
Benzimidazole-Based Derivatives as Apoptotic Antiproliferative Agents: Design, Synthesis, Docking, and Mechanistic Studies

 ,
,  ,
,  ,
,
Abstract
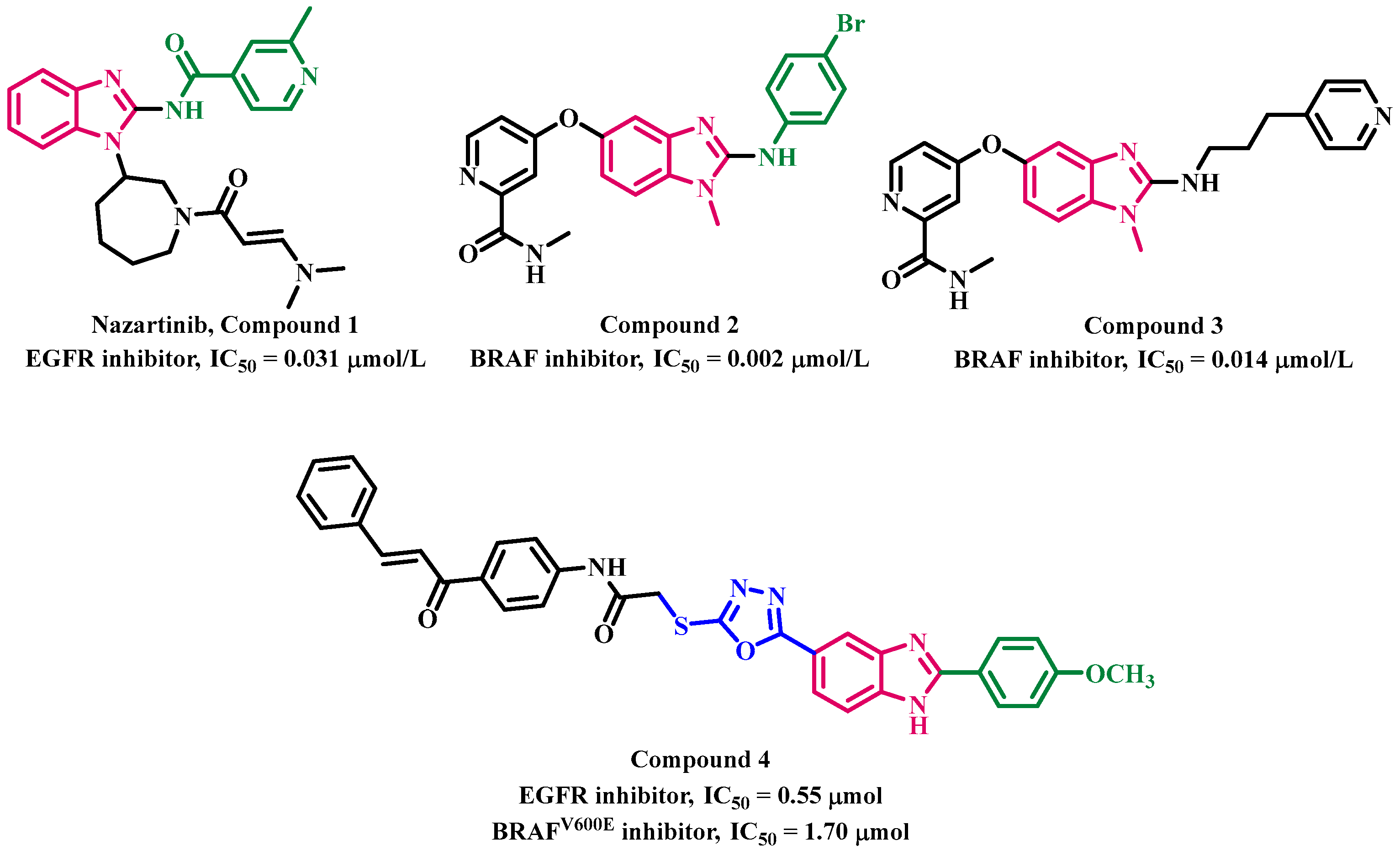
1. Introduction
2. Results and Discussion
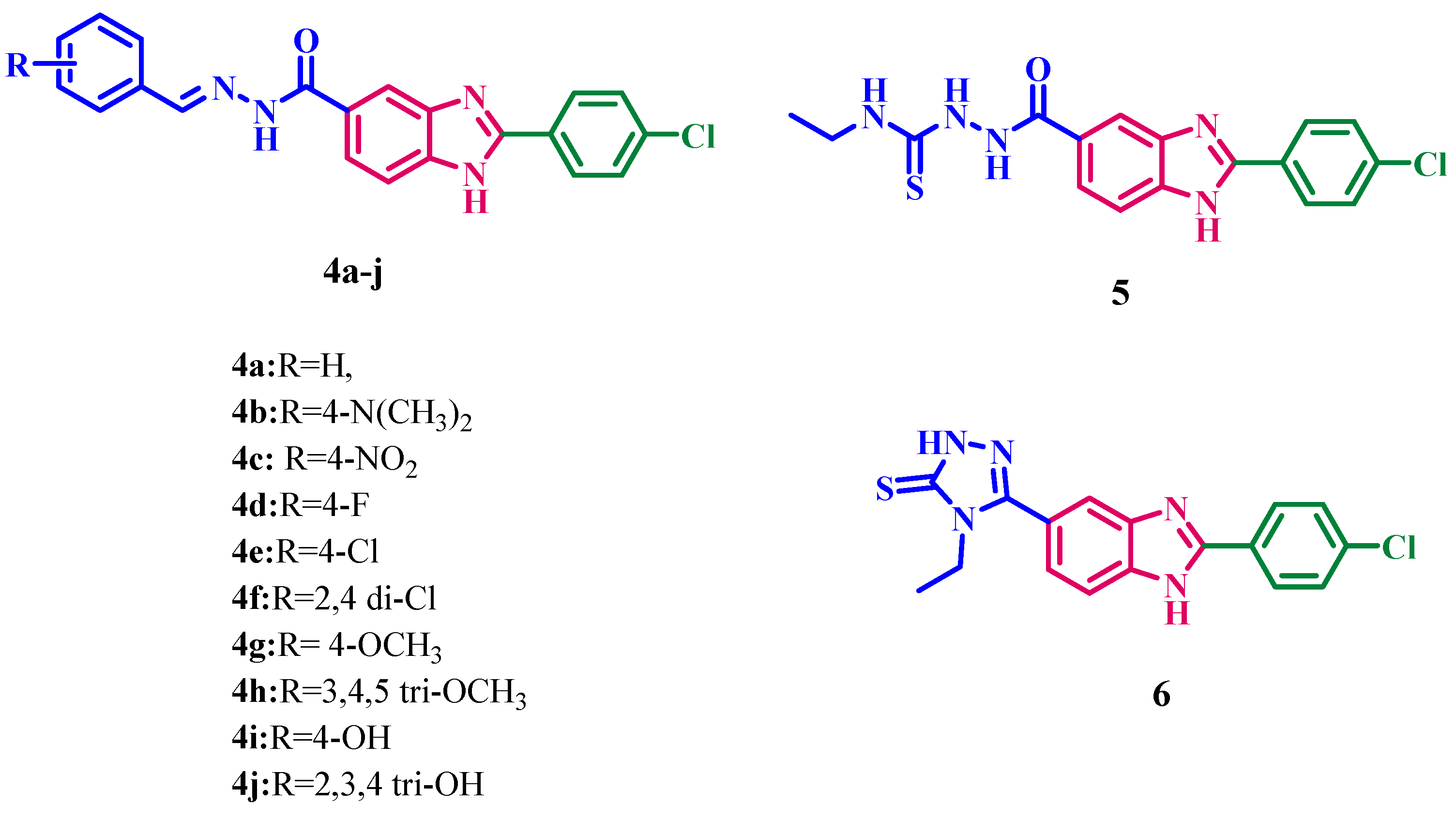
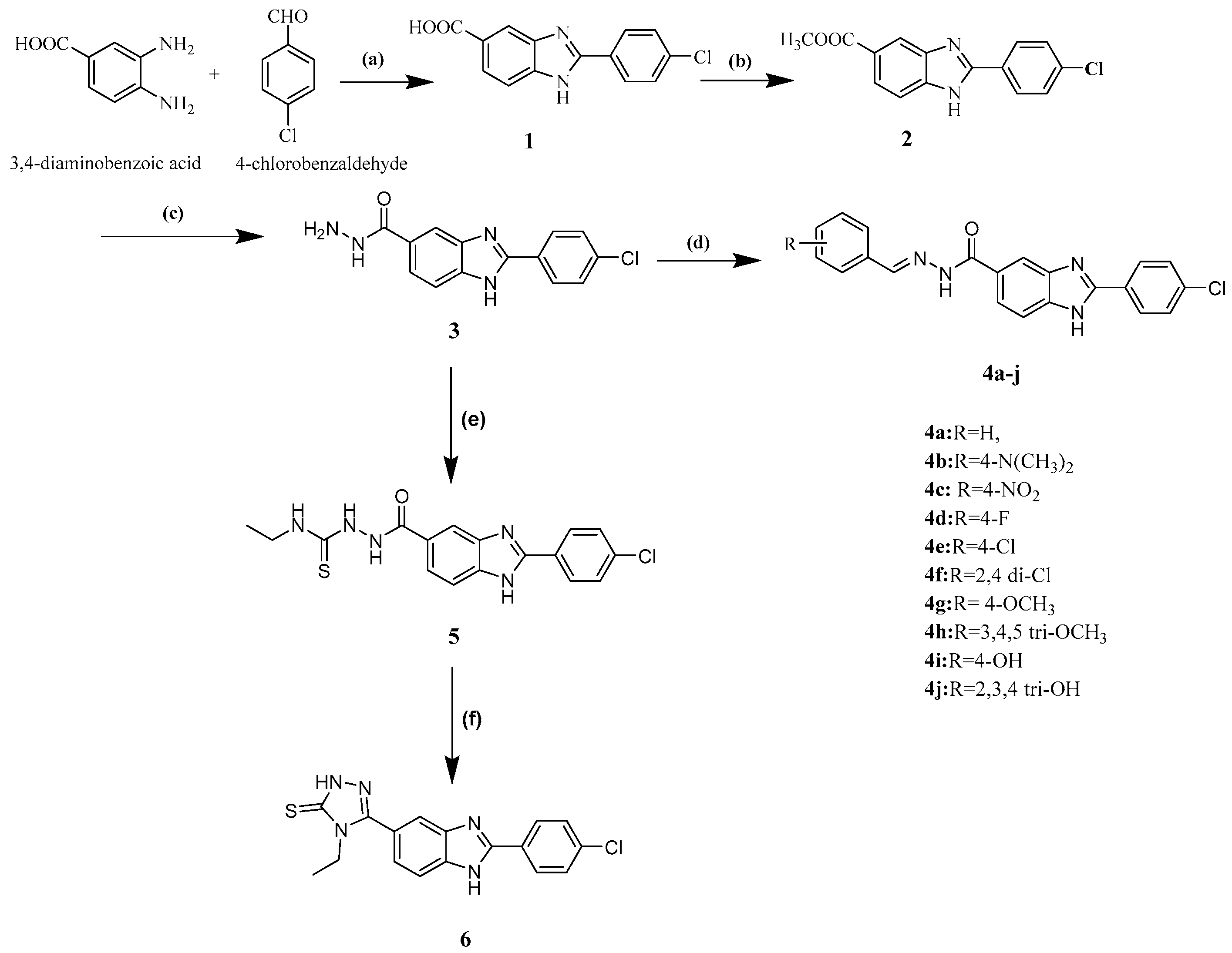
2.1. Chemistry
2.2. Biology
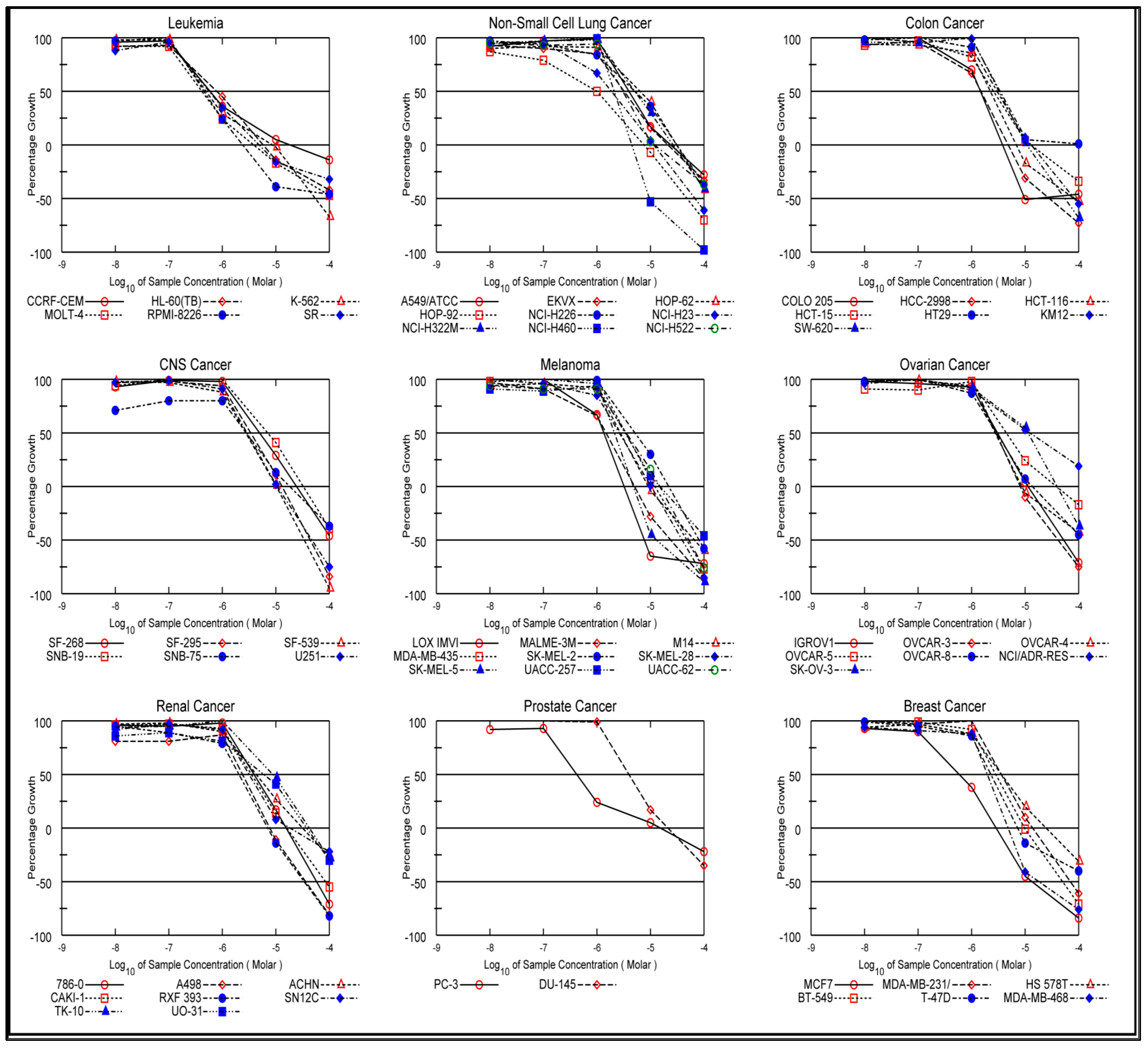
2.2.1. In Vitro Anti-Cancer Activity
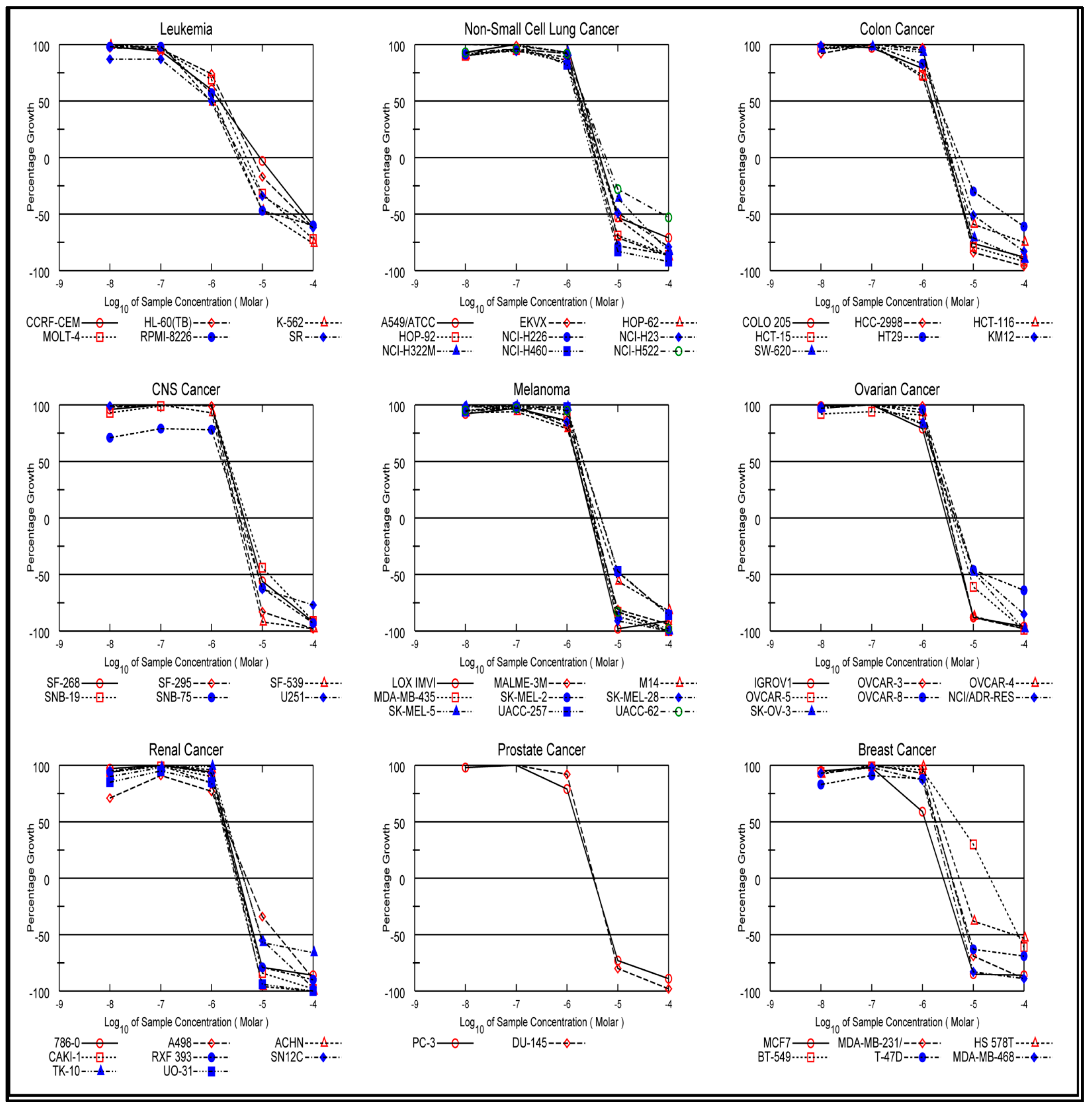
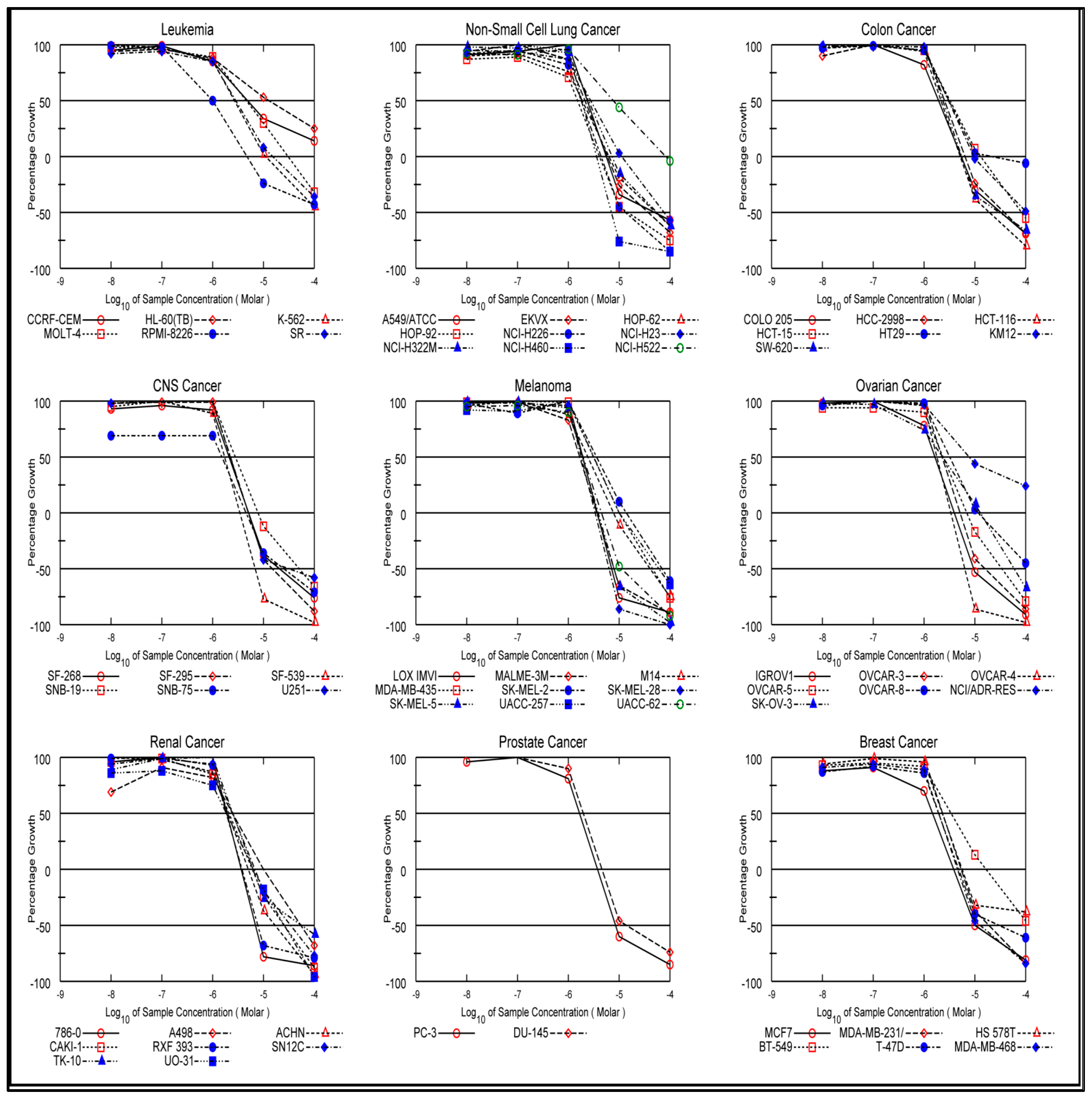
In Vitro One-Dose Assay
In Vitro Five-Dose Assay
2.2.2. EGFR Inhibitory Assay
2.2.3. BRAFV600E Inhibitory Assay
2.2.4. Apoptosis Induction Assays
Assay for Caspase-3 Activation
Assays for Caspase-8, Bax, and Bcl2 Levels
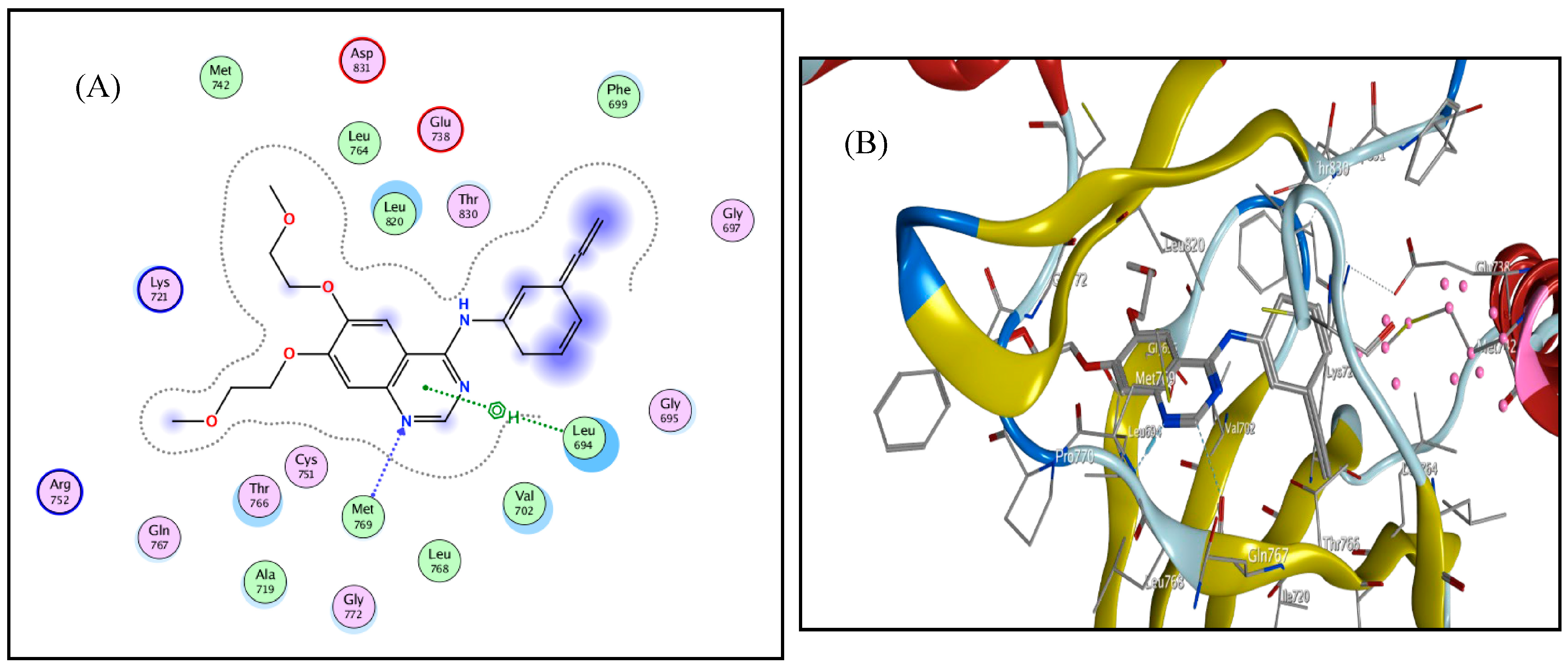
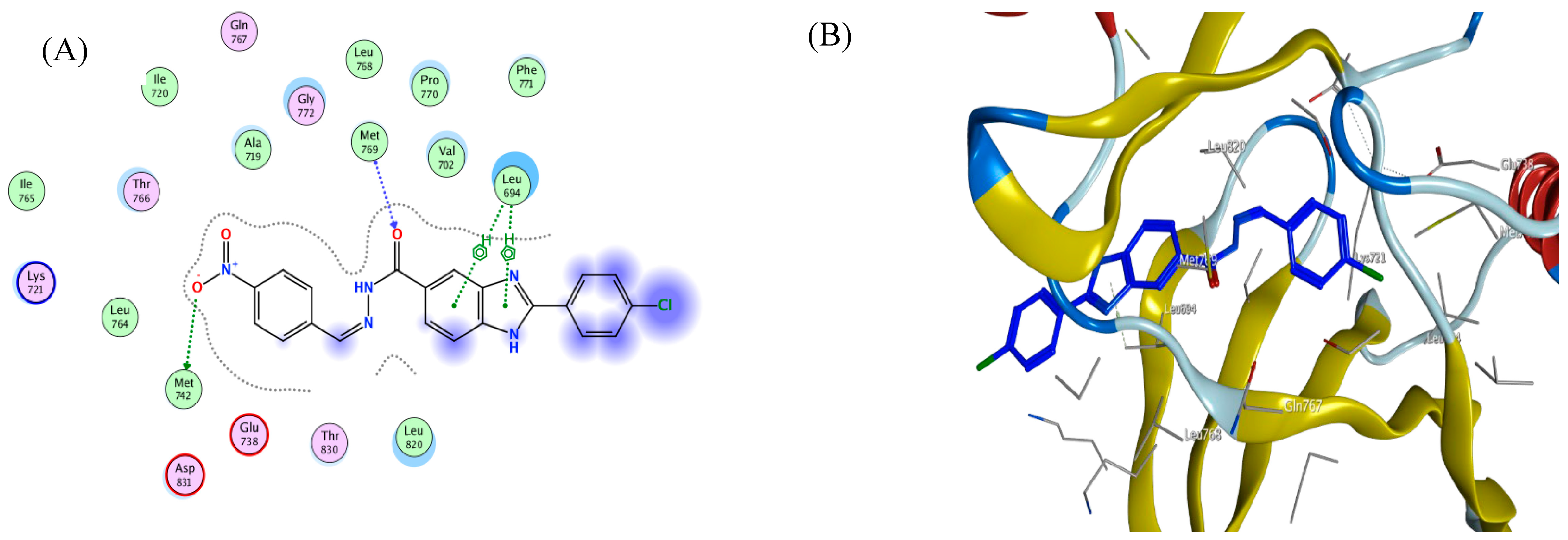
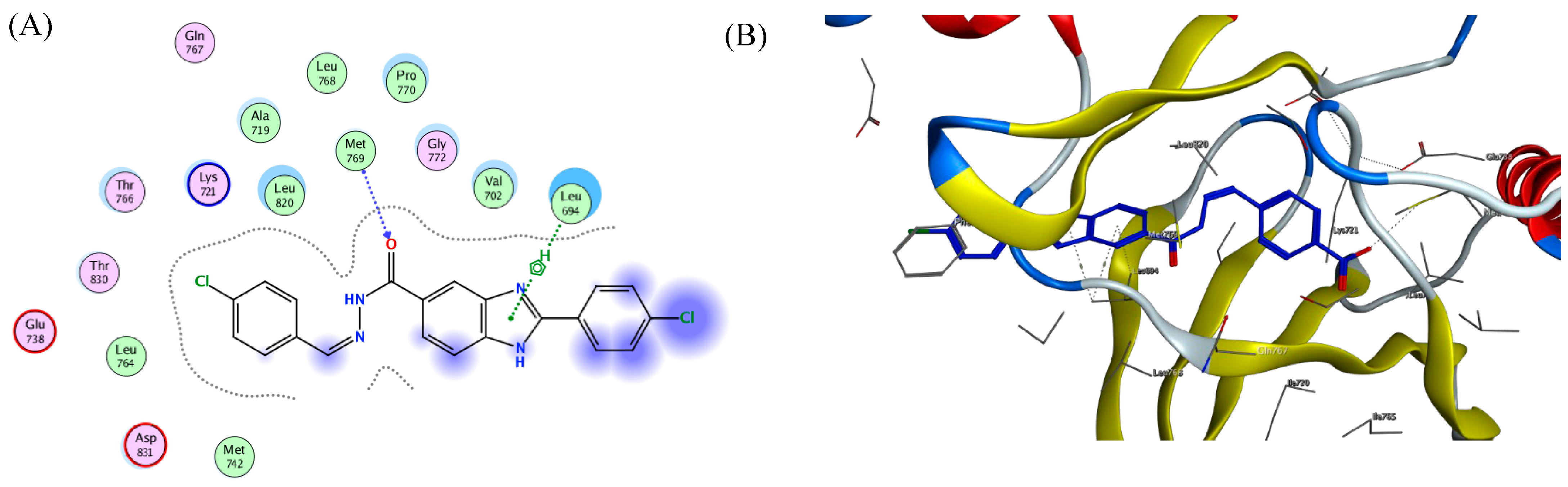
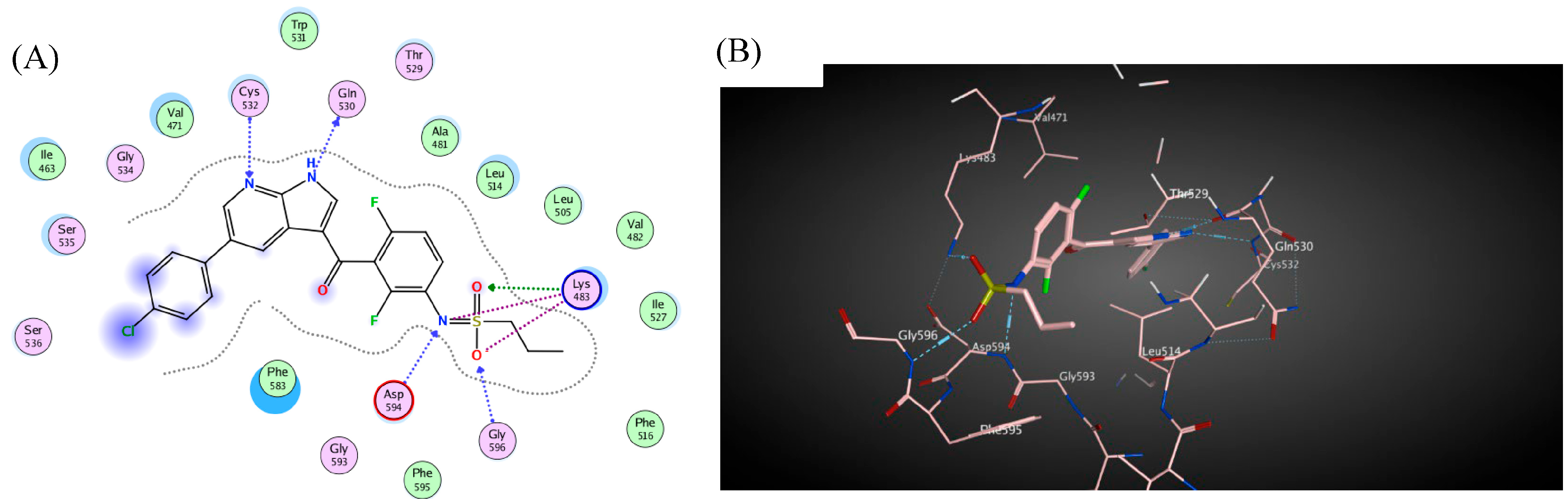
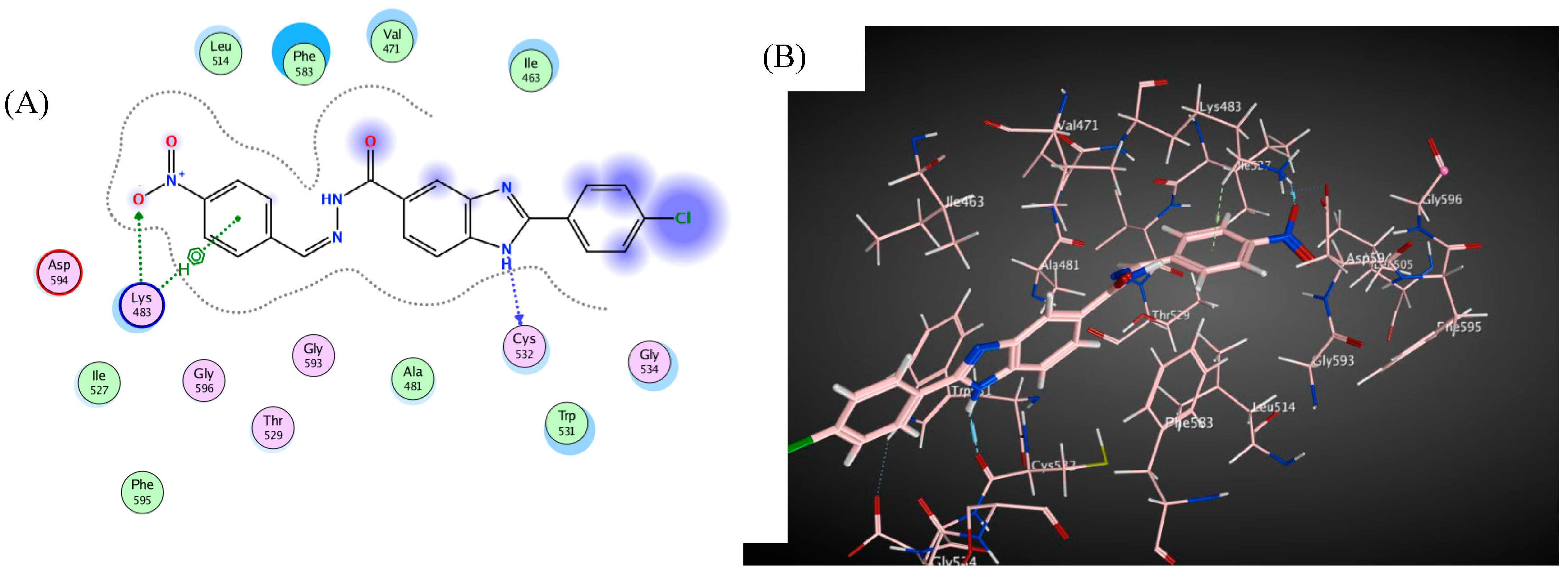
3. Docking Study
4. Conclusions
5. Experimental
5.1. Chemistry
5.1.1. General Procedure for Synthesis of N′-(substituted benzylidene)-2-(4-chlorophenyl)-1H-benzimidazole-5-carbohydrazide (4a–j)
N′-Benzylidene-2-(4-chlorophenyl)-1H-benzimidazole-5-carbohydrazide (4a)
N′-(4-(Dimethylamino)benzylidene)-2-(4-chlorophenyl)-1H-benzimidazole-5-carbohydrazide (4b)
N′-(4-Nitrobenzylidene)-2-(4-chlorophenyl)-1H-benzimidazole-5-carbohydrazide (4c)
N′-(4-Fluorobenzylidene)-2-(4-chlorophenyl)-1H-benzimidazole-5-carbohydrazide (4d)
N′-(4-Chlorobenzylidene)-2-(4-chlorophenyl)-1H-benzimidazole-5-carbohydrazide (4e)
N′-(2,4-Dichlorobenzylidene)-2-(4-chlorophenyl)-1H-benzimidazole-5-carbohydrazide (4f)
N′-(4-Methoxybenzylidene)-2-(4-chlorophenyl)-1H-benzimidazole-5-carbohydrazide (4g)
N′-(3,4,5-Trimethoxybenzylidene)-2-(4-chlorophenyl)-1H-benzimidazole-5 carbohydrazide (4h)
N′-(4-Hydroxybenzylidene)-2-(4-chlorophenyl)-1H-benzimidazole-5- carbohydrazide (4i)
N′-(3,4,5-Trihydroxybenzylidene)-2-(4-nitrophenyl)-1H-benzimidazole-5 carbohydrazide (4j)
5.1.2. General procedure for the synthesis of 2-aryl-1H-benzimidazole-5-carbonyl hydrazine-1-carbothioamide (5a)
5.1.3. General procedure for the synthesis of 4-ethyl-5-(2-(4-chlorophenyl)-1H-benzimidazol-5-yl)-2,4-dihydro-3H-1,2,4triazole-3-thione (6a)
5.2. Biology
5.2.1. In Vitro Antiproliferative Activity of Compounds Selected by the NCI
5.2.2. Assay for EGFR Inhibitory Effect
5.2.3. Assay for BRAFV600E Inhibitory Action
5.2.4. Apoptotic Markers Assays
5.3. Docking Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, X.; Yu, H. Cancer issue: Global burden of cancer. Yale J. Biol. Med. 2006, 79, 85. [Google Scholar]
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349.e15. [Google Scholar] [CrossRef]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Sargent, D.J.; Grothey, A.; Kerbel, R.S. Drug rechallenge and treatment beyond progression—Implications for drug resistance. Nat. Rev. Clin. Oncol. 2013, 10, 571–587. [Google Scholar] [CrossRef]
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol. Oncol. 2012, 6, 155–176. [Google Scholar] [CrossRef]
- Kong, X.; Xu, J.; Yang, X.; Zhai, Y.; Ji, J.; Zhai, G. Progress in tumour-targeted drug delivery based on cell-penetrating peptides. J. Drug Target. 2021, 30, 46–60. [Google Scholar] [CrossRef]
- Hodson, R. Precision medicine. Nature 2016, 537, S49. [Google Scholar] [CrossRef]
- Tan, Y.J.; Lee, Y.T.; Petersen, S.H.; Kaur, G.; Kono, K.; Tan, S.C.; Majid, A.M.S.A.; Oon, C.E. BZD9L1 sirtuin inhibitor as a potential adjuvant for sensitization of colorectal cancer cells to 5-fluorouracil. Ther. Adv. Med. Oncol. 2019, 11, 1758835919878977. [Google Scholar] [CrossRef]
- Hagar, F.F.; Abbas, S.H.; Abdelhamid, D.; Gomaa, H.A.M.; Youssif, B.G.M.; Abdel-Aziz, M. New 1,3,4-oxadiazole-chalcone/benzimidazole hybrids as potent antiproliferative agents. Arch. Pharm. 2022, 356, e2200357. [Google Scholar] [CrossRef]
- Hagar, F.F.; Abbas, S.H.; Gomaa, H.A.M.; Youssif, B.G.M.; Sayed, A.M.; Abdelhamid, D.; Abdel-Aziz, M. Chalcone/1,3,4-Oxadiazole/Benzimidazole hybrids as novel anti-proliferative agents inducing apoptosis and inhibiting EGFR & BRAFV600E. BMC Chem. 2023, 17, 116. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US FDA approved pharmaceuticals: Miniperspective. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Gaba, M.; Mohan, C. Development of drugs based on imidazole and benzimidazole bioactive heterocycles: Recent advances and future directions. Med. Chem. Res. 2015, 25, 173–210. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef]
- Subramanian, P.; Rudolf, G.C.; Kaliappan, K.P. Recent Trends in Copper-Catalyzed C−H Amination Routes to Biologically Important Nitrogen Scaffolds. Chem. Asian J. 2015, 11, 168–192. [Google Scholar] [CrossRef]
- Tan, Y.J.; Lee, Y.T.; Yeong, K.Y.; Petersen, S.H.; Kono, K.; Tan, S.C.; Oon, C.E. Anti-cancer activities of a benzimidazole compound through sirtuin inhibition in colorectal cancer. Future Med. Chem. 2018, 10, 2039–2057. [Google Scholar] [CrossRef]
- Shrivastava, N.; Naim, M.J.; Alam, M.J.; Nawaz, F.; Ahmed, S.; Alam, O. Benzimidazole scaffold as anti-cancer agent: Synthetic approaches and structure–activity relationship. Arch. Pharm. 2017, 350, e201700040. [Google Scholar] [CrossRef]
- Shimomura, I.; Yokoi, A.; Kohama, I.; Kumazaki, M.; Tada, Y.; Tatsumi, K.; Ochiya, T.; Yamamoto, Y. Drug library screen reveals benzimidazole derivatives as selective cytotoxic agents for KRAS-mutant lung cancer. Cancer Lett. 2019, 451, 11–22. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Benzimidazole and its derivatives as cancer therapeutics: The potential role from traditional to precision medicine. Acta Pharm. Sin. B 2023, 13, 478–497. [Google Scholar] [CrossRef]
- Tsatsanis, C.; Spandidos, D.A. The role of oncogenic kinases in human cancer. Int. J. Mol. Med. 2000, 5, 583–673. [Google Scholar] [CrossRef]
- Tan, D.S.-W.; Leighl, N.B.; Riely, G.J.; Yang, J.C.-H.; Sequist, L.V.; Wolf, J.; Seto, T.; Felip, E.; Aix, S.P.; Jonnaert, M.; et al. Safety and efficacy of nazartinib (EGF816) in adults with EGFR-mutant non-small-cell lung carcinoma: A multicentre, open-label, phase 1 study. Lancet Respir. Med. 2020, 8, 561–572. [Google Scholar] [CrossRef]
- Masuzawa, K.; Yasuda, H.; Hamamoto, J.; Nukaga, S.; Hirano, T.; Kawada, I.; Naoki, K.; Soejima, K.; Betsuyaku, T. Characterization of the efficacies of osimertinib and nazartinib against cells expressing clinically relevant epidermal growth factor receptor mutations. Oncotarget 2017, 8, 105479–105491. [Google Scholar] [CrossRef]
- Lelais, G.; Epple, R.; Marsilje, T.H.; Long, Y.O.; McNeill, M.; Chen, B.; Lu, W.; Anumolu, J.; Badiger, S.; Bursulaya, B. Discovery of (R, E)-N-(7-Chloro-1-(1-[4-(dimethylamino) but-2-enoyl] azepan-3-yl)-1 H-benzo [d] imidazol-2-yl)-2-methylisonicotinamide (EGF816), a Novel, Potent, and WT Sparing Covalent Inhibitor of Oncogenic (L858R, ex19del) and Resistant (T790M) EGFR Mutants for the Treatment of EGFR Mutant Non-Small-Cell Lung Cancers. J. Med. Chem. 2016, 59, 6671–6689. [Google Scholar] [PubMed]
- Kim, D.-W.; Tan, D.S.-W.; Aix, S.P.; Sequist, L.V.; Smit, E.F.; Hida, T.; Yang, J.C.-H.; Felip, E.; Seto, T.; Grohé, C. Preliminary Phase II results of a multicenter, open-label study of nazartinib (EGF816) in adult patients with treatment-naive EGFR-mutant non-small cell lung cancer (NSCLC). Am. Soc. Clin. Oncol. 2018, 36, 9094. [Google Scholar] [CrossRef]
- Jia, Y.; Juarez, J.; Li, J.; Manuia, M.; Niederst, M.J.; Tompkins, C.; Timple, N.; Vaillancourt, M.-T.; Pferdekamper, A.C.; Lockerman, E.L. EGF816 exerts anti-cancer effects in non–small cell lung cancer by irreversibly and selectively targeting primary and acquired activating mutations in the EGF receptor. Cancer Res. 2016, 76, 1591–1602. [Google Scholar] [CrossRef]
- Ramurthy, S.; Subramanian, S.; Aikawa, M.; Amiri, P.; Costales, A.; Dove, J.; Fong, S.; Jansen, J.M.; Levine, B.; Ma, S.; et al. Design and Synthesis of Orally Bioavailable Benzimidazoles as Raf Kinase Inhibitors. J. Med. Chem. 2008, 51, 7049–7052. [Google Scholar] [CrossRef] [PubMed]
- Alshammari, M.B.; Aly, A.A.; Youssif, B.G.M.; Bräse, S.; Ahmad, A.; Brown, A.B.; Ibrahim, M.A.A.; Mohamed, A.H. Design and synthesis of new thiazolidinone/uracil derivatives as antiproliferative agents targeting EGFR and/or BRAFV600E. Front. Chem. 2022, 10, 1076383. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Abou-Zied, H.A.; Beshr, E.A.; Youssif, B.G.; Hayallah, A.M.; Abdel-Aziz, M. Design, Synthesis, Antiproliferative Actions, and DFT Studies of New Bis–Pyrazoline Derivatives as Dual EGFR/BRAFV600E Inhibitors. Int. J. Mol. Sci. 2023, 24, 9104. [Google Scholar] [CrossRef] [PubMed]
- Al-Wahaibi, L.H.; El-Sheref, E.M.; Hammouda, M.M.; Youssif, B.G. One-Pot Synthesis of 1-Thia-4-azaspiro [4.4/5] alkan-3-ones via Schiff Base: Design, Synthesis, and Apoptotic Antiproliferative Properties of Dual EGFR/BRAFV600E Inhibitors. Pharmaceuticals 2023, 16, 467. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Gouda, A.M.; Abou-Ghadir, O.F.; Salem, O.I.; Ali, A.T.; Farghaly, H.S.; Abdelrahman, M.H.; Trembleau, L.; Abdu-Allah, H.H.; Youssif, B.G. Design and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAFV600E dual inhibitors. Bioorg. Chem. 2020, 104, 104260. [Google Scholar] [CrossRef] [PubMed]
- Al-Wahaibi, L.H.; Mahmoud, M.A.; Mostafa, Y.A.; Raslan, A.E.; Youssif, B.G. Novel piperine-carboximidamide hybrids: Design, synthesis, and antiproliferative activity via a multi-targeted inhibitory pathway. J. Enzym. Inhib. Med. Chem. 2023, 38, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.A.; Alshammari, M.B.; Ahmad, A.; Gomaa, H.A.M.; Youssif, B.G.M.; Bräse, S.; Ibrahim, M.A.A.; Mohamed, A.H. Design, synthesis, docking and mechanistic studies of new thiazolyl/thiazolidinylpyrimidine-2,4-dione antiproliferative agents. Arab. J. Chem. 2023, 16, 104612. [Google Scholar] [CrossRef]
- El-Lateef, H.M.A.; Elbastawesy, M.A.; Ibrahim, T.M.A.; Khalaf, M.M.; Gouda, M.; Wahba, M.G.; Zaki, I.; Morcoss, M.M. Design, Synthesis, Docking Study, and Antiproliferative Evaluation of Novel Schiff Base–Benzimidazole Hybrids with VEGFR-2 Inhibitory Activity. Molecules 2023, 28, 481. [Google Scholar] [CrossRef] [PubMed]
- Galal, S.A.; Hegab, K.H.; Kassab, A.S.; Rodriguez, M.L.; Kerwin, S.M.; El-Khamry, A.-M.A.; El Diwani, H.I. New transition metal ion complexes with benzimidazole-5-carboxylic acid hydrazides with antitumor activity. Eur. J. Med. Chem. 2009, 44, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Morcoss, M.M.; Abdelhafez, E.S.; Abdel-Rahman, H.M.; Abdel-Aziz, M.; El-Ella, A.; Dalal, A. Novel Benzimidazole/Hydrazone Derivatives as Promising Anti-cancer Lead Compounds: Design, Synthesis, and Molecular Docking Study. J. Adv. Biomed. Pharm. Sci. 2020, 3, 45–52. [Google Scholar]
- Abuilaiwi, F.A.; Laoui, T.; Al-Harthi, M.; Atieh, M. Modification and functionalization of multiwalled carbon nanotube (MWCNT) via fischer esterification. Arab. J. Sci. Eng. 2010, 35, 37–48. [Google Scholar]
- Morcoss, M.M.; Abdelhafez, E.S.M.; Ibrahem, R.A.; Abdel-Rahman, H.M.; Abdel-Aziz, M.; El-Ella, D.A.A. Design, synthesis, mechanistic studies and in silico ADME predictions of benzimidazole derivatives as novel antifungal agents. Bioorg. Chem. 2020, 101, 103956. [Google Scholar] [CrossRef]
- Al-Warhi, T.; Alqahtani, L.S.; Alsharif, G.; Abualnaja, M.; Ali, O.A.A.; Qahl, S.H.; Althagafi, H.A.E.; Alharthi, F.; Jafri, I.; Elsaid, F.G. Design, Synthesis, and Investigation of Cytotoxic Activity of cis-Vinylamide-Linked Combretastatin Analogues as Potential Anti-cancer Agents. Symmetry 2022, 14, 2088. [Google Scholar] [CrossRef]
- El-Sherief, H.A.; Youssif, B.G.; Bukhari, S.N.A.; Abdelazeem, A.H.; Abdel-Aziz, M.; Abdel-Rahman, H.M. Synthesis, anti-cancer activity and molecular modeling studies of 1, 2, 4-triazole derivatives as EGFR inhibitors. Eur. J. Med. Chem. 2018, 156, 774–789. [Google Scholar] [CrossRef]
- Wong, C.C.; Sagineedu, S.R.; Sumon, S.H.; Sidik, S.M.; Phillips, R.; Lajis, N.H.; Stanslas, J. NCI in vitro and in silico anticancer screen, cell cycle pertubation and apoptosis-inducing potential of new acylated, benzylidene and isopropylidene derivatives of andrographolide. Environ. Toxicol. Pharmacol. 2014, 38, 489–501. [Google Scholar] [CrossRef]
- Ali, A.R.; El-Bendary, E.R.; Ghaly, M.A.; Shehata, I.A. Synthesis, in vitro anti-cancer evaluation and in silico studies of novel imidazo [2,1-b] thiazole derivatives bearing pyrazole moieties. Eur. J. Med. Chem. 2014, 75, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.R.; El-Bendary, E.R.; Ghaly, M.A.; Shehata, I.A. Novel acetamidothiazole derivatives: Synthesis and in vitro anti-cancer evaluation. Eur. J. Med. Chem. 2013, 69, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Kryshchyshyn, A.; Atamanyuk, D.; Lesyk, R. Fused Thiopyrano[2,3-d]thiazole Derivatives as Potential Anti-cancer Agents. Sci. Pharm. 2012, 80, 509–530. [Google Scholar] [CrossRef]
- Mahmoud, M.A.; Mohammed, A.F.; Salem, O.I.; Gomaa, H.A.; Youssif, B.G. New 1, 3, 4-oxadiazoles linked with the 1, 2, 3-triazole moiety as antiproliferative agents targeting the EGFR tyrosine kinase. Arch. Pharm. 2022, 355, 2200009. [Google Scholar] [CrossRef] [PubMed]
- Zubair, T.; Bandyopadhyay, D. Small Molecule EGFR Inhibitors as Anti-Cancer Agents: Discovery, Mechanisms of Action, and Opportunities. Int. J. Mol. Sci. 2023, 24, 2651. [Google Scholar] [CrossRef]
- Youssif, B.G.; Gouda, A.M.; Moustafa, A.H.; Abdelhamid, A.A.; Gomaa, H.A.; Kamal, I.; Marzouk, A.A. Design and synthesis of new triarylimidazole derivatives as dual inhibitors of BRAFV600E/p38α with potential antiproliferative activity. J. Mol. Struct. 2021, 1253, 132218. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 21. [Google Scholar] [CrossRef]
- Villa-Pulgarin, J.A.; Gajate, C.; Botet, J.; Jimenez, A.; Justies, N.; Varela-M, R.E.; Cuesta-Marban, A.; Müller, I.; Modolell, M.; Revuelta, J.L. Mitochondria and lipid raft-located FOF1-ATP synthase as major therapeutic targets in the antileishmanial and anti-cancer activities of ether lipid edelfosine. PLoS Neglected Trop. Dis. 2017, 11, e0005805. [Google Scholar] [CrossRef]
- Bao, H.; Zhang, Q.; Zhu, Z.; Xu, H.; Ding, F.; Wang, M.; Du, S.; Du, Y.; Yan, Z. BHX, a novel pyrazoline derivative, inhibits breast cancer cell invasion by reversing the epithelial-mesenchymal transition and down-regulating Wnt/β-catenin signalling. Sci. Rep. 2017, 7, 9153. [Google Scholar] [CrossRef]
- Martin, S. Caspases: Executioners of apoptosis. In Pathobiology of Human Disease; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Youssif, B.G.; Mohamed, A.M.; Osman, E.E.A.; Abou-Ghadir, O.F.; Elnaggar, D.H.; Abdelrahman, M.H.; Treamblu, L.; Gomaa, H.A. 5-Chlorobenzofuran-2-carboxamides: From allosteric CB1 modulators to potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 177, 1–11. [Google Scholar] [CrossRef] [PubMed]
- AboulWafa, O.M.; Daabees, H.M.; Badawi, W.A. 2-Anilinopyrimidine derivatives: Design, synthesis, in vitro anti-proliferative activity, EGFR and ARO inhibitory activity, cell cycle analysis and molecular docking study. Bioorg. Chem. 2020, 99, 103798. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, H.A.; Shaker, M.E.; Alzarea, S.I.; Hendawy, O.; Mohamed, F.A.; Gouda, A.M.; Ali, A.T.; Morcoss, M.M.; Abdelrahman, M.H.; Trembleau, L.; et al. Optimization and SAR investigation of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg. Chem. 2022, 120, 105616. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.S.; Bokhtia, R.M.; Al-Mahmoudy, A.M.; Taher, E.S.; AlAwadh, M.A.; Elagawany, M.; Abdel-Aal, E.H.; Panda, S.; Gouda, A.M.; Asfour, H.Z.; et al. Design, synthesis and biological evaluation of novel 5-((substituted quinolin-3-yl/1-naphthyl) methylene)-3-substituted imidazolidin-2,4-dione as HIV-1 fusion inhibitors. Bioorg. Chem. 2020, 99, 103782. [Google Scholar] [CrossRef]
- Shaykoon, M.S.; Marzouk, A.A.; Soltan, O.M.; Wanas, A.S.; Radwan, M.M.; Gouda, A.M.; Youssif, B.G.; Abdel-Aziz, M. Design, synthesis and antitrypanosomal activity of heteroaryl-based 1,2,4-triazole and 1,3,4-oxadiazole derivatives. Bioorg. Chem. 2020, 100, 103933. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subpanel Cancer Cell Lines | Cell Inhibition Percent | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4a | 4b | 4c | 4d | 4e | 4f | 4g | 4h | 4i | 4j | 5 | 6 | |
| Leukemia | ||||||||||||
| CCRF-CEM | 11.20 | 6.50 | 90.13 | 78.63 | 84.49 | 48.15 | 34.69 | 63.44 | 13.58 | 45.33 | 86.90 | 0 |
| RPMI-8226 | 13.83 | 5.27 | 89.22 | 32.55 | 97.17 | 15.43 | 40.78 | 80.13 | 1.52 | 5.54 | 95.85 | 0.68 |
| HL-60(TB) | 0 | 0 | 93.42 | 62.62 | 94.23 | 5.98 | 3.67 | 55.12 | 0 | 0 | 0 | 0 |
| K-562 | 7.37 | 5.96 | 90.32 | 61.21 | 91.33 | 53.70 | 68.14 | 67.33 | 0.25 | 0 | 88.44 | 1.43 |
| MOLT-4 | 12.90 | 3.18 | 95.76 | 76.45 | 95.55 | 72.64 | 31.79 | 58.45 | 15.60 | 17.16 | 90.05 | 2.85 |
| SR | 18.05 | 2.54 | 108.49 | 79.32 | −124.18 | 54.59 | 99.35 | 54.32 | 2.26 | 51.12 | 88.91 | 17.53 |
| Non-Small Cell Lung Cancer | ||||||||||||
| A549/ATCC | 0 | 0 | 65.46 | 82.16 | 126.66 | 4.70 | 88.04 | 14.22 | 0 | 0 | 6.17 | 0 |
| EKVX | 1.38 | 2.85 | 50.97 | 26.79 | 91.77 | 13.67 | 70.56 | 4.23 | 0 | 82.89 | 18.21 | 0.44 |
| HOP-62 | 0 | 2.51 | 55.22 | 39.48 | 91.95 | 5.89 | 69.68 | 18.33 | 0 | 59.21 | 0.87 | 1.71 |
| HOP-92 | 0 | 0 | 40.32 | 15.44 | 71.87 | 0 | 64.03 | 28.33 | 0 | 92.98 | 1.09 | 0.32 |
| NCI-H226 | 5.74 | 4.67 | 45.22 | 6.86 | 0.67 | 18.84 | 26.17 | 24.03 | 20.60 | 51.83 | 9.79 | 1.97 |
| NCI-H23 | 1.85 | 1.51 | 83.75 | 38.62 | 100.37 | 4.26 | 58.45 | 46.27 | 4.44 | 93.41 | 5.79 | 4.53 |
| NCI-H322M | 0 | 0.24 | 29.93 | 22.45 | 72.31 | 0 | 44.09 | 12.45 | 0.47 | 59.69 | 3.36 | 0 |
| NCI-H460 | 0 | 2.77 | 94.56 | 63.77 | 133.56 | 1.37 | 94.09 | 17.19 | 0 | 97.46 | 1.06 | 0 |
| NCI-H522 | 1.00 | 3.02 | 66.97 | 3.87 | 36.76 | 7.79 | 17.38 | 21.72 | 9.85 | 88.43 | 5.4 | 1.85 |
| Colon Cancer | ||||||||||||
| COLO 205 | 0.76 | 0 | 97.72 | 58.71 | 123.92 | 0 | 77.14 | 54.34 | 0.44 | 0 | 0 | 0 |
| HCC-2998 | 0 | 6.31 | 85.09 | 20.98 | 96.31 | 3.25 | 48.89 | 20.02 | 0 | 14.96 | 5.54 | 0 |
| HCT-116 | 0 | 4.70 | 85.40 | 51.35 | 101.88 | 8.63 | 81.02 | 63.83 | 0 | 5.85 | 0 | 0 |
| HCT-15 | 4.81 | 0 | 70.12 | 33.19 | 95.31 | 0 | 42.48 | 3.98 | 0 | 4.51 | 5.90 | 1.62 |
| HT29 | 3.67 | 2.26 | 91.75 | 95.67 | 98.33 | 2.18 | 78.10 | 60.07 | 0 | 0 | 2.33 | 2.07 |
| KM12 | 3.80 | 2.41 | 86.97 | 35.11 | 129.12 | 1.75 | 82.72 | 38.17 | 2.85 | 13.12 | 7.97 | 0 |
| SW-620 | 0 | 0 | 87.34 | 53.71 | 128.29 | 4.30 | 75.58 | 21.33 | 0 | 5.64 | 0 | 0 |
| SF-268 | 0 | 0.73 | 51.50 | 21.80 | 62.03 | 3.63 | 80.07 | 20.31 | 13.65 | |||
| SF-295 | 13.18 | 1.06 | 78.32 | 68.72 | 149.27 | 1.19 | 138.85 | 55.97 | 0 | 56.38 | 1.33 | 1.04 |
| Continued | 56.23 | 1.95 | 12.11 | 4.31 | 0.56 | |||||||
| SF-539 | 9.57 | 5.22 | 58.34 | 12.04 | 62.90 | 5.73 | 57.39 | 29.47 | 12.25 | 3.69 | ||
| SNB-19 | 0 | 0 | 47.95 | 39.59 | 95.65 | 7.76 | 75.84 | 28.97 | 0 | 20.22 | 2.37 | 0 |
| SNB-75 | 10.65 | 0.61 | 30.71 | 2.77 | 1.60 | 12.39 | 8.15 | 13.37 | 0 | 9.91 | 7.77 | 3.91 |
| U251 | 4.35 | 0 | 77.67 | 98.38 | 115.25 | 41.02 | 92.04 | 46.76 | 0 | 43.95 | 2.36 | 0 |
| Melanoma | ||||||||||||
| LOX IMVI | 7.23 | 3.70 | 100.91 | 65.96 | 155.86 | 18.88 | 99.12 | 49.36 | 11.95 | 33.15 | 12.38 | 0 |
| MALME-3M | 1.28 | 12.86 | 92.21 | 88.22 | 158.65 | 9.55 | 93.47 | 71.57 | 21.36 | 19.90 | 1.54 | 2.41 |
| M14 | 8.89 | 3.58 | 74.09 | 39.75 | 80.33 | 17.14 | 73.78 | 57.97 | 0.44 | 15.66 | 1.67 | 0 |
| MDA-MB-435 | 0 | 0 | 52.42 | 6.12 | 61.19 | 0 | 31.49 | 35.98 | 0 | 1.11 | 0.91 | 0 |
| SK-MEL-2 | 0 | 0 | 33.28 | 16.72 | 61.09 | 2.90 | 35.90 | 16.41 | 0 | 13.30 | 0 | 0 |
| SK-MEL-28 | 0 | 0 | 54.65 | 28.10 | 109.23 | 0 | 87.53 | 53.48 | 0 | 20.39 | 0 | 0 |
| SK-MEL-5 | 4.85 | 5.02 | 97.42 | 11.02 | 63.84 | 2.92 | 66.34 | 58.08 | 4.32 | 7.75 | 9.57 | 2.04 |
| UACC-257 | 0 | 0 | 74.92 | 0 | 68.26 | 6.59 | 45.62 | 45.65 | 0 | 0 | 0 | 0 |
| UACC-62 | 0 | 1.06 | 56.35 | 11.85 | 83.15 | 0 | 69.11 | 71.28 | 5.20 | 26.16 | 1.88 | 1.04 |
| IGROV1 | 4.80 | 14.96 | 80.88 | 59.83 | 127.35 | 31.01 | 85.41 | 31.57 | 17.03 | |||
| OVCAR-3 | 0 | 0 | 88.68 | 29.34 | 152.88 | 0 | 79.87 | 38.44 | 0 | 26.51 | 14.29 | 0.16 |
| OVCAR-4 | 0 | 0 | 76.99 | 63.60 | 117.88 | 0 | 130.63 | 0 | 0 | 32.23 | 0 | 0 |
| OVCAR-5 | 0 | 0 | 46.76 | 10.84 | 76.28 | 0 | 22.66 | 0 | 0 | 25.74 | 5.13 | 0.60 |
| OVCAR-8 | 1.37 | 5.81 | 78.40 | 48.73 | 85.77 | 17.10 | 68.70 | 39.48 | 0 | 11.87 | 3.26 | 0 |
| NCI/ADR-RES | 0 | 0.43 | 22.73 | 22.98 | 58.78 | 0 | 26.43 | 0 | 0 | 22.28 | 6.74 | 0 |
| SK-OV-3 | 4.70 | 0 | 39.50 | 1.74 | 50.18 | 7.23 | 27.70 | 12.22 | 5.09 | 0 | 1.07 | 0 |
| Renal Cancer | 21.76 | 0.37 | 0 | |||||||||
| 786-0 | 11.58 | 0 | 50.97 | 87.83 | 142.12 | 0 | 96.85 | 48.80 | 0 | |||
| A498 | 22.08 | 12.86 | 100.50 | 27.11 | 43.85 | 36.67 | 33.43 | 101.95 | 5.34 | 0 | 0 | 0 |
| ACHN | 3.26 | 1.36 | 67.71 | 64.33 | 120.18 | 9.01 | 78.11 | 32.27 | 4.33 | 22.48 | 57.69 | 56.86 |
| CAKI-1 | 1.57 | 4.40 | 57.25 | 15.06 | 38.77 | 9.61 | 39.51 | 24.00 | 1.91 | 0.62 | 6.31 | 0 |
| RXF 393 | 15.54 | 7.79 | 68.40 | 22.63 | 81.08 | 12.49 | 56.23 | 63.67 | 0 | 10.39 | 17.05 | 7.78 |
| SN12C | 3.99 | 1.47 | 80.35 | 58.81 | 136.90 | 1.23 | 93.76 | 31.47 | 0 | 7.62 | 7.61 | 13.89 |
| TK-10 | 0 | 0 | 10.16 | 5.63 | 104.33 | 0 | 60.87 | 22.64 | 0 | 0 | 10.09 | 0 |
| UO-31 | 8.73 | 13.31 | 57.44 | 24.61 | 78.42 | 27.84 | 18.07 | 11.66 | 15.86 | 0 | 0 | 0 |
| Prostate Cancer | 25.13 | 6.39 | 16.76 | |||||||||
| PC-3 | 0 | 4.81 | 78.57 | 53.21 | 124.90 | 7.03 | 104.40 | 40.06 | 0 | |||
| DU-145 | 3.83 | 0 | 57.97 | 14.62 | 69.46 | 0 | 20.74 | 2.57 | 0 | 18.38 | 7.83 | 7.76 |
| Breast Cancer | 5.98 | 0 | 0 | |||||||||
| MCF7 | 23.51 | 23.84 | 99.28 | 82.56 | 119.97 | 13.41 | 83.33 | 87.88 | 25.12 | |||
| MDA-MB 231 | 0 | 15.58 | 23.15 | 36.06 | 90.52 | 11.40 | 60.16 | 0 | 2.75 | 11.50 | 22.10 | 14.58 |
| HS 578T | 7.92 | 6.32 | 63.24 | 30.02 | 67.05 | 10.43 | 67.67 | 51.70 | 5.94 | 27.19 | 20.49 | 2.36 |
| BT-549 | 4.12 | 0 | 85.96 | 0 | 0 | 0 | 43.07 | 29.28 | 0 | 16.99 | 9.75 | 1.05 |
| T-47D | 18.22 | 14.13 | 89.50 | 40.63 | 113.10 | 12.30 | 76.71 | 84.11 | 24.20 | 15.72 | 0 | 7.91 |
| MDA-MB-468 | 0 | 0 | 138.86 | 23.87 | 144.13 | 0 | 75.82 | 0 | 0 | 19.77 | 18.43 | 0.30 |
| 90.44 | 0 | 0 | ||||||||||
| Panel Name | Cell Name | GI50 | TGI b | LC50 | ||
|---|---|---|---|---|---|---|
| Conc. per Cell Line | Subpanel MID b | Selectivity Ratio (MID a/MID b) | ||||
| Leukemia | CCRF-CEM | 0.591 | 0.55 | 5.96 | 17.70 | >100 |
| HL-60(TB) | 0.797 | 5.73 | >100 | |||
| K-562 | 0.499 | 8.44 | 54.60 | |||
| MOLT-4 | 0.421 | 3.85 | >100 | |||
| RPMI-8226 | 0.461 | 2.41 | >100 | |||
| SR | 0.566 | 4.87 | >100 | |||
| Non-Small Cell Lung Cancer | A549/ATCC | 3.98 | 3.52 | 0.93 | 24.10 | >100 |
| EKVX | 3.19 | 21.20 | >100 | |||
| HOP-62 | 6.42 | 30.80 | >100 | |||
| HOP-92 | 0.985 | 7.52 | 47.60 | |||
| NCI-H226 | 5.03 | 31.20 | >100 | |||
| NCI-H23 | 1.85 | 11.50 | 67.60 | |||
| NCI-H322M | 5.04 | 26.40 | >100 | |||
| NCI-H460 | 2.10 | 4.49 | 9.61 | |||
| NCI-H522 | 3.05 | 11.70 | >100 | |||
| Colon Cancer | COLO 205 | 1.46 | 2.53 | 1.30 | 3.76 | - |
| HCC-2998 | 1.48 | 4.81 | 28.10 | |||
| HCT-116 | 2.25 | 6.90 | 82.30 | |||
| HCT-15 | 2.60 | 13.30 | >100 | |||
| HT29 | 3.02 | >100 | >100 | |||
| KM12 | 3.43 | 13.00 | 82.60 | |||
| SW-620 | 3.44 | 10.80 | 55.60 | |||
| CNS Cancer | SF-268 | 4.97 | 4.05 | 0.81 | 24.30 | >100 |
| SF-295 | 3.40 | 13.10 | 43.80 | |||
| SF-539 | 2.73 | 10.10 | 33.80 | |||
| SNB-19 | 7.48 | 32.40 | >100 | |||
| SNB-75 | 2.78 | 18.00 | >100 | |||
| U251 | 2.92 | 10.80 | 48.00 | |||
| Melanoma | LOX IMVI | 1.34 | 2.87 | 1.14 | 3.22 | 7.74 |
| MALME-3M | 1.48 | 5.00 | 23.60 | |||
| M14 | 2.73 | 9.13 | 67.10 | |||
| MDA-MB-435 | 3.57 | 12.20 | 47.60 | |||
| SK-MEL-2 | 5.10 | 21.90 | 81.30 | |||
| SK-MEL-28 | 2.60 | 10.30 | 39.00 | |||
| SK-MEL-5 | 2.11 | 4.78 | 12.80 | |||
| UACC-257 | 3.37 | 15.40 | >100 | |||
| UACC-62 | 3.51 | 14.90 | 52.20 | |||
| Ovarian Cancer | IGROV1 | 3.04 | 5.77 | 0.57 | 11.40 | 52.40 |
| OVCAR-3 | 3.25 | 8.23 | 40.90 | |||
| OVCAR-4 | 2.75 | 8.64 | >100 | |||
| OVCAR-5 | 4.43 | 37.80 | >100 | |||
| OVCAR-8 | 2.89 | 13.70 | >100 | |||
| NCI/ADR-RES | 12.60 | >100 | >100 | |||
| SK-OV-3 | 11.40 | 39.60 | >100 | |||
| Renal Cancer | 786-0 | 3.92 | 4.30 | 0.76 | 15.70 | 58.20 |
| A498 | 2.40 | 7.75 | 36.40 | |||
| ACHN | 4.36 | 31.90 | >100 | |||
| CAKI-1 | 3.49 | 16.10 | 85.40 | |||
| RXF 393 | 2.03 | 7.07 | 34.00 | |||
| SN12C | 3.14 | 18.10 | >100 | |||
| TK-10 | 8.99 | 40.23 | >100 | |||
| UO-31 | 6.05 | 38.30 | >100 | |||
| Prostate Cancer | PC-3 | 0.420 | 2.20 | 1.49 | 15.30 | >100 |
| DU-145 | 3.99 | 21.40 | >100 | |||
| Breast Cancer | MCF7 | 0.582 | 2.50 | 1.31 | 2.85 | 13.30 |
| MDA-MB-231 | 3.75 | 13.90 | 69.10 | |||
| HS 578T | 3.58 | 24.40 | >100 | |||
| BT-549 | 2.84 | 9.70 | 50.50 | |||
| T-47D | 2.29 | 7.21 | >100 | |||
| MDA-MB-468 | 1.97 | 4.79 | 17.70 | |||
| MID a | 3.28 | |||||
| Panel Name | Cell Name | GI50 | TGI b | LC50 | ||
|---|---|---|---|---|---|---|
| Conc. per Cell Line | Subpanel MID b | Selectivity Ratio (MID a/MID b) | ||||
| Leukemia | CCRF-CEM | 1.43 | 1.33 | 1.35 | 9.03 | 65.80 |
| HL-60(TB) | 1.85 | 6.53 | 54.80 | |||
| K-562 | 0.969 | 3.23 | 12.30 | |||
| MOLT-4 | 1.55 | 4.81 | 28.20 | |||
| RPMI-8226 | 1.16 | 3.52 | 16.50 | |||
| SR | 1.01 | 3.95 | 37.00 | |||
| Non-Small Cell Lung Cancer | A549/ATCC | 2.16 | 1.9 | 0.94 | 4.53 | 9.52 |
| EKVX | 1.82 | 3.68 | 7.43 | |||
| HOP-62 | 1.80 | 4.10 | 9.32 | |||
| HOP-92 | 1.67 | 3.53 | 7.47 | |||
| NCI-H226 | 1.77 | 3.48 | 6.86 | |||
| NCI-H23 | 1.93 | 4.43 | 10.80 | |||
| NCI-H322M | 2.15 | 5.26 | 19.80 | |||
| NCI-H460 | 1.56 | 3.13 | 6.30 | |||
| NCI-H522 | 2.24 | 5.85 | 77.90 | |||
| Colon Cancer | COLO 205 | 1.55 | 1.72 | 1.04 | 3.25 | 8.82 |
| HCC-2998 | 1.82 | 3.44 | 6.50 | |||
| HCT-116 | 1.45 | 3.51 | 8.52 | |||
| HCT-15 | 1.42 | 3.03 | 6.46 | |||
| HT29 | 1.97 | 5.47 | 44.10 | |||
| KM12 | 2.03 | 4.46 | 9.80 | |||
| SW-620 | 1.83 | 3.69 | 7.43 | |||
| CNS Cancer | SF-268 | 2.08 | 1.94 | 0.92 | 4.35 | 9.09 |
| SF-295 | 1.86 | 3.50 | 6.56 | |||
| SF-539 | 1.70 | 3.17 | 5.93 | |||
| SNB-19 | 2.34 | 5.06 | 13.30 | |||
| SNB-75 | 1.59 | 3.62 | 8.22 | |||
| U251 | 2.10 | 4.18 | 8.35 | |||
| Melanoma | LOX IMVI | 1.57 | 1.77 | 1.01 | 2.94 | 5.50 |
| MALME-3M | 1.55 | 3.15 | 6.41 | |||
| M14 | 1.65 | 3.84 | 8.96 | |||
| MDA-MB-435 | 1.73 | 3.35 | 6.49 | |||
| SK-MEL-2 | 2.09 | 4.64 | 11.40 | |||
| SK-MEL-28 | 1.58 | 3.04 | 5.86 | |||
| SK-MEL-5 | 1.81 | 3.39 | 6.33 | |||
| UACC-257 | 2.14 | 4.72 | 11.60 | |||
| UACC-62 | 1.78 | 3.40 | 6.49 | |||
| Ovarian Cancer | IGROV1 | 1.50 | 1.81 | 0.99 | 2.98 | 5.92 |
| OVCAR-3 | 1.84 | 3.39 | 6.28 | |||
| OVCAR-4 | 1.74 | 3.29 | 6.23 | |||
| OVCAR-5 | 1.85 | 3.97 | 8.52 | |||
| OVCAR-8 | 2.11 | 4.74 | 16.50 | |||
| NCI/ADR-RES | 1.83 | 4.43 | 12.70 | |||
| SK-OV-3 | 1.79 | 4.31 | 11.00 | |||
| Renal Cancer | 786-0 | 1.79 | 1.77 | 1.01 | 3.48 | 6.76 |
| A498 | 1.74 | 4.94 | 19.60 | |||
| ACHN | 1.73 | 3.17 | 5.78 | |||
| CAKI-1 | 1.70 | 3.29 | 6.37 | |||
| RXF 393 | 1.61 | 3.27 | 6.64 | |||
| SN12C | 1.96 | 4.26 | 9.26 | |||
| TK-10 | 2.06 | 4.30 | 8.98 | |||
| UO-31 | 1.57 | 2.99 | 5.67 | |||
| Prostate Cancer | PC-3 | 1.55 | 1.65 | 1.08 | 3.30 | 7.05 |
| DU-145 | 1.76 | 3.44 | 6.70 | |||
| Breast Cancer | MCF7 | 1.15 | 2.27 | 0.79 | 2.56 | 5.73 |
| MDA-MB-231 | 1.85 | 3.77 | 7.67 | |||
| HS 578T | 2.28 | 5.27 | 63.20 | |||
| BT-549 | 4.93 | 2.13 | 75.80 | |||
| T-47D | 1.79 | 3.83 | 8.21 | |||
| MDA-MB-468 | 1.64 | 3.23 | 6.36 | |||
| MID a | 1.79 | |||||
| Panel Name | Cell Name | GI50 | TGI b | LC50 | ||
|---|---|---|---|---|---|---|
| Conc. per Cell Line | Subpanel MID b | Selectivity Ratio (MID a/MID b) | ||||
| Leukemia | CCRF-CEM | 4.90 | 2.88 | 0.87 | >100 | >100 |
| HL-60(TB) | 1.30 | >100 | >100 | |||
| K-562 | 2.69 | 11.20 | >100 | |||
| MOLT-4 | 4.53 | 30.40 | >100 | |||
| RPMI-8226 | 0.997 | 4.77 | >100 | |||
| SR | 2.87 | 15.40 | >100 | |||
| Non-Small Cell Lung Cancer | A549/ATCC | 2.46 | 2.71 | 0.93 | 5.65 | 48.10 |
| EKVX | 2.11 | 5.84 | 36.60 | |||
| HOP-62 | 1.88 | 6.37 | 53.60 | |||
| HOP-92 | 1.52 | 4.11 | 15.10 | |||
| NCI-H226 | 1.79 | 4.43 | 13.50 | |||
| NCI-H23 | 2.74 | 11.20 | 75.90 | |||
| NCI-H322M | 2.49 | 7.20 | 55.40 | |||
| NCI-H460 | 1.83 | 3.59 | 7.05 | |||
| NCI-H522 | 7.60 | 81.50 | >100 | |||
| Colon Cancer | COLO 205 | 1.94 | 2.63 | 0.96 | 5.43 | 33.00 |
| HCC-2998 | 2.57 | 6.44 | 37.80 | |||
| HCT-116 | 2.14 | 5.12 | 18.90 | |||
| HCT-15 | 3.24 | 12.80 | 82.00 | |||
| HT29 | 3.09 | 22.10 | >100 | |||
| KM12 | 3.03 | 9.58 | >100 | |||
| SW-620 | 2.41 | 5.58 | 31.00 | |||
| CNS Cancer | SF-268 | 2.09 | 2.19 | 1.15 | 5.10 | 20.80 |
| SF-295 | 2.23 | 5.05 | 15.00 | |||
| SF-539 | 1.73 | 3.46 | 6.92 | |||
| SNB-19 | 3.13 | 7.95 | 49.60 | |||
| SNB-75 | 1.53 | 4.57 | 25.70 | |||
| U251 | 2.43 | 5.22 | 30.40 | |||
| Melanoma | LOX IMVI | 2.19 | 2.43 | 1.04 | 4.01 | 7.34 |
| MALME-3M | 1.67 | 3.63 | 7.90 | |||
| M14 | 2.41 | 7.76 | 40.50 | |||
| MDA-MB-435 | 3.11 | 9.84 | 45.50 | |||
| SK-MEL-2 | 3.59 | 13.80 | 69.70 | |||
| SK-MEL-28 | 1.79 | 3.37 | 6.35 | |||
| SK-MEL-5 | 1.89 | 3.88 | 7.95 | |||
| UACC-257 | 3.27 | 10.10 | 60.00 | |||
| UACC-62 | 1.94 | 4.47 | 10.90 | |||
| Ovarian Cancer | IGROV1 | 1.65 | 3.05 | 0.83 | 3.97 | 9.57 |
| OVCAR-3 | 2.28 | 5.14 | 16.00 | |||
| OVCAR-4 | 1.79 | 3.36 | 6.33 | |||
| OVCAR-5 | 2.35 | 6.89 | 33.50 | |||
| OVCAR-8 | 3.19 | 11.50 | >100 | |||
| NCI/ADR-RES | 7.81 | >100 | >100 | |||
| SK-OV-3 | 2.30 | 12.80 | 59.10 | |||
| Renal Cancer | 786-0 | 1.93 | 2.20 | 1.14 | 3.67 | 6.98 |
| A498 | 2.45 | 9.87 | 54.60 | |||
| ACHN | 2.00 | 5.04 | 16.90 | |||
| CAKI-1 | 2.10 | 6.03 | 25.60 | |||
| RXF 393 | 1.86 | 3.80 | 7.76 | |||
| SN12C | 2.46 | 6.79 | 33.90 | |||
| TK-10 | 2.95 | 6.59 | 56.70 | |||
| UO-31 | 1.86 | 6.38 | 25.70 | |||
| Prostate Cancer | PC-3 | 1.65 | 1.81 | 1.39 | 3.76 | 8.55 |
| DU-145 | 1.98 | 4.61 | 14.00 | |||
| Breast Cancer | MCF7 | 1.47 | 2.24 | 1.12 | 3.84 | 10.00 |
| MDA-MB-231 | 2.42 | 5.45 | 18.80 | |||
| HS 578T | 2.30 | 5.64 | >100 | |||
| BT-549 | 3.36 | 1.64 | >100 | |||
| T-47D | 1.94 | 4.83 | 30.00 | |||
| MDA-MB-468 | 1.94 | 4.55 | 12.60 | |||
| MID a | 2.52 | |||||
| Compd. No. | EGFR Inhibition IC50 ± SEM (µM) | BRAFV600E Inhibition IC50 ± SEM (µM) |
|---|---|---|
| 4c | 0.11 ± 0.01 | 0.31 ± 0.07 |
| 4d | 0.68 ± 0.05 | 0.95 ± 0.07 |
| 4e | 0.09 ± 0.01 | 0.20 ± 0.02 |
| 4g | 0.27 ± 0.03 | 0.60 ± 0.05 |
| 4h | 0.56 ± 0.05 | 0.82 ± 0.06 |
| Erlotinib | 0.08 ± 0.04 | 0.06 ± 0.02 |
| Vemurafenib | -- | 0.03 ± 0.01 |
| Compd. No. | Caspase-3 | Caspase-8 | Bax | Bcl-2 | ||||
|---|---|---|---|---|---|---|---|---|
| Conc (Pg/mL) | Fold Change | Conc (ng/mL) | Fold Change | Conc (Pg/mL) | Fold Change | Conc (ng/mL) | Fold Reduction | |
| 4c | 365 ± 12 | 7 | 1.90 | 16 | 185 | 26 | 0.80 | 4 |
| 4e | 470 ± 15 | 9 | 2.20 | 18 | 220 | 31 | 0.75 | 4 |
| Staurosporine | 260 ± 5 | 5 | 1.65 | 14 | 170 | 24 | 1.00 | 3 |
| Control | 52 | 1 | 0.12 | 1 | 7 | 1 | 3.00 | 1 |
| EGFR (PDB: 1 M17) | ||||
|---|---|---|---|---|
| Compound | Binding Affinity (kcal mol−1) | Distance from the Main Residue (in Å) | Interaction Type | Amino Acids Residue |
| 4c | −0.5 | 3.60 | H-donor | MET 742 |
| 3.30 | H-acceptor | MET 769 | ||
| 4.09 | pi-H | LEU 694 | ||
| 4.36 | pi-H | LEU 694 | ||
| 4e | −1.5 | 3.35 | H-acceptor | MET 769 |
| 3.69 | pi-H | LEU 694 | ||
| Erlotinib | −1.9 | 3.35 | H-acceptor | MET 769 |
| 3.69 | pi-H | LEU 694 | ||
| BRAFV600E (PDB: 3OG7) | ||||
|---|---|---|---|---|
| Compd. | Binding Affinity (kcal mol−1) | Distance from the Main Residue (Å) | Interaction Type | Amino Acids Residue |
| 4c | −5.4 | 2.92 | H-donor | CYS 532 |
| 3.12 | H-acceptor | LYS 483 | ||
| 3.66 | pi-H | LYS 483 | ||
| 4e | −1.9 | 3.07 | H-acceptor | CYS 532 |
| 3.52 | pi-H | CYS 532 | ||
| 3.54 | pi-H | SER 535 | ||
| Vemurafenib | −3.5 | 3.15 | H-donor | GLN 530 |
| 3.14 | H-acceptor | CYS 532 | ||
| 3.15 | H-acceptor | ASP 594 | ||
| 3.08 | H-acceptor | LYS 483 | ||
| 2.98 | H-acceptor | GLY 596 | ||
| 3.23 | Ionic | LYS 483 | ||
| 3.08 | Ionic | LYS 483 | ||
| 3.88 | Ionic | LYS 483 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Youssif, B.G.M.; Morcoss, M.M.; Bräse, S.; Abdel-Aziz, M.; Abdel-Rahman, H.M.; Abou El-Ella, D.A.; Abdelhafez, E.S.M.N. Benzimidazole-Based Derivatives as Apoptotic Antiproliferative Agents: Design, Synthesis, Docking, and Mechanistic Studies. Molecules 2024, 29, 446. https://doi.org/10.3390/molecules29020446
Youssif BGM, Morcoss MM, Bräse S, Abdel-Aziz M, Abdel-Rahman HM, Abou El-Ella DA, Abdelhafez ESMN. Benzimidazole-Based Derivatives as Apoptotic Antiproliferative Agents: Design, Synthesis, Docking, and Mechanistic Studies. Molecules. 2024; 29(2):446. https://doi.org/10.3390/molecules29020446
Chicago/Turabian StyleYoussif, Bahaa G. M., Martha M. Morcoss, Stefan Bräse, Mohamed Abdel-Aziz, Hamdy M. Abdel-Rahman, Dalal A. Abou El-Ella, and El Shimaa M. N. Abdelhafez. 2024. "Benzimidazole-Based Derivatives as Apoptotic Antiproliferative Agents: Design, Synthesis, Docking, and Mechanistic Studies" Molecules 29, no. 2: 446. https://doi.org/10.3390/molecules29020446
APA StyleYoussif, B. G. M., Morcoss, M. M., Bräse, S., Abdel-Aziz, M., Abdel-Rahman, H. M., Abou El-Ella, D. A., & Abdelhafez, E. S. M. N. (2024). Benzimidazole-Based Derivatives as Apoptotic Antiproliferative Agents: Design, Synthesis, Docking, and Mechanistic Studies. Molecules, 29(2), 446. https://doi.org/10.3390/molecules29020446