google Scholar] [CrossRef]
- Wild, C.; Oas, T.; McDanal, C.; Bolognesi, D.; Matthews, T. A synthetic peptide inhibitor of human immunodeficiency virus replication: Correlation between solution structure and viral inhibition. Proc. Natl. Acad. Sci. USA 1992, 89, 10537–10541. [Google Scholar] [CrossRef]
- Lu, M.; Blacklow, S.C.; Kim, P.S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 1995, 2, 1075–1082. [Google Scholar] [CrossRef]
- Ingallinella, P.; Bianchi, E.; Ladwa, N.A.; Wang, Y.J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.J.; Moore, J.P.; Miller, M.D.; et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. USA 2009, 106, 5801–5806. [Google Scholar] [CrossRef]
- He, Y.; Xiao, Y.; Song, H.; Liang, Q.; Ju, D.; Chen, X.; Lu, H.; Jing, W.; Jiang, S.; Zhang, L. Design and Evaluation of Sifuvirtide, a Novel HIV-1 Fusion Inhibitor*. J. Biol. Chem. 2008, 283, 11126–11134. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Izumi, K.; Kodama, E.; Sakagami, Y.; Kajiwara, K.; Nishikawa, H.; Watanabe, K.; Sarafianos, S.G.; Oishi, S.; Fujii, N.; et al. SC29EK, a peptide fusion inhibitor with enhanced alpha-helicity, inhibits replication of human immunodeficiency virus type 1 mutants resistant to enfuvirtide. Antimicrob. Agents Chemother. 2009, 53, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.J.; Wilson, K.L.; Davison, D.K.; Freel, S.A.; Seedorff, J.E.; Wring, S.A.; Tvermoes, N.A.; Matthews, T.J.; Greenberg, M.L.; Delmedico, M.K. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. USA 2007, 104, 12772–12777. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Cai, L.; Lu, H.; Lu, L.; Jiang, S. A Novel Chimeric Protein-based HIV-1 Fusion Inhibitor Targeting gp41 Glycoprotein with High Potency and Stability*. J. Biol. Chem. 2011, 286, 28425–28434. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Yao, C.; Wang, L.; Min, W.; Xu, J.; Xiao, J.; Huang, M.; Chen, B.; Liu, B.; Li, X.; et al. An albumin-conjugated peptide exhibits potent anti-HIV activity and long in vivo half-life. Antimicrob. Agents Chemother. 2010, 54, 191–196. [Google Scholar] [CrossRef]
- He, Y.; Cheng, J.; Lu, H.; Li, J.; Hu, J.; Qi, Z.; Liu, Z.; Jiang, S.; Dai, Q. Potent HIV fusion inhibitors against Enfuvirtide-resistant HIV-1 strains. Proc. Natl. Acad. Sci. USA 2008, 105, 16332–16337. [Google Scholar] [CrossRef]
- Xiong, S.; Borrego, P.; Ding, X.; Zhu, Y.; Martins, A.; Chong, H.; Taveira, N.; He, Y. A Helical Short-Peptide Fusion Inhibitor with Highly Potent Activity against Human Immunodeficiency Virus Type 1 (HIV-1), HIV-2, and Simian Immunodeficiency Virus. J. Virol. 2016, 91, 10-1128. [Google Scholar] [CrossRef]
- Zhu, X.; Zhu, Y.; Ye, S.; Wang, Q.; Xu, W.; Su, S.; Sun, Z.; Yu, F.; Liu, Q.; Wang, C.; et al. Improved Pharmacological and Structural Properties of HIV Fusion Inhibitor AP3 over Enfuvirtide: Highlighting Advantages of Artificial Peptide Strategy. Sci. Rep. 2015, 5, 13028. [Google Scholar] [CrossRef]
- Su, S.; Ma, Z.; Hua, C.; Li, W.; Lu, L.; Jiang, S. Adding an Artificial Tail—Anchor to a Peptide-Based HIV-1 Fusion Inhibitor for Improvement of Its Potency and Resistance Profile. Molecules 2017, 22, 1996. [Google Scholar] [CrossRef]
- Su, S.; Rasquinha, G.; Du, L.; Wang, Q.; Xu, W.; Li, W.; Lu, L.; Jiang, S. A Peptide-Based HIV-1 Fusion Inhibitor with Two Tail-Anchors and Palmitic Acid Exhibits Substantially Improved In Vitro and Ex Vivo Anti-HIV-1 Activity and Prolonged In Vivo Half-Life. Molecules 2019, 24, 1134. [Google Scholar] [CrossRef]
- Su, S.; Zhu, Y.; Ye, S.; Qi, Q.; Xia, S.; Ma, Z.; Yu, F.; Wang, Q.; Zhang, R.; Jiang, S.; et al. Creating an Artificial Tail Anchor as a Novel Strategy To Enhance the Potency of Peptide-Based HIV Fusion Inhibitors. J. Virol. 2017, 91, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Zhang, X.; Chong, H.; Zhu, Y.; Wei, H.; Wu, X.; He, J.; Wang, X.; He, Y. Enfuvirtide (T20)-Based Lipopeptide Is a Potent HIV-1 Cell Fusion Inhibitor: Implications for Viral Entry and Inhibition. J. Virol. 2017, 91, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Zhu, Y.; Yu, D.; He, Y. Structural and Functional Characterization of Membrane Fusion Inhibitors with Extremely Potent Activity against Human Immunodeficiency Virus Type 1 (HIV-1), HIV-2, and Simian Immunodeficiency Virus. J. Virol. 2018, 92, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, X.; Li, J.; Li, Q.; Feng, S.; Lu, L.; Wang, C.; Jiang, S. Design of artificial α-helical peptides targeting both gp41 deep pocket and subpocket as potent HIV-1 fusion inhibitors. Eur. J. Med. Chem. 2022, 236, 114336. [Google Scholar] [CrossRef]
- Welch, B.D.; Francis, J.N.; Redman, J.S.; Paul, S.; Weinstock, M.T.; Reeves, J.D.; Lie, Y.S.; Whitby, F.G.; Eckert, D.M.; Hill, C.P.; et al. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J. Virol. 2010, 84, 11235–11244. [Google Scholar] [CrossRef]
- Cimbro, R.; Peterson, F.C.; Liu, Q.; Guzzo, C.; Zhang, P.; Miao, H.; Van Ryk, D.; Ambroggio, X.; Hurt, D.E.; De Gioia, L.; et al. Tyrosine-sulfated V2 peptides inhibit HIV-1 infection via coreceptor mimicry. EBioMedicine 2016, 10, 45–54. [Google Scholar] [CrossRef]
- Zhu, R.; Sang, X.; Zhou, J.; Meng, Q.; Huang, L.S.M.; Xu, Y.; An, J.; Huang, Z. CXCR4 Recognition by L- and D-Peptides Containing the Full-Length V3 Loop of HIV-1 gp120. Viruses 2023, 15, 1084. [Google Scholar] [CrossRef]
- Schauenburg, D.; Zech, F.; Heck, A.J.; von Maltitz, P.; Harms, M.; Führer, S.; Alleva, N.; Münch, J.; Kuan, S.L.; Kirchhoff, F.; et al. Peptide Bispecifics Inhibiting HIV-1 Infection by an Orthogonal Chemical and Supramolecular Strategy. Bioconjug Chem. 2023, 34, 1645–1652. [Google Scholar] [CrossRef]
- Aneja, R.; Grigoletto, A.; Nangarlia, A.; Rashad, A.A.; Wrenn, S.; Jacobson, J.M.; Pasut, G.; Chaiken, I. Pharmacokinetic stability of macrocyclic peptide triazole HIV-1 inactivators alone and in liposomes. J. Pept. Sci. 2019, 25, e3155. [Google Scholar] [CrossRef]
- Münch, J.; Ständker, L.; Adermann, K.; Schulz, A.; Schindler, M.; Chinnadurai, R.; Pöhlmann, S.; Chaipan, C.; Biet, T.; Peters, T.; et al. Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell 2007, 129, 263–275. [Google Scholar] [CrossRef]
- Müller, J.A.; Glöckle, A.; Gawanbacht, A.; Geyer, M.; Münch, J.; Kirchhoff, F. Reduced Susceptibility to VIRIP-Based HIV-1 Entry Inhibitors Has a High Genetic Barrier and Severe Fitness Costs. J. Virol. 2018, 92, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Gómara, M.J.; Sánchez-Merino, V.; Paús, A.; Merino-Mansilla, A.; Gatell, J.M.; Yuste, E.; Haro, I. Definition of an 18-mer Synthetic Peptide Derived from the GB virus C E1 Protein as a New HIV-1 Entry Inhibitor. Biochim. Biophys. Acta 2016, 1860, 1139–1148. [Google Scholar] [CrossRef]
- Borrego, P.; Calado, R.; Marcelino, J.M.; Pereira, P.; Quintas, A.; Barroso, H.; Taveira, N. An ancestral HIV-2/simian immunodeficiency virus peptide with potent HIV-1 and HIV-2 fusion inhibitor activity. Aids 2013, 27, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Hayn, M.; Blötz, A.; Rodríguez, A.; Vidal, S.; Preising, N.; Ständker, L.; Wiese, S.; Stürzel, C.M.; Harms, M.; Gross, R.; et al. Natural cystatin C fragments inhibit GPR15-mediated HIV and SIV infection without interfering with GPR15L signaling. Proc. Natl. Acad. Sci. USA 2021, 118, e2023776118. [Google Scholar] [CrossRef]
- Mani, S.; Bhatt, S.B.; Vasudevan, V.; Prabhu, D.; Rajamanikandan, S.; Velusamy, P.; Ramasamy, P.; Raman, P. The Updated Review on Plant Peptides and Their Applications in Human Health. Int. J. Pept. Res. Ther. 2022, 28, 135. [Google Scholar] [CrossRef]
- Lucchese, G.; Stufano, A.; Calabro, M.; Kanduc, D. Charting the peptide crossreactome between HIV-1 and the human proteome. Front. Biosci. 2011, 3, 1385–1400. [Google Scholar] [CrossRef]
- Méndez-Samperio, P. The human cathelicidin hCAP18/LL-37: A multifunctional peptide involved in mycobacterial infections. Peptides 2010, 31, 1791–1798. [Google Scholar] [CrossRef]
- Apellániz, B.; Nieva, J.L. The Use of Liposomes to Shape Epitope Structure and Modulate Immunogenic Responses of Peptide Vaccines Against HIV MPER. Adv. Protein Chem. Struct. Biol. 2015, 99, 15–54. [Google Scholar]
- Kardani, K.; Hashemi, A.; Bolhassani, A. Comparison of HIV-1 Vif and Vpu accessory proteins for delivery of polyepitope constructs harboring Nef, Gp160 and P24 using various cell penetrating peptides. PLoS ONE 2019, 14, e0223844. [Google Scholar] [CrossRef]
- Davoodi, S.; Bolhassani, A.; Namazi, F. In vivo delivery of a multiepitope peptide and Nef protein using novel cell-penetrating peptides for development of HIV-1 vaccine candidate. Biotechnol. Lett. 2021, 43, 547–559. [Google Scholar] [CrossRef]
- Rostami, B.; Irani, S.; Bolhassani, A.; Cohan, R.A. Gene and protein delivery using four cell penetrating peptides for HIV-1 vaccine development. IUBMB Life 2019, 71, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Chavali, S.S.; Mali, S.M.; Bonn, R.; Saseendran Anitha, A.; Bennett, R.P.; Smith, H.C.; Fasan, R.; Wedekind, J.E. Cyclic peptides with a distinct arginine-fork motif recognize the HIV trans-activation response RNA in vitro and in cells. J. Biol. Chem. 2021, 297, 101390. [Google Scholar] [CrossRef]
- Ali, N.; Khalil, R.; Nur, E.A.M.; Ahmed, S.; Ul-Haq, Z. Probing the mechanism of peptide binding to REV response element RNA of HIV-1; MD simulations and free energy calculations. J. Biomol. Struct. Dyn. 2022, 40, 4399–4408. [Google Scholar] [CrossRef]
- Bivalkar-Mehla, S.; Mehla, R.; Chauhan, A. Chimeric peptide-mediated siRNA transduction to inhibit HIV-1 infection. J. Drug Target. 2017, 25, 307–319. [Google Scholar] [CrossRef]
- Mizuguchi, T.; Ohashi, N.; Matsumoto, D.; Hashimoto, C.; Nomura, W.; Yamamoto, N.; Murakami, T.; Tamamura, H. Development of anti-HIV peptides based on a viral capsid protein. Biopolymers 2017, 108, e22920. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Curreli, F.; Waheed, A.A.; Mercredi, P.Y.; Mehta, M.; Bhargava, P.; Scacalossi, D.; Tong, X.; Lee, S.; Cooper, A.; et al. Dual-acting stapled peptides target both HIV-1 entry and assembly. Retrovirology 2013, 10, 136. [Google Scholar] [CrossRef]
- Dewan, V.; Liu, T.; Chen, K.M.; Qian, Z.; Xiao, Y.; Kleiman, L.; Mahasenan, K.V.; Li, C.; Matsuo, H.; Pei, D.; et al. Cyclic peptide inhibitors of HIV-1 capsid-human lysyl-tRNA synthetase interaction. ACS Chem. Biol. 2012, 7, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Bocanegra, R.; Nevot, M.; Doménech, R.; López, I.; Abián, O.; Rodríguez-Huete, A.; Cavasotto, C.N.; Velázquez-Campoy, A.; Gómez, J.; Martínez, M.; et al. Rationally designed interfacial peptides are efficient in vitro inhibitors of HIV-1 capsid assembly with antiviral activity. PLoS ONE 2011, 6, e23877. [Google Scholar] [CrossRef]
- Sticht, J.; Humbert, M.; Findlow, S.; Bodem, J.; Müller, B.; Dietrich, U.; Werner, J.; Kräusslich, H.G. A peptide inhibitor of HIV-1 assembly in vitro. Nat. Struct. Mol. Biol. 2005, 12, 671–677. [Google Scholar] [CrossRef]
- Harms, M.; Hayn, M.; Zech, F.; Kirchhoff, F.; Münch, J. Endogenous Peptide Inhibitors of HIV Entry. Adv. Exp. Med. Biol. 2022, 1366, 65–85. [Google Scholar]
- Hu, H.; Di, B.; Tolbert, W.D.; Gohain, N.; Yuan, W.; Gao, P.; Ma, B.; He, Q.; Pazgier, M.; Zhao, L.; et al. Systematic mutational analysis of human neutrophil α-defensin HNP4. Biochim. Biophys. Acta Biomembr. 2019, 1861, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Quiñones-Mateu, M.E.; Lederman, M.M.; Feng, Z.; Chakraborty, B.; Weber, J.; Rangel, H.R.; Marotta, M.L.; Mirza, M.; Jiang, B.; Kiser, P.; et al. Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. Aids 2003, 17, F39–F48. [Google Scholar] [CrossRef] [PubMed]
- Schröder, J.M.; Harder, J. Human beta-defensin-2. Int. J. Biochem. Cell Biol. 1999, 31, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Shafee, T.M.; Lay, F.T.; Hulett, M.D.; Anderson, M.A. The Defensins Consist of Two Independent, Convergent Protein Superfamilies. Mol. Biol. Evol. 2016, 33, 2345–2356. [Google Scholar] [CrossRef]
- Wu, Z.; Cocchi, F.; Gentles, D.; Ericksen, B.; Lubkowski, J.; Devico, A.; Lehrer, R.I.; Lu, W. Human neutrophil alpha-defensin 4 inhibits HIV-1 infection in vitro. FEBS Lett. 2005, 579, 162–166. [Google Scholar] [CrossRef]
- Vera-Cruz, A.; Tanphaichitr, N.; Angel, J.B. Antimicrobial Peptide, LL-37, And Its Potential As An Anti-HIV Agent. Clin. Investig. Med. 2021, 44, E64–E71. [Google Scholar] [CrossRef]
- Agerberth, B.; Gunne, H.; Odeberg, J.; Kogner, P.; Boman, H.G.; Gudmundsson, G.H. FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc. Natl. Acad. Sci. USA 1995, 92, 195–199. [Google Scholar] [CrossRef]
- Oren, Z.; Lerman, J.C.; Gudmundsson, G.H.; Agerberth, B.; Shai, Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: Relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 1999, 341 Pt 3, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Steinstraesser, L.; Tippler, B.; Mertens, J.; Lamme, E.; Homann, H.-H.; Lehnhardt, M.; Wildner, O.; Steinau, H.-U.; Überla, K. Inhibition of early steps in the lentiviral replication cycle by cathelicidin host defense peptides. Retrovirology 2005, 2, 1–12. [Google Scholar] [CrossRef]
- Wong, J.H.; Legowska, A.; Rolka, K.; Ng, T.B.; Hui, M.; Cho, C.H.; Lam, W.W.; Au, S.W.; Gu, O.W.; Wan, D.C. Effects of cathelicidin and its fragments on three key enzymes of HIV-1. Peptides 2011, 32, 1117–1122. [Google Scholar] [CrossRef]
- Huang, Y.H.; Du, Q.; Craik, D.J. Cyclotides: Disulfide-rich peptide toxins in plants. Toxicon 2019, 172, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Jacob, B.; Vogelaar, A.; Cadenas, E.; Camarero, J.A. Using the Cyclotide Scaffold for Targeting Biomolecular Interactions in Drug Development. Molecules 2022, 27, 6430. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Natural antimicrobial peptides as promising anti-HIV candidates. Curr. Top. Pept. Protein Res. 2012, 13, 93–110. [Google Scholar]
- Ireland, D.C.; Wang, C.K.; Wilson, J.A.; Gustafson, K.R.; Craik, D.J. Cyclotides as natural anti-HIV agents. Biopolymers 2008, 90, 51–60. [Google Scholar] [CrossRef]
- Daly, N.L.; Rosengren, K.J.; Craik, D.J. Discovery, structure and biological activities of cyclotides. Adv. Drug Deliv. Rev. 2009, 61, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Conzelmann, C.; Muratspahić, E.; Tomašević, N.; Münch, J.; Gruber, C.W. In vitro Inhibition of HIV-1 by Cyclotide-Enriched Extracts of Viola tricolor. Front. Pharmacol. 2022, 13, 888961. [Google Scholar] [CrossRef]
- Velásquez, J.E.; van der Donk, W.A. Genome mining for ribosomally synthesized natural products. Curr. Opin. Chem. Biol. 2011, 15, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Hellinger, R.; Koehbach, J.; Soltis, D.E.; Carpenter, E.J.; Wong, G.K.; Gruber, C.W. Peptidomics of Circular Cysteine-Rich Plant Peptides: Analysis of the Diversity of Cyclotides from Viola tricolor by Transcriptome and Proteome Mining. J. Proteome Res. 2015, 14, 4851–4862. [Google Scholar] [CrossRef]
- Azmi, S.; Mustafa, M.; Shoaib, S.; Hussain, M.K. Structures, Functions and Therapeutic Potential of Cyclotides. J. Explor. Res. Pharmacol. 2022, 7, 234–242. [Google Scholar] [CrossRef]
- Aboye, T.L.; Ha, H.; Majumder, S.; Christ, F.; Debyser, Z.; Shekhtman, A.; Neamati, N.; Camarero, J.A. Design of a novel cyclotide-based CXCR4 antagonist with anti-human immunodeficiency virus (HIV)-1 activity. J. Med. Chem. 2012, 55, 10729–10734. [Google Scholar] [CrossRef]
- Lesniak, W.G.; Aboye, T.; Chatterjee, S.; Camarero, J.A.; Nimmagadda, S. In vivo evaluation of an engineered cyclotide as specific CXCR4 imaging reagent. Chemistry 2017, 23, 14469–14475. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, D.; Lu, T.; Jacob, B.; Abraham, S.; Shankar, P.; Poss, M.A.; Neamati, N.; Camarero, J.A. Lipidation of a bioactive cyclotide-based CXCR4 antagonist greatly improves its pharmacokinetic profile in vivo. J. Control Release 2023, 359, 26–32. [Google Scholar] [CrossRef]
- Gustafson, K.R.; Sowder, R.C., II; Henderson, L.E.; Parsons, I.C.; Kashman, Y.; Cardellina, J.H., II; McMahon, J.B.; Buckheit, R.W., Jr.; Pannell, L.K.; Boyd, M.R. Circulins A and B. Novel human immunodeficiency virus (HIV)-inhibitory macrocyclic peptides from the tropical tree Chassalia parvifolia. J. Am. Chem. Soc. 1994, 116, 9337–9338. [Google Scholar] [CrossRef]
- Seetaha, S.; Hannongbua, S.; Rattanasrisomporn, J.; Choowongkomon, K. Novel peptides with HIV-1 reverse transcriptase inhibitory activity derived from the fruits of Quercus infectoria. Chem. Biol. Drug Des. 2021, 97, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Chhikara, B.S.; Singh, N.; Poonam; Bazard, P.; Varma, R.S.; Parang, K. Nanotherapeutics and HIV: Four decades of infection canvass the quest for drug development using nanomedical technologies. Appl. NanoMedicine 2022, 2, 354. [Google Scholar]
- Shuli, Z.; Linlin, L.; Li, G.; Yinghu, Z.; Nan, S.; Haibin, W.; Hongyu, X. Bioinformatics and Computer Simulation Approaches to the Discovery and Analysis of Bioactive Peptides. Curr. Pharm. Biotechnol. 2022, 23, 1541–1555. [Google Scholar]
- Premeaux Thomas, A.; Mediouni, S.; Leda, A.; Furler Robert, L.; Valente Susana, T.; Fine Howard, A.; Nixon Douglas, F.; Ndhlovu Lishomwa, C. Next-Generation Human Cerebral Organoids as Powerful Tools To Advance NeuroHIV Research. mBio 2021, 12, 10-1128. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
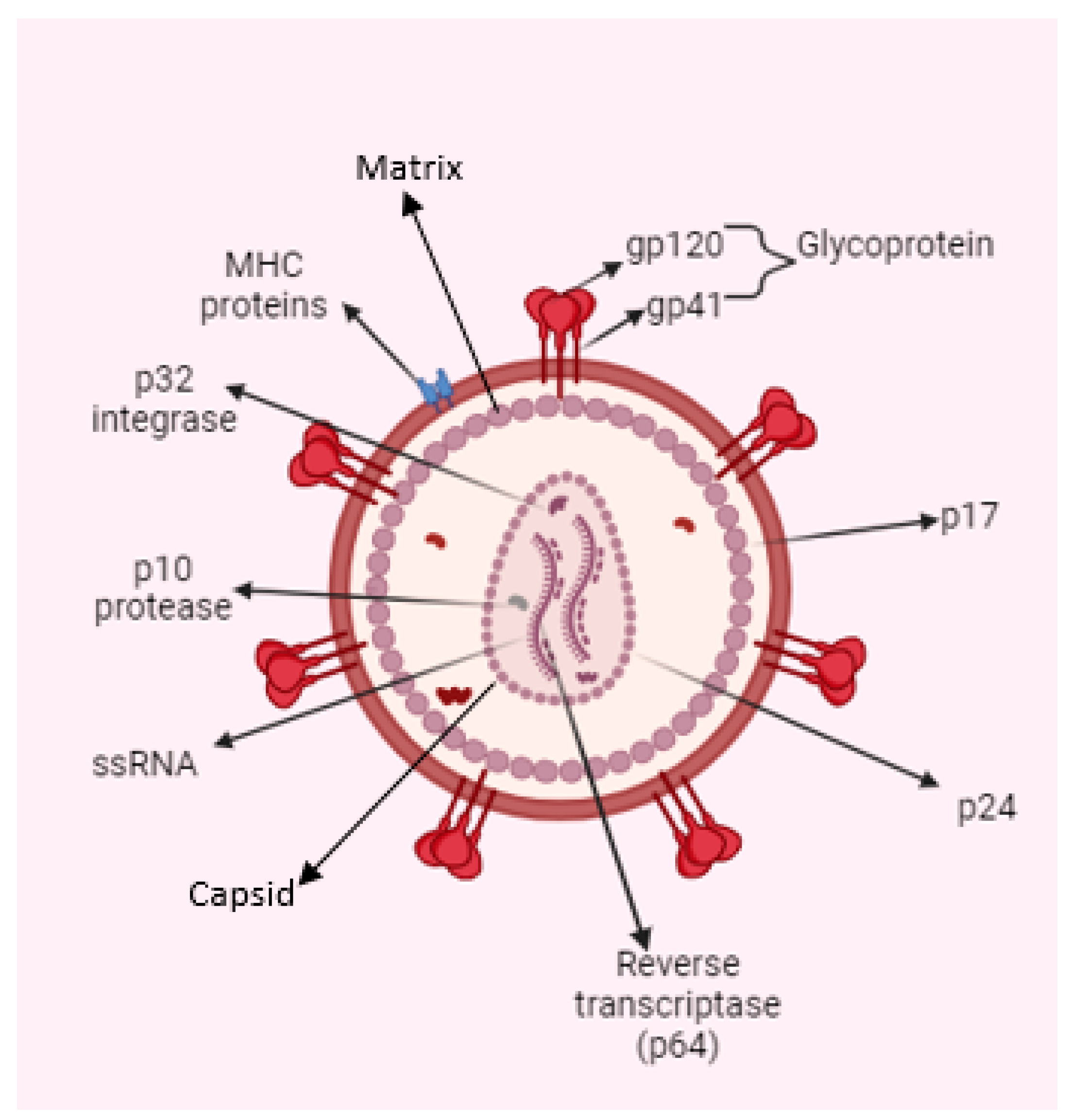


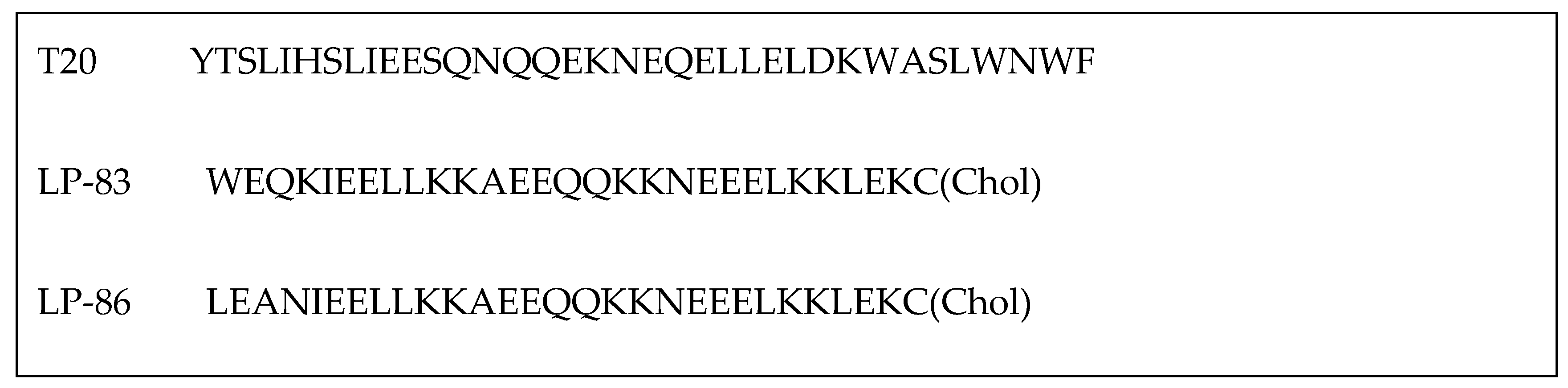

| Country | New Infections (per 1000 Uninfected Population) | AIDS-Related Deaths | People Living with HIV |
|---|---|---|---|
| Global | 0.17 (0.13–0.23) | 630,000 (480,000–880,000) | 39,000,000 (33,100,000–45,700,000) |
| African Region | 0.57 (0.41–0.8) | 380,000 (300,000–540,000) | 25,600,000 (21,600,000–30,000,000) |
| Eastern and Southern Africa | 1.07 (0.78–1.45) | 240,000 (190,000–360,000) | 20,400,000 (17,200,000–24,100,000) |
| Western and Central Africa | 0.26 (0.17–0.39) | 140,000 (110,000–180,000) | 5,100,000 (4,500,000–5,900,000) |
| Region of the Americas | 0.16 (0.13–0.19) | 41,000 (31,000–54,000) | 3,800,000 (3,400,000–4,300,000) |
| Southeast Asia Region | 0.06 (0.04–0.08) | 85,000 (62,000–120,000) | 3,900,000 (3,400,000–4,600,000) |
| European Region | 0.2 (0.16–0.23) | 52,000 (40,000–65,000) | 3,000,000 (2,600,000–3,300,000) |
| Eastern Mediterranean region | 0.07 (0.06–0.1) | 20,000 (16,000–27,000) | 490,000 (420,000–60,0000) |
| Western Pacific Region | 0.07 (0.05–0.1) | 51,000 (30,000–80,000) | 2,200,000 (1,700,000–2,800,000) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helmy, N.M.; Parang, K. The Role of Peptides in Combatting HIV Infection: Applications and Insights. Molecules 2024, 29, 4951. https://doi.org/10.3390/molecules29204951
Helmy NM, Parang K. The Role of Peptides in Combatting HIV Infection: Applications and Insights. Molecules. 2024; 29(20):4951. https://doi.org/10.3390/molecules29204951
Chicago/Turabian StyleHelmy, Naiera M., and Keykavous Parang. 2024. "The Role of Peptides in Combatting HIV Infection: Applications and Insights" Molecules 29, no. 20: 4951. https://doi.org/10.3390/molecules29204951
APA StyleHelmy, N. M., & Parang, K. (2024). The Role of Peptides in Combatting HIV Infection: Applications and Insights. Molecules, 29(20), 4951. https://doi.org/10.3390/molecules29204951