
Design, Synthesis, and Anti-Hepatocellular Carcinoma Evaluation of Sesquiterpene Lactone Epimers Trilobolide-6-O-isobutyrate Analogs

Abstract
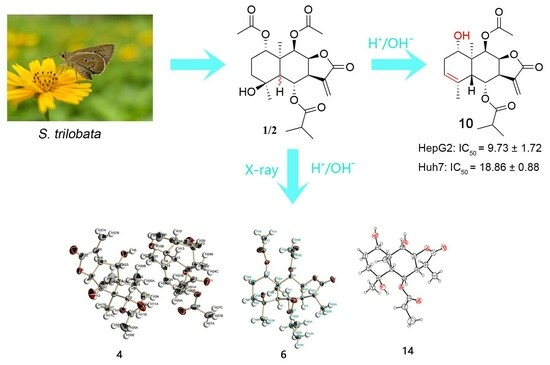
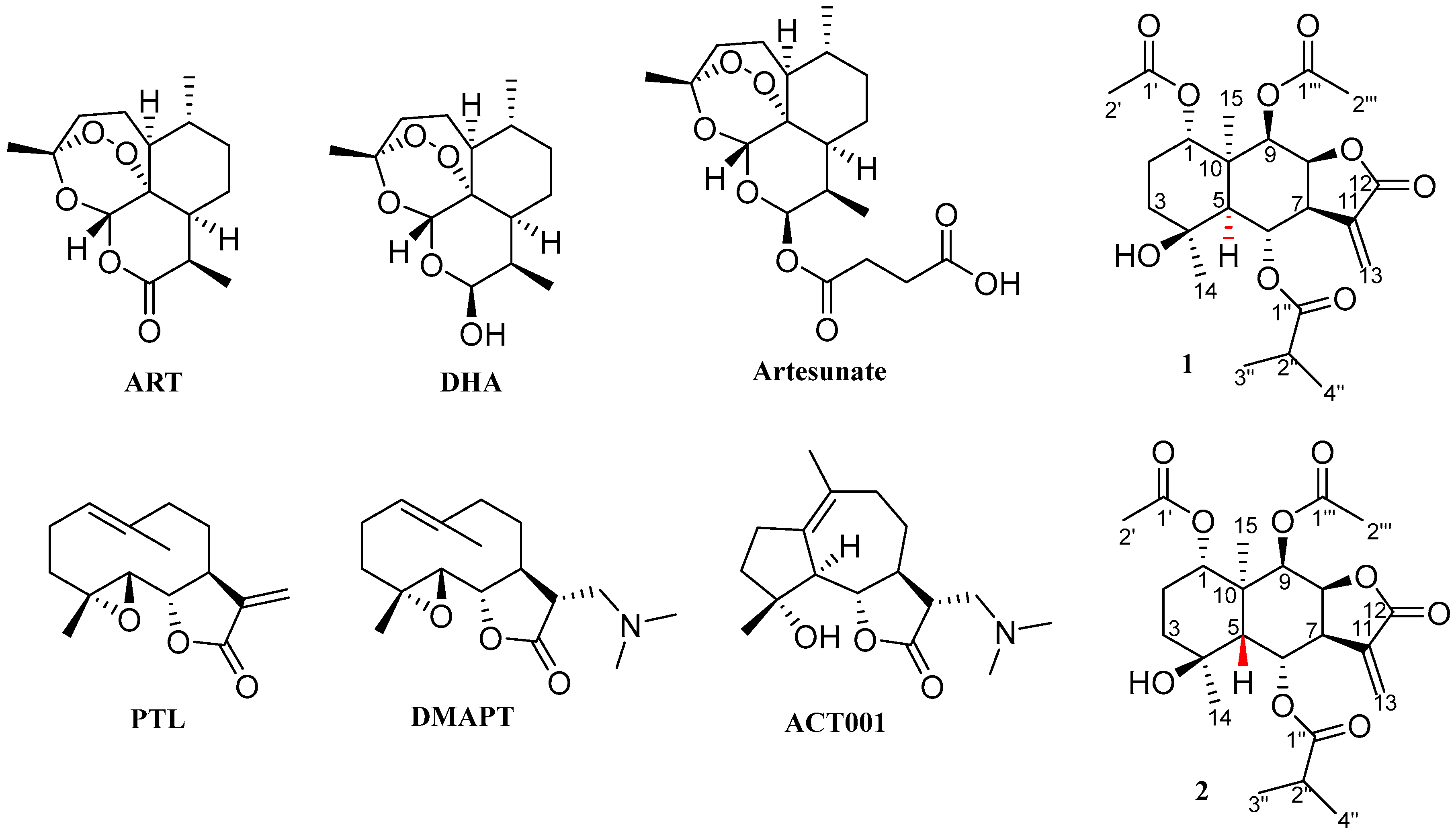
1. Introduction
2. Results and Discussion
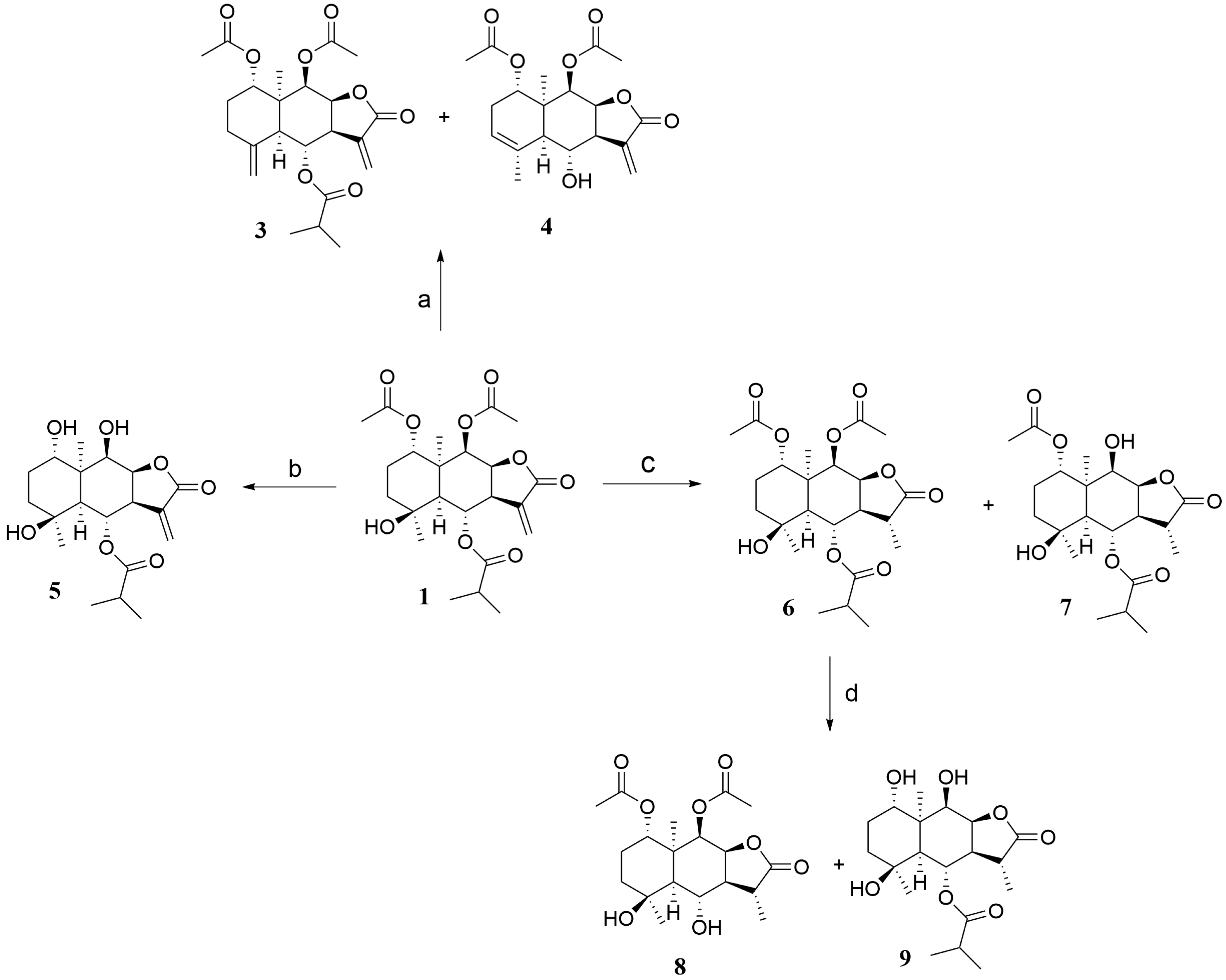
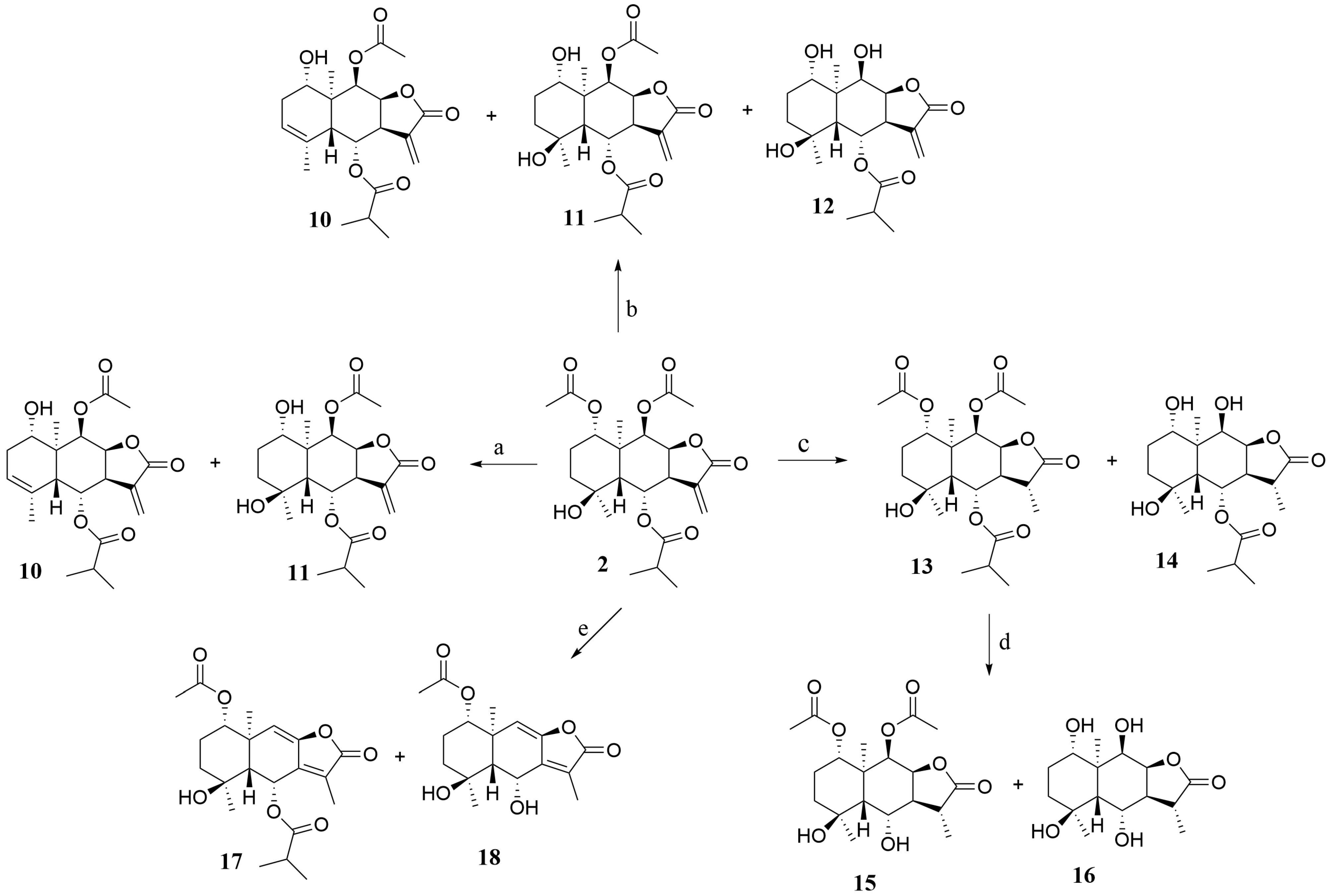
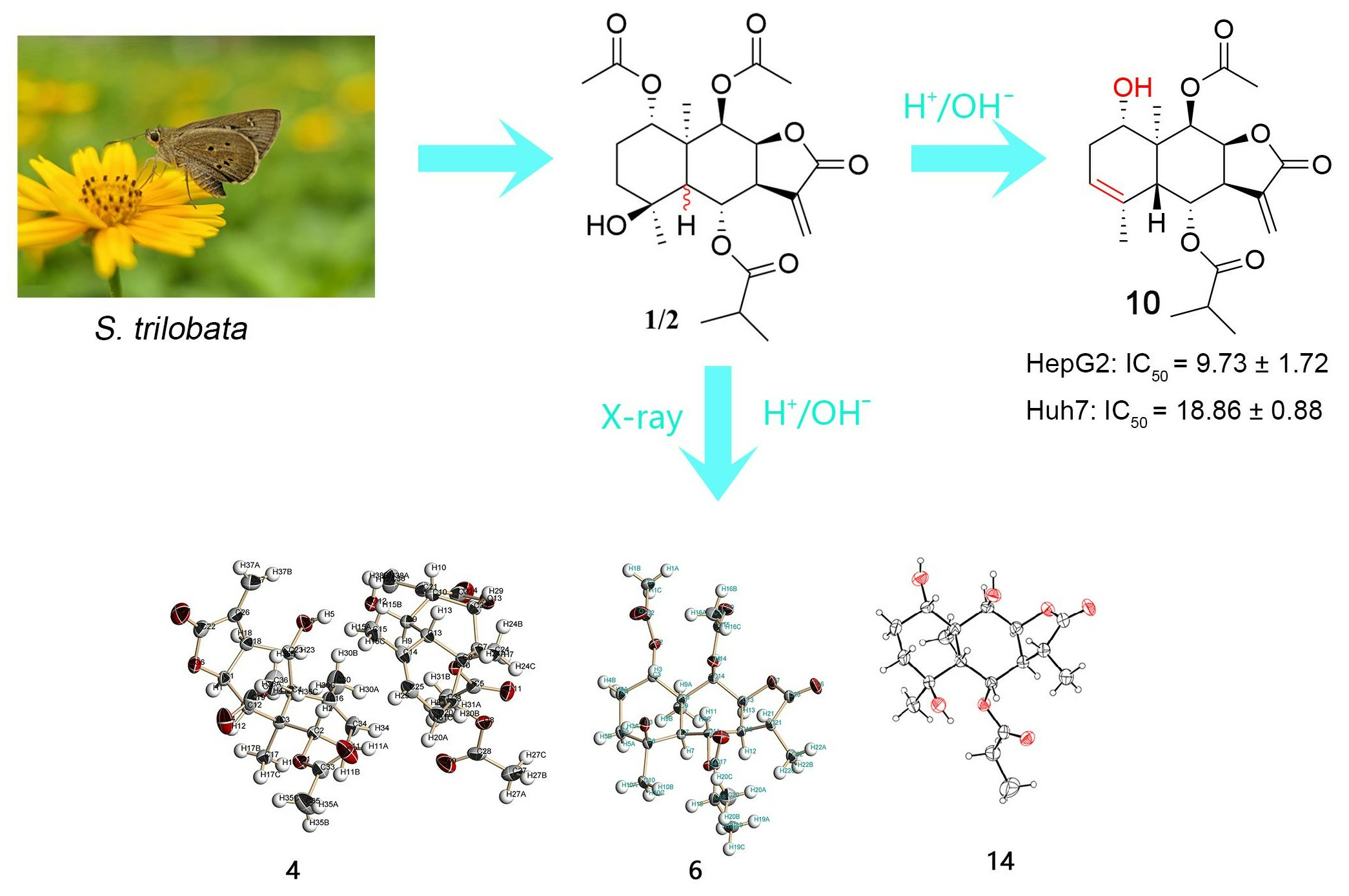
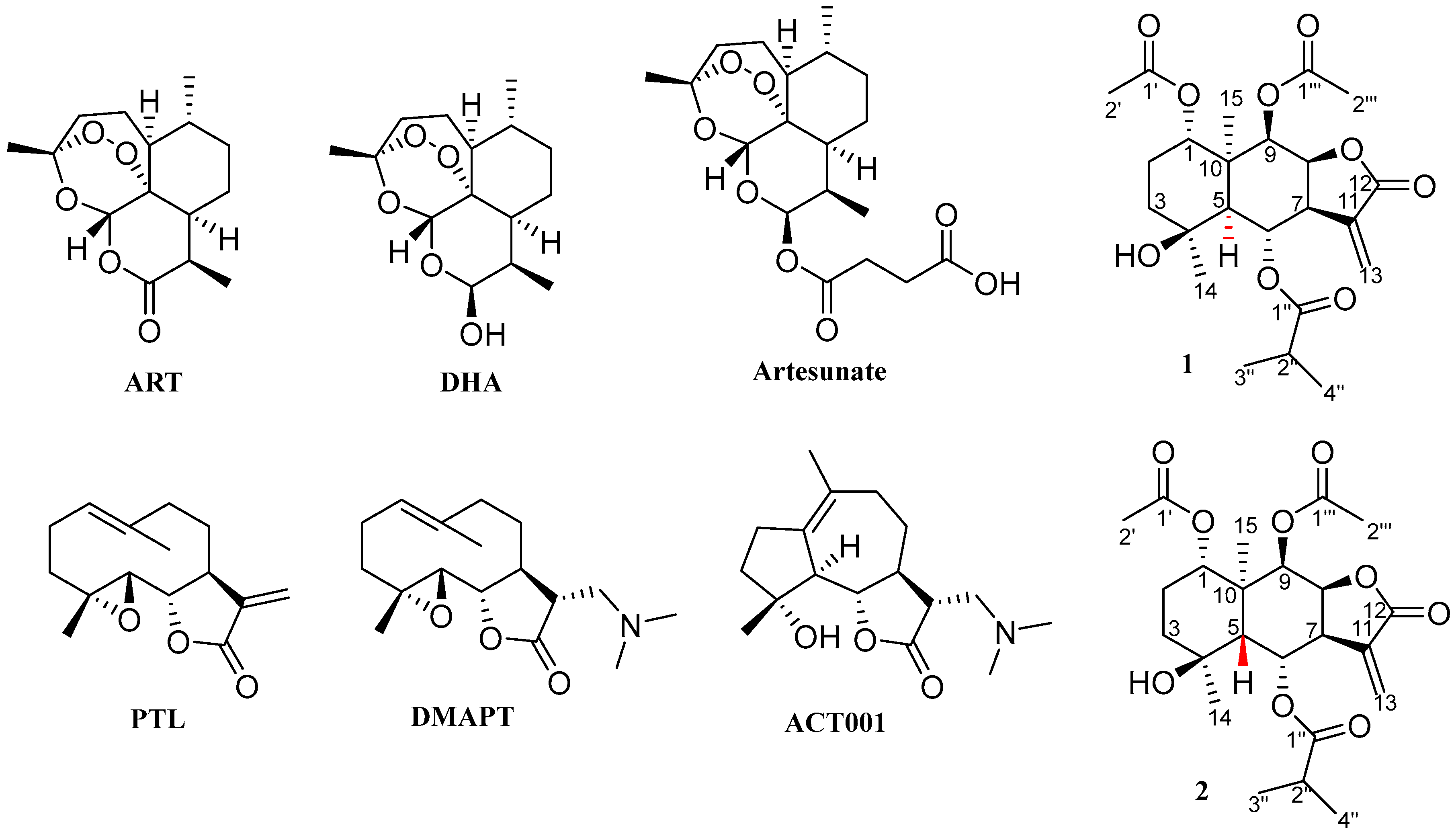
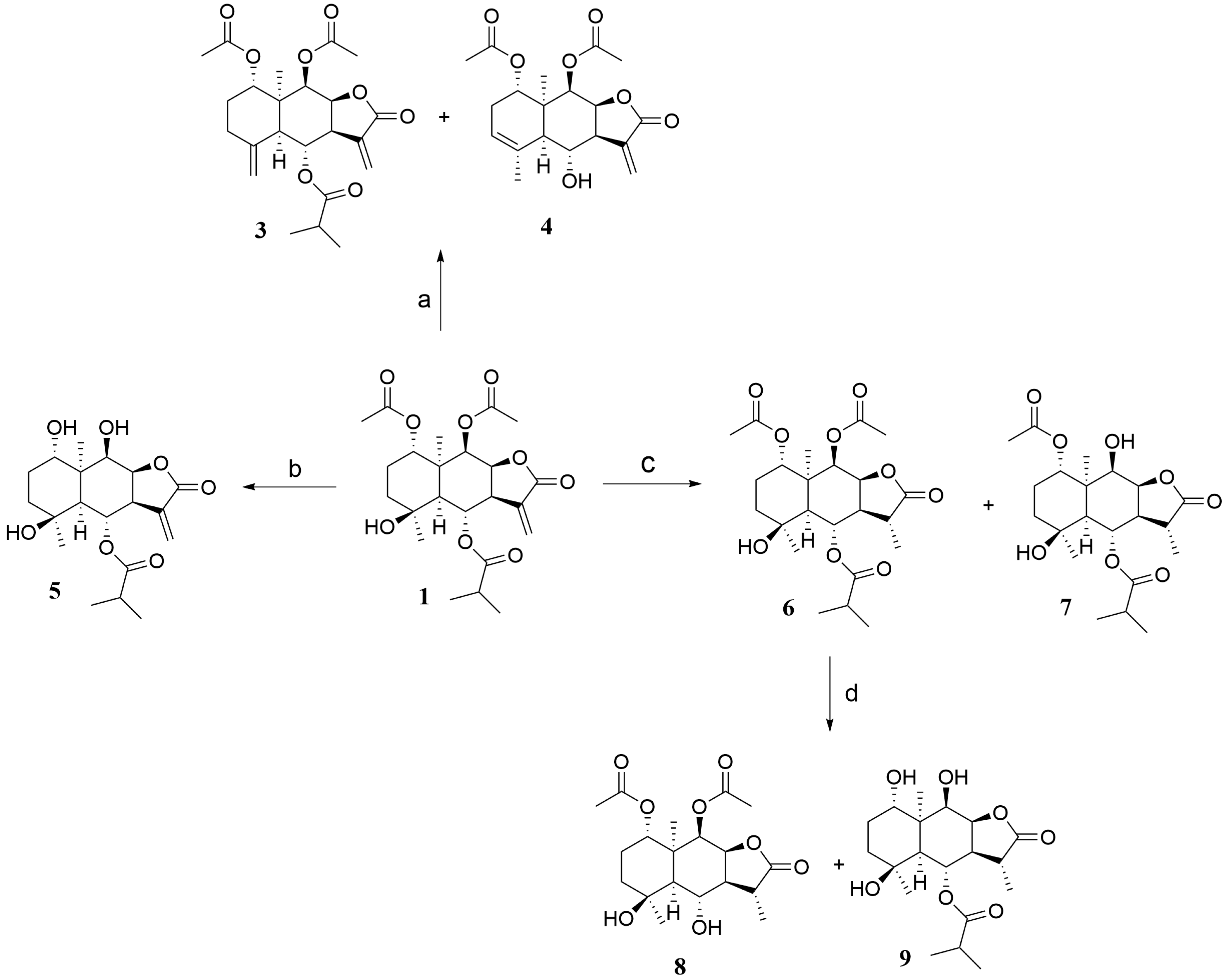
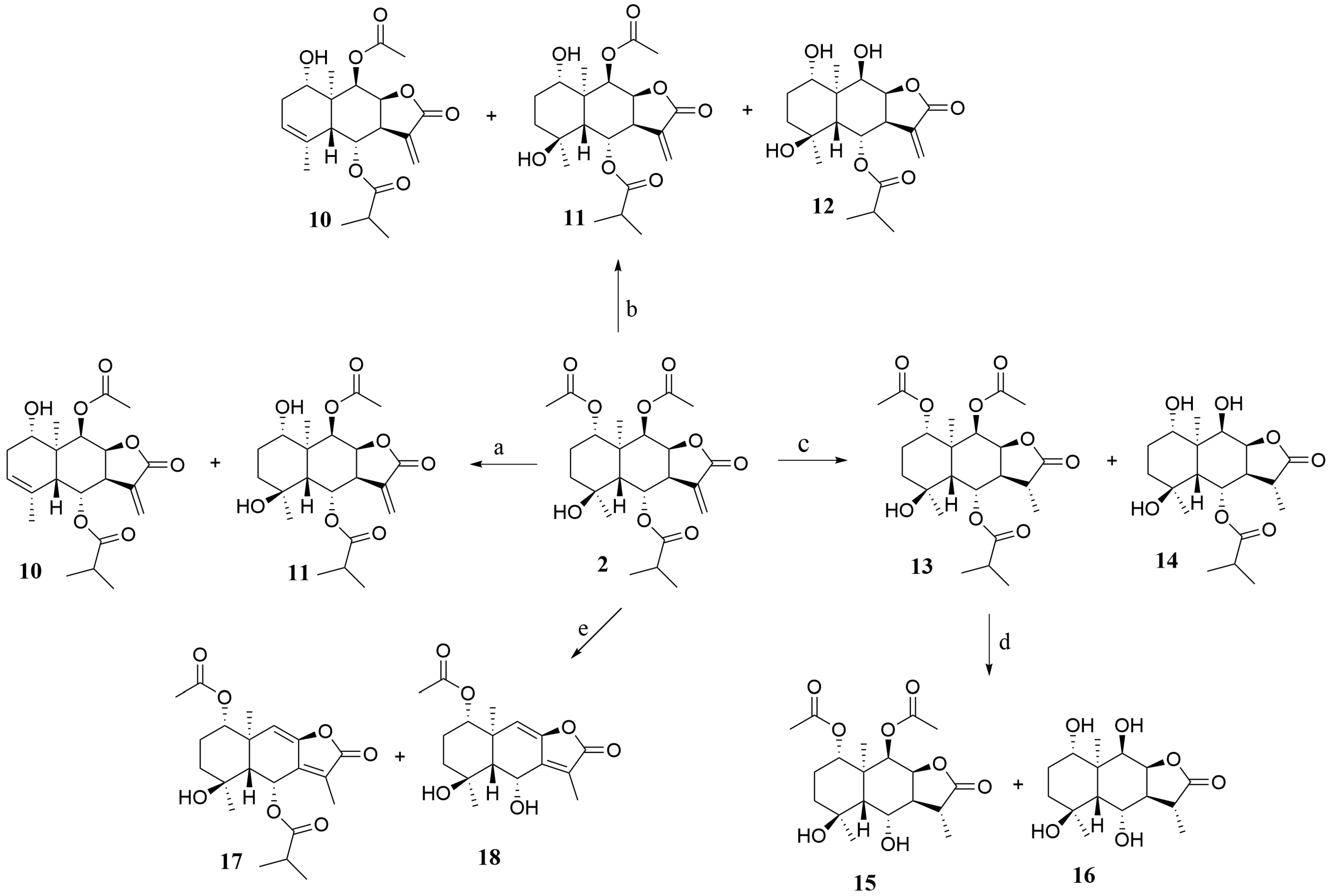
2.1. Synthesis
2.2. Biological Evaluation
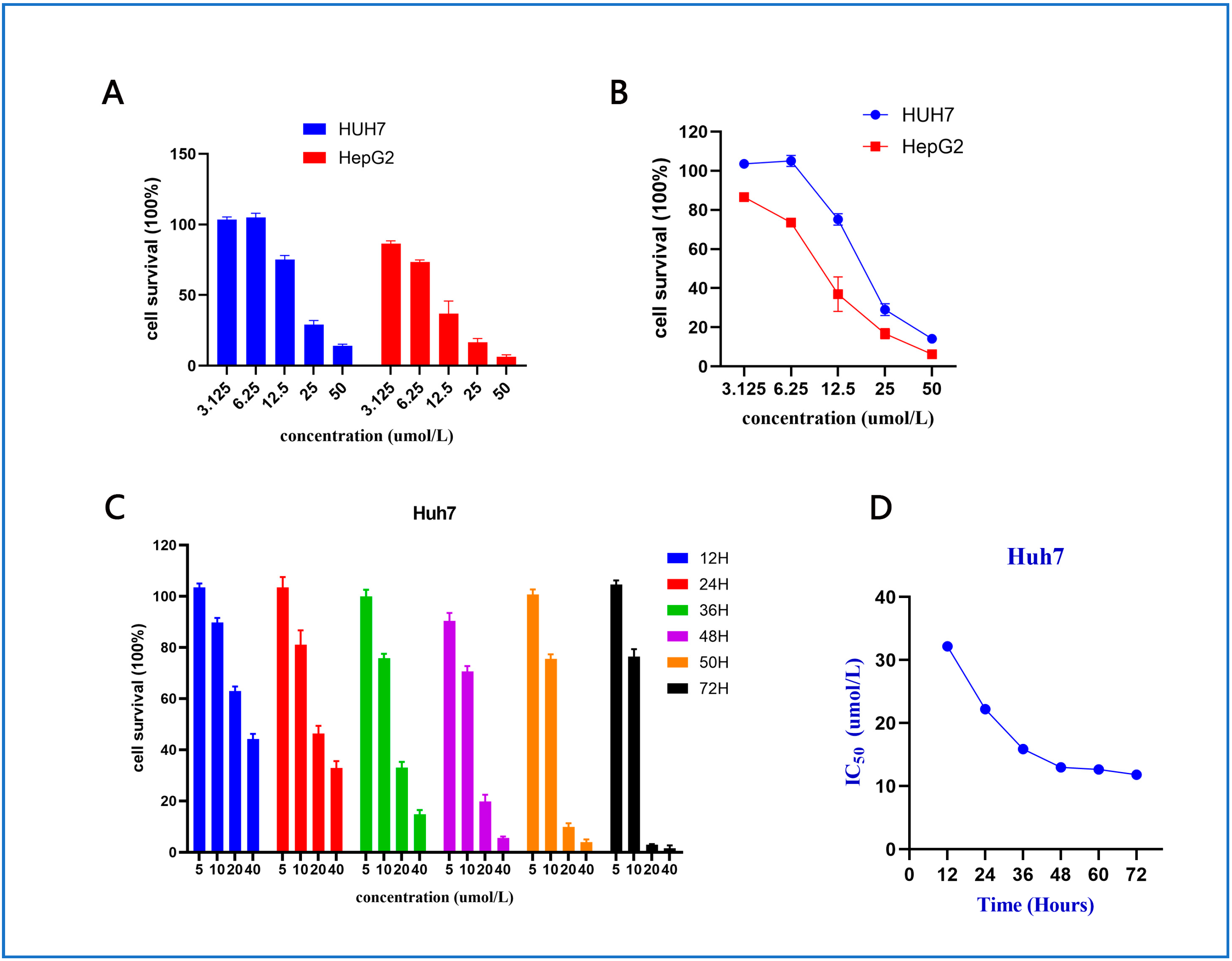
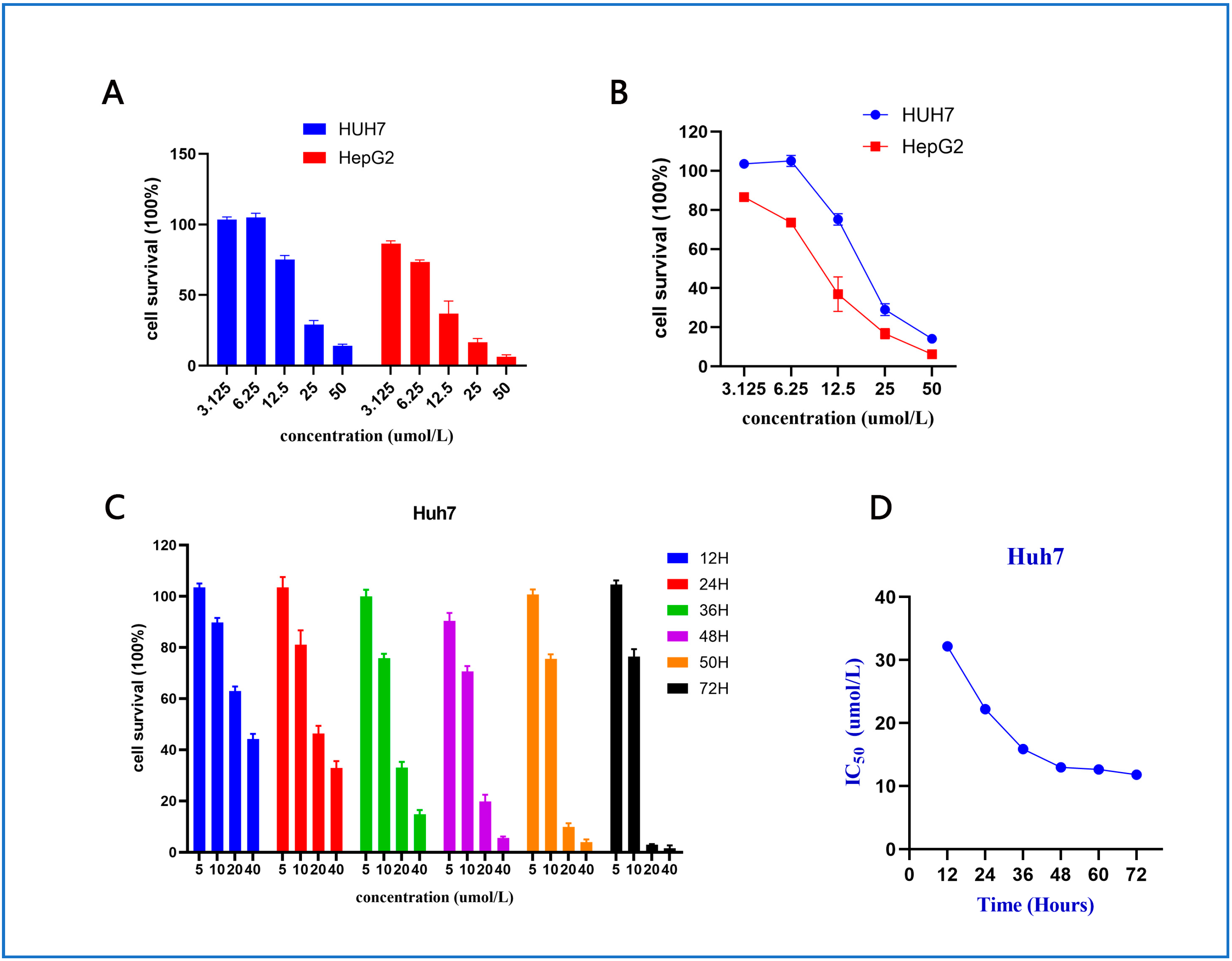
2.2.1. Cell Viability Assay and Structure–Activity Relationships (SARs)
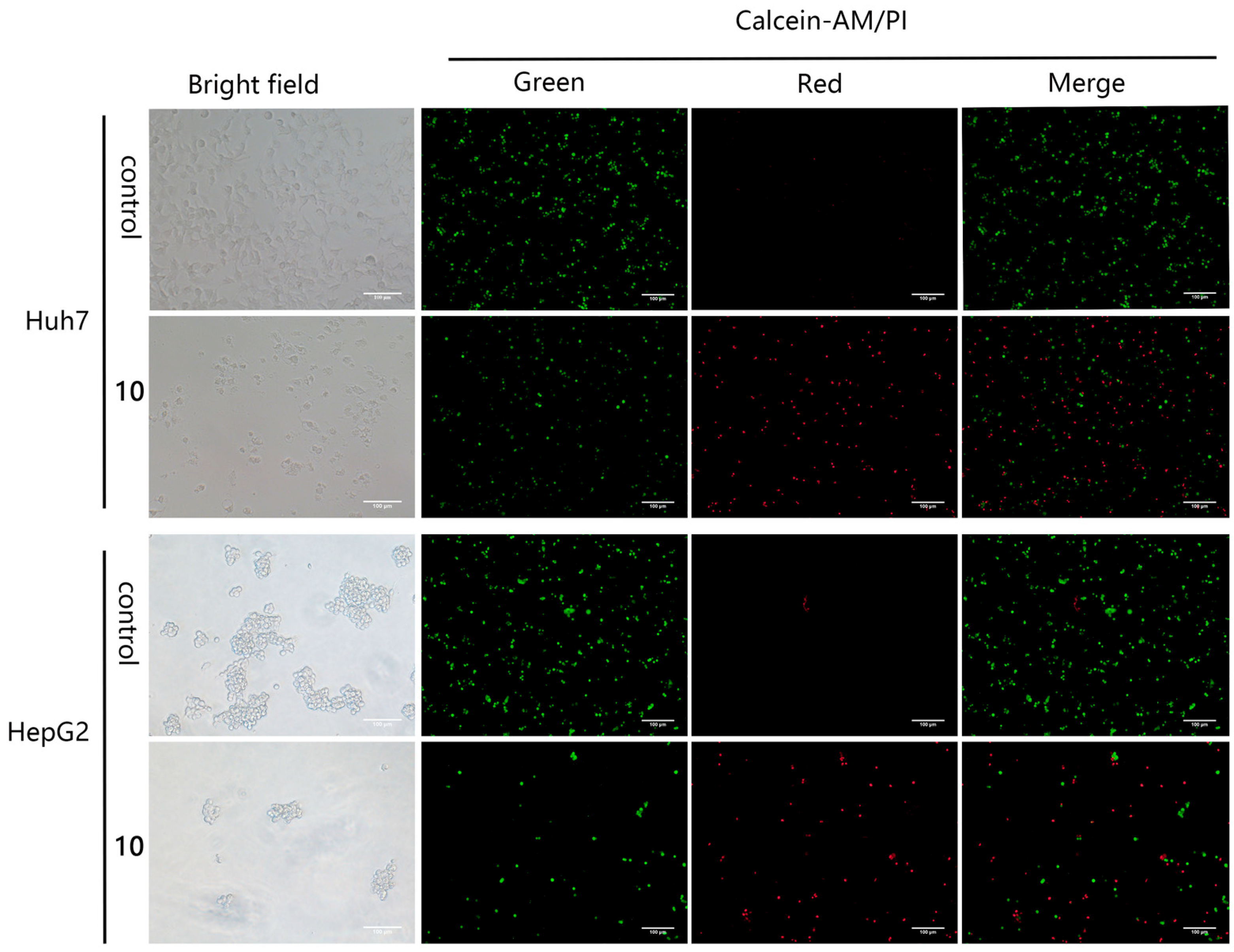
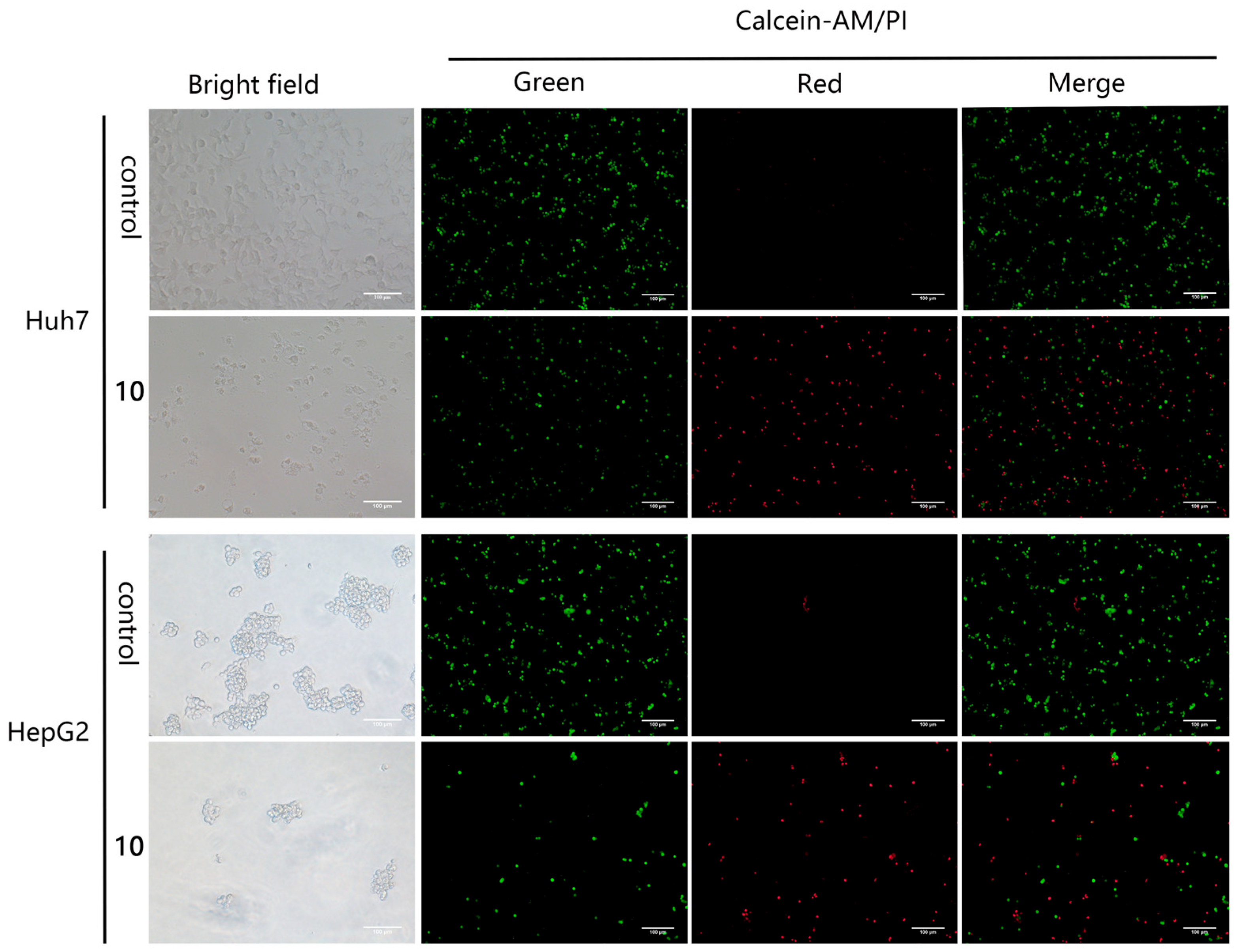
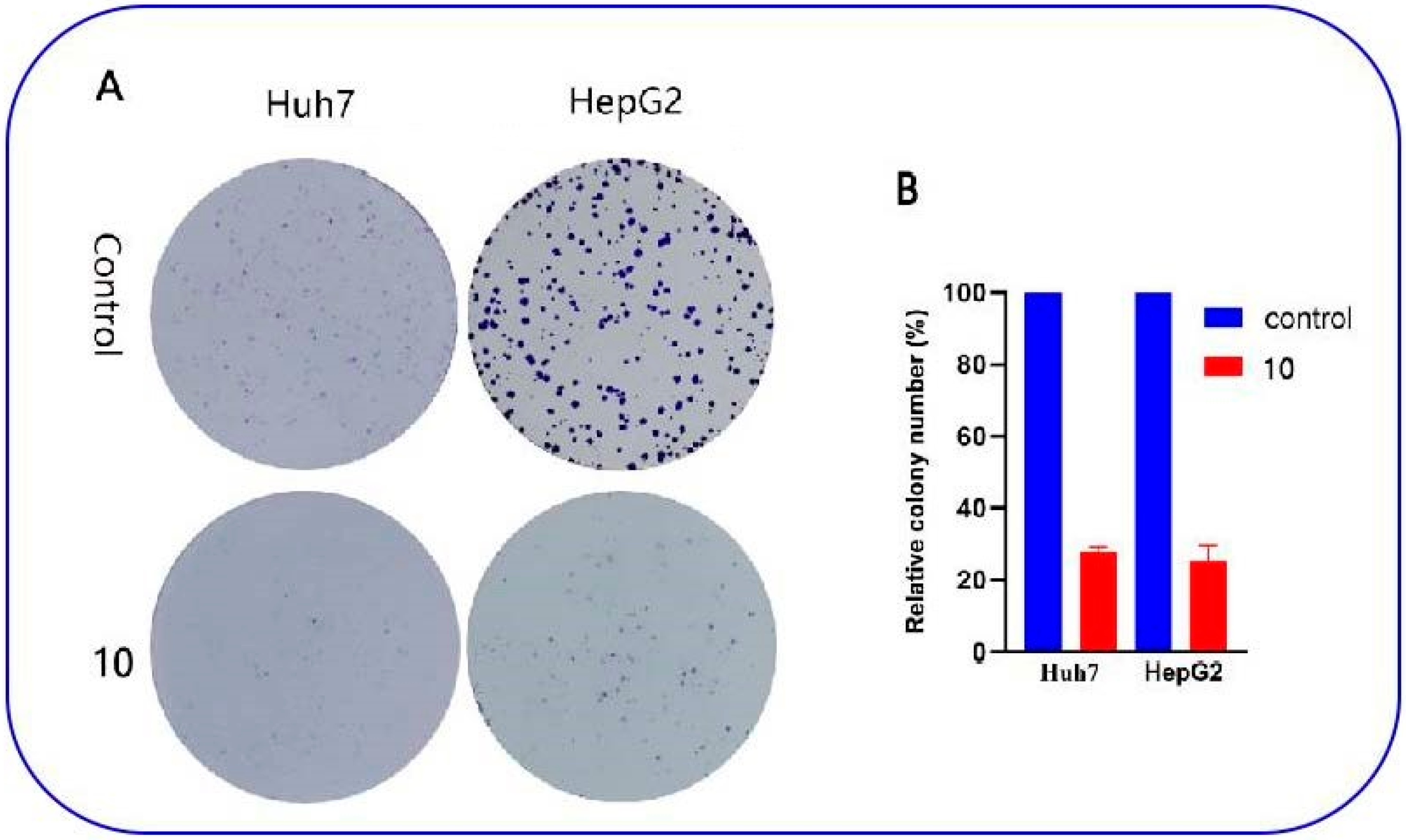
2.2.2. Anti-Proliferative Effect of Compound 10 on Huh7 and HepG2 Cells
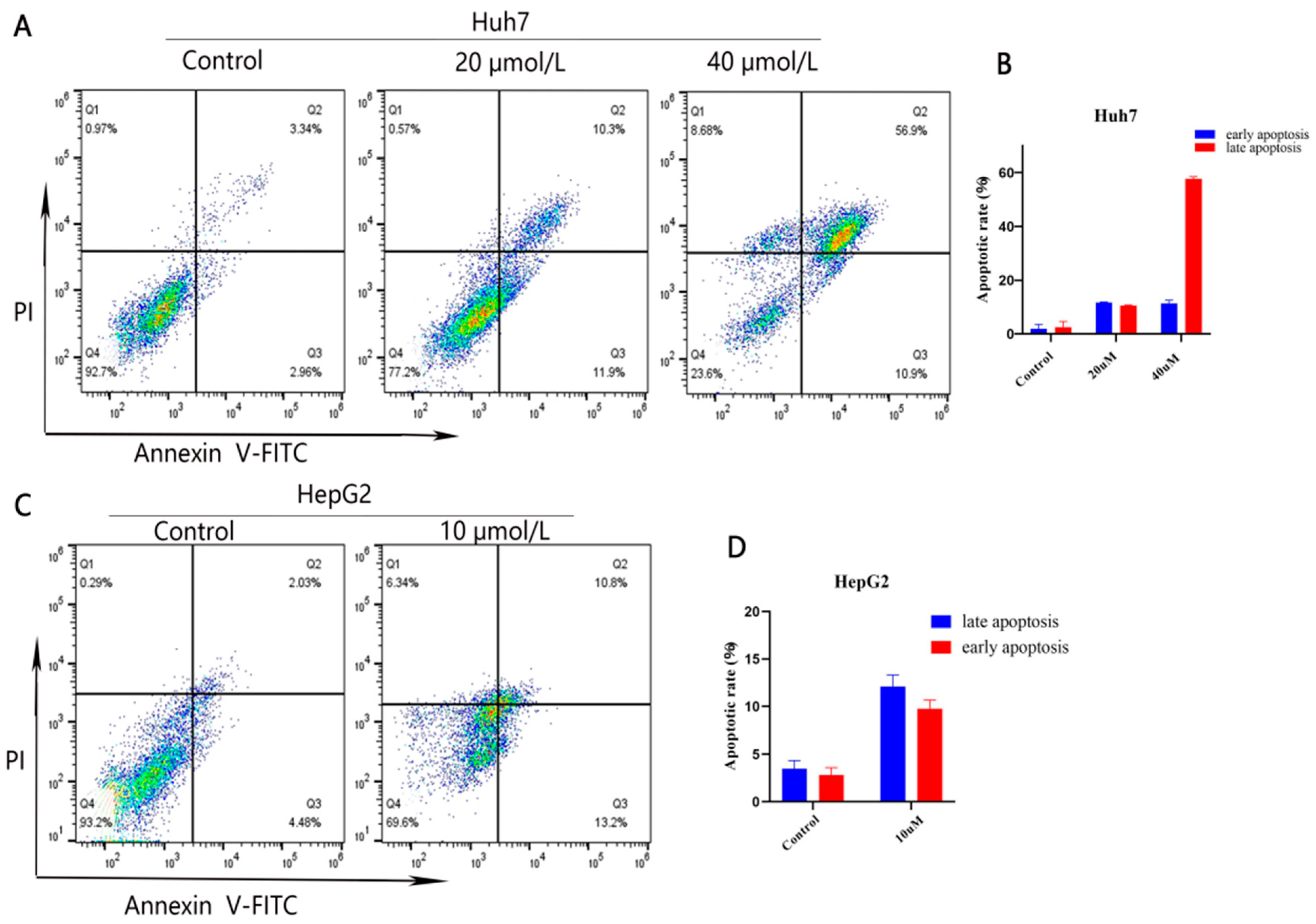
2.2.3. Compound 10 Promoted Cell Apoptosis of Huh7 and HepG2 Cells
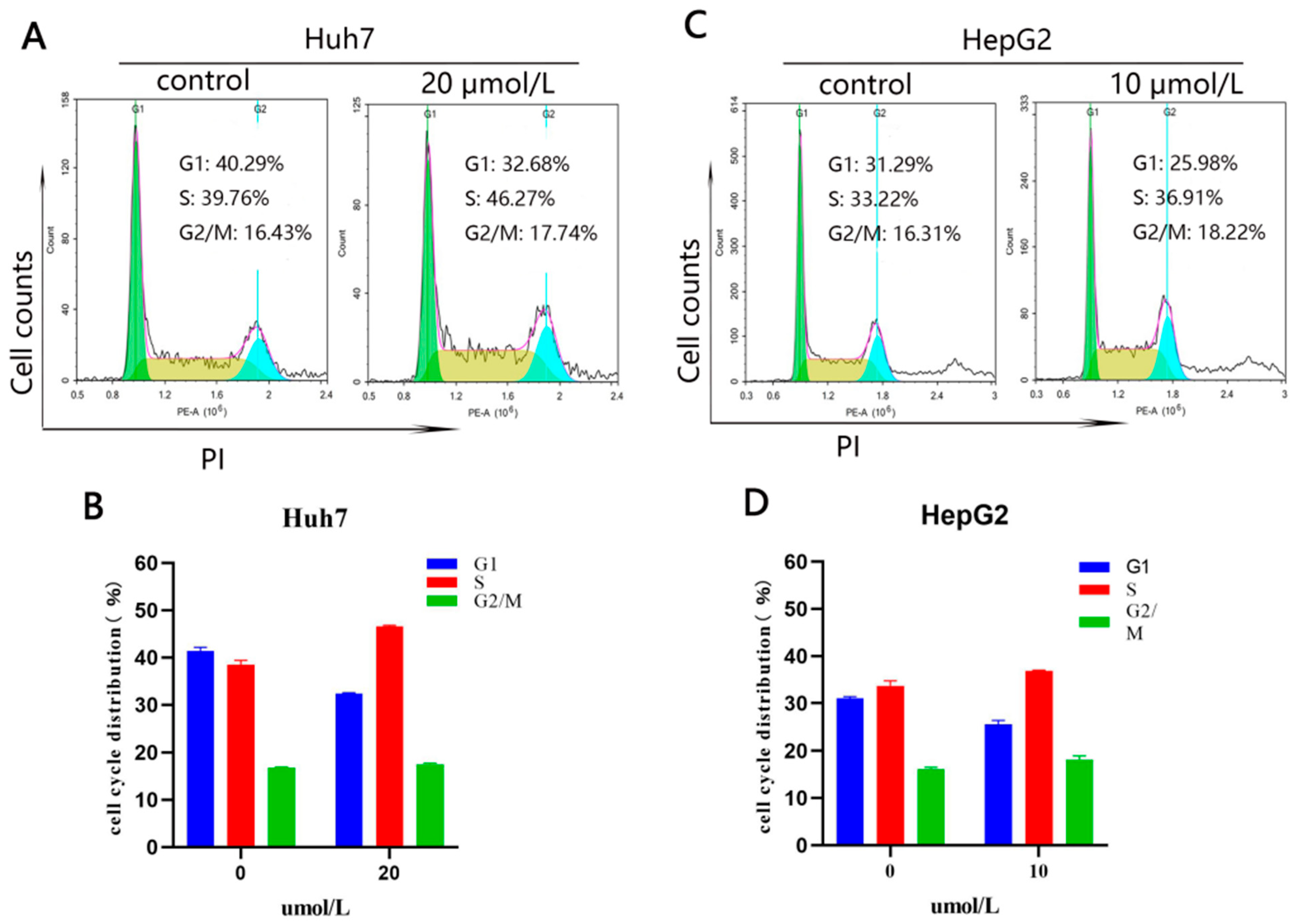
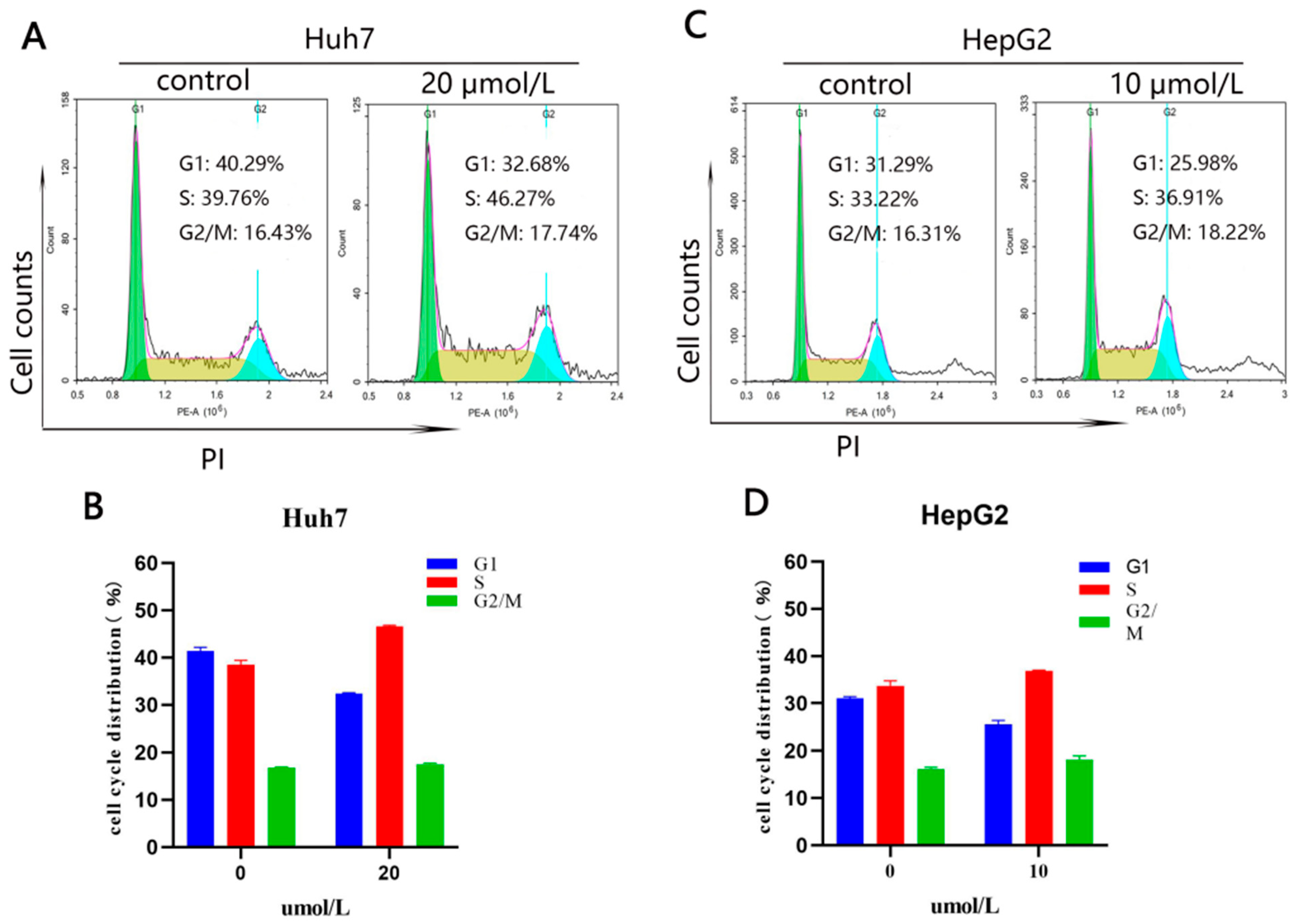
2.2.4. Compound 10 Arrested the Cell Cycle in the S Phase in Huh7 and HepG2 Cells
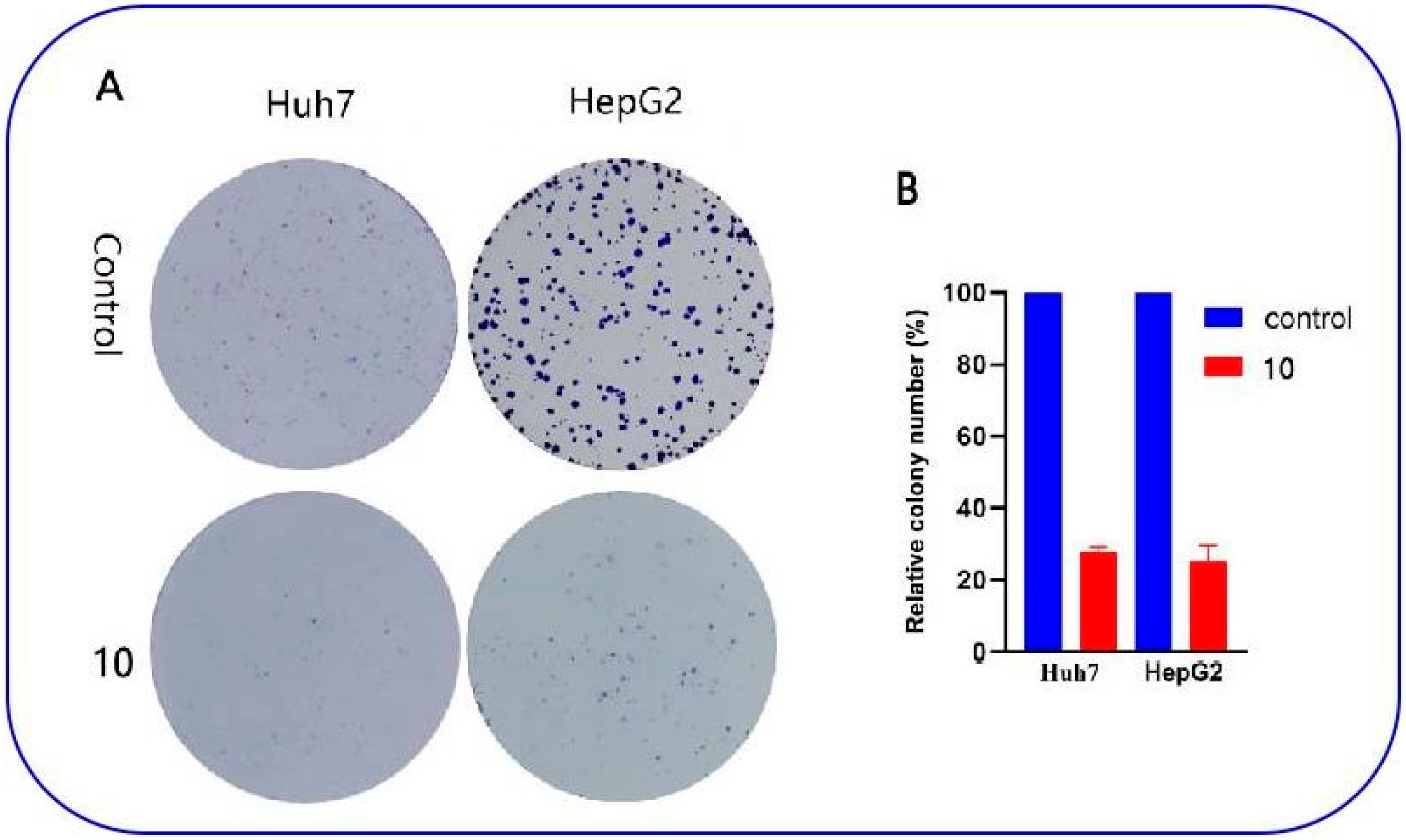
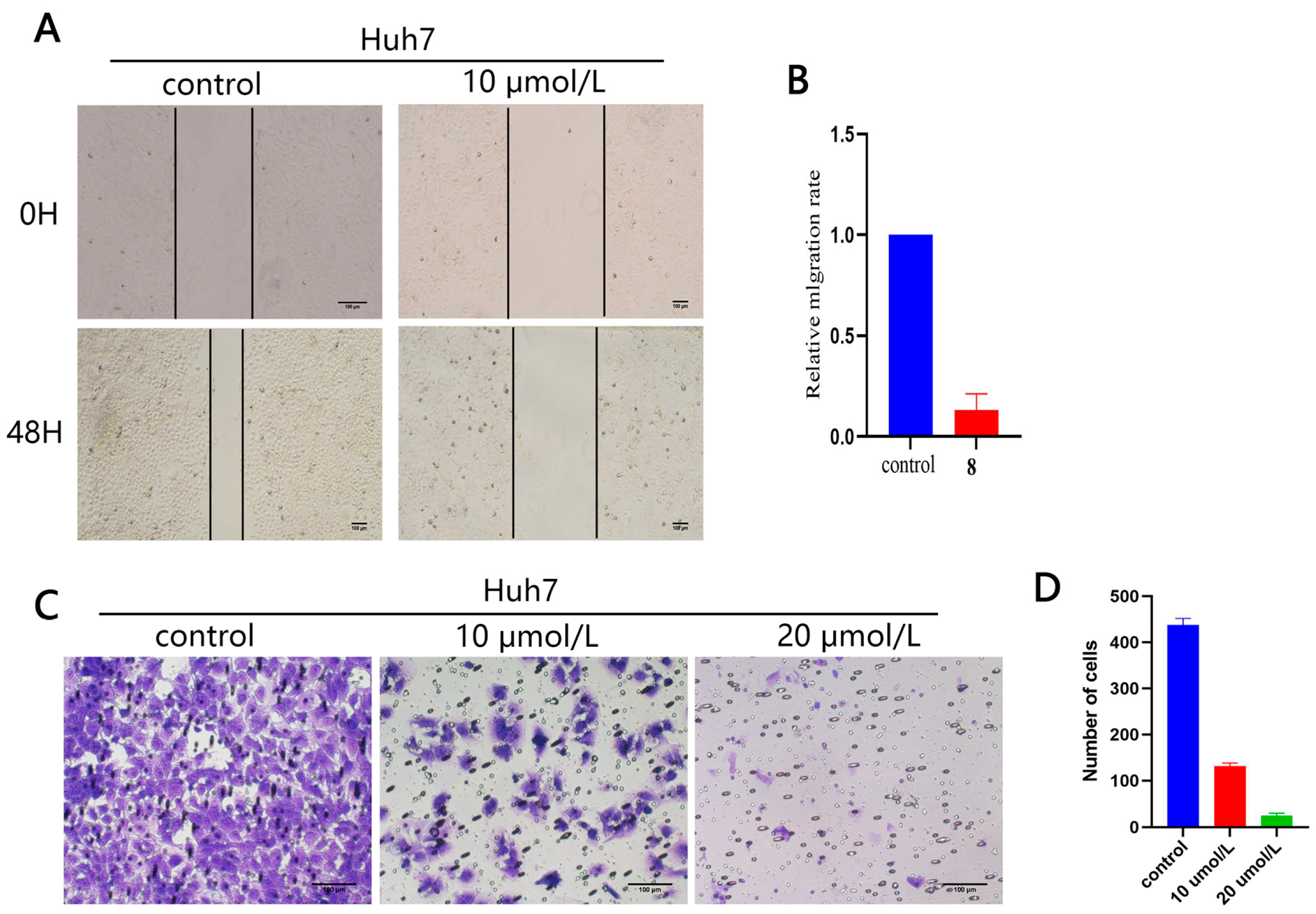
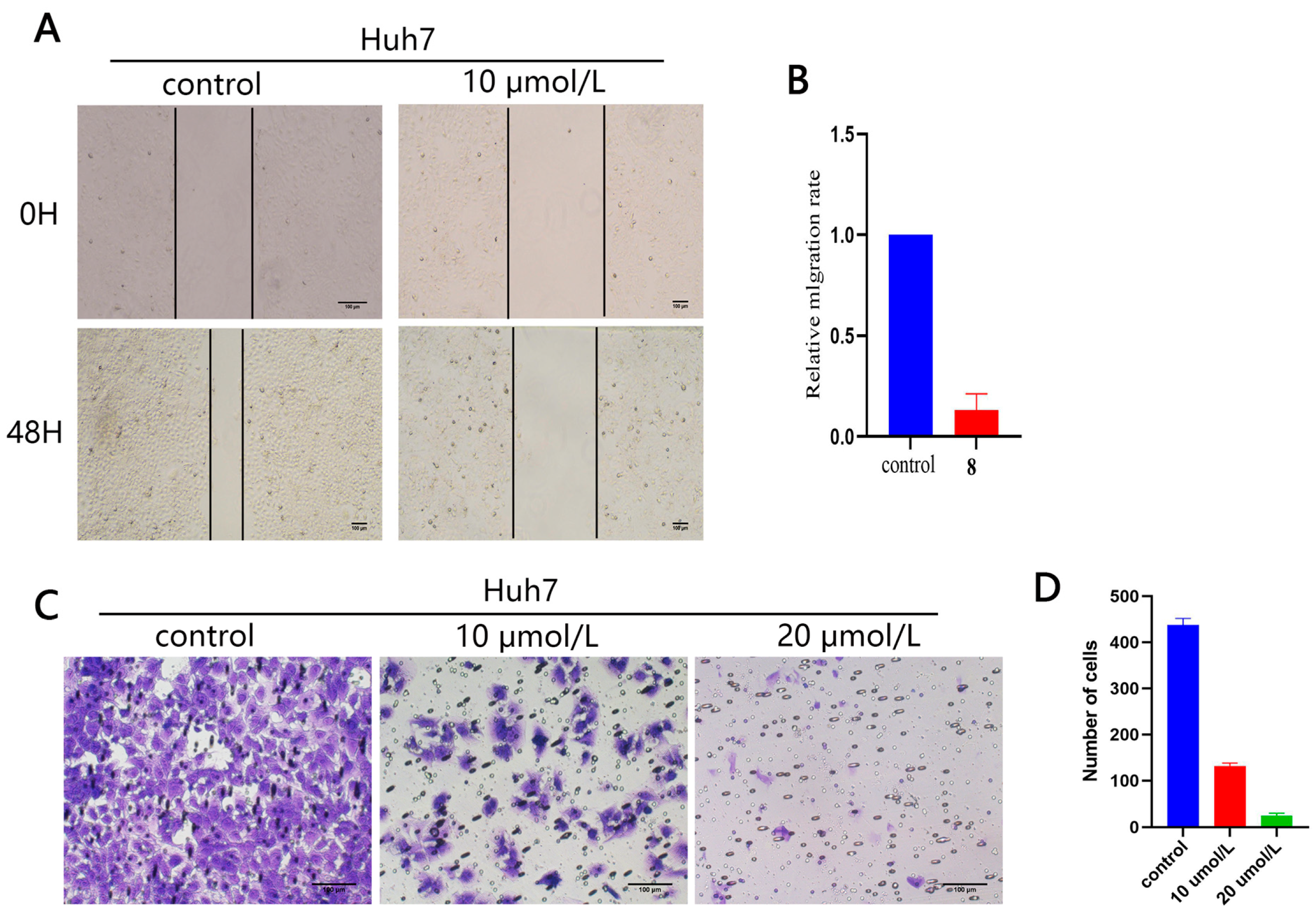
2.2.5. Effect of Compound 10 on Cell Migration
2.2.6. ADME Prediction
3. Materials and Methods
3.1. General Experimental Procedures
3.2. The Separation of SLs 1 and 2
3.3. General Procedure for the Synthesis of 1 Hydrolysis Derivatives 3–4
3.4. General Procedure for the Synthesis of 1 Hydrolysis Derivative 5
3.5. General Procedure for the Synthesis of 1 Hydrolysis Derivatives 6–9
3.6. General Procedure for the Synthesis of 2 Hydrolysis Derivatives 10–12
3.7. General Procedure for the Synthesis of 2 Hydrolysis Derivatives 13–16
3.8. General Procedure for the Synthesis of 2 Hydrolysis Derivatives 17–18
3.9. X-ray Crystallographic Analysis
3.10. Biological Evaluation
3.10.1. Cell Lines and Cell Culture
3.10.2. Cell Cytotoxicity Assay
3.10.3. Calcein-AM/PI Costaining
3.10.4. Colony Formation Assay
3.10.5. Cell Apoptosis Analysis
3.10.6. Cell Cycle Arrest Analysis
3.10.7. Wound Healing Assay
3.10.8. Cell Migration Assays
3.10.9. ADME Prediction
3.10.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mao, J.J.; Pillai, G.G.; Andrade, C.J.; Ligibel, J.A.; Basu, P.; Cohen, L.; Khan, I.A.; Mustian, K.M.; Puthiyedath, R.; Dhiman, K.S. Integrative oncology: Addressing the global challenges of cancer prevention and treatment. CA Cancer J. Clin. 2022, 72, 144–164. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Forner, A.; Gilabert, M.; Bruix, J.; Raoul, J.-L. Treatment of intermediate-stage hepatocellular carcinoma. Hepatobil. Surg. Nutr. 2014, 11, 525–535. [Google Scholar] [CrossRef]
- Sperandio, R.C.; Pestana, R.C.; Miyamura, B.V.; Kaseb, A.O. Hepatocellular carcinoma immunotherapy. Annu. Rev. Med. 2022, 73, 267–278. [Google Scholar] [CrossRef]
- Ling, Y.; Gao, W.J.; Ling, C.C.; Liu, J.; Meng, C.; Qian, J.Q.; Liu, S.Q.; Gan, H.L.; Wu, H.M.; Tao, J.H.; et al. β-carboline and N-hydroxycinnamamide hybrids as anticancer agents for drug-resistant hepatocellular carcinoma. Eur. J. Med. Chem. 2019, 168, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Ruwizhi, N.; Aderibigbe, B.A. Cinnamic acid derivatives and their biological efficacy. Int. J. Mol. Sci. 2020, 21, 5712. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jones, A.X.; Lei, X.G. Synthesis and mode of action of oligomeric sesquiterpene lactones. Nat. Prod. Rep. 2016, 33, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.L.; Yu, J.H.; Kinghorn, D.A. Development of anticancer agents from plant-derived sesquiterpene lactones. Curr. Med. Chem. 2016, 23, 2397–2420. [Google Scholar] [CrossRef]
- Yang, Z.J.; Ge, W.Z.; Li, Q.Y.; Lu, Y.X.; Gong, J.M.; Kuang, B.J.; Xi, X.N.; Wu, H.T.; Zhang, Q.; Chen, Y. Syntheses and biological evaluation of costunolide, parthenolide, and their fluorinated analogues. J. Med. Chem. 2015, 58, 7007–7020. [Google Scholar] [CrossRef]
- Chen, H.; Wu, G.Z.; Gao, S.; Guo, R.H.; Zhao, Z.; Yuan, H.; Liu, S.X.; Wu, J.; Lu, X.L.; Yuan, X.; et al. Discovery of potent small-molecule inhibitors of ubiquitin-conjugating enzyme UbcH5c from α-santonin derivatives. J. Med. Chem. 2017, 60, 6828–6852. [Google Scholar] [CrossRef]
- Janganati, V.; Ponder, J.; Jordan, C.T.; Borrelli, M.J.; NPenthala, A.R.; Crooks, P.A. Dimers of melampomagnolide B exhibit potent anticancer activity against hematological and solid tumor cells. J. Med. Chem. 2015, 58, 8896–8906. [Google Scholar] [CrossRef] [PubMed]
- Zhangabylov, N.S.; Dederer, L.Y.; Gorbacheva, L.B.; Vasil’eva, S.V.; Terekhov, V.S.; Adekenov, S.M. Sesquiterpene lactone arglabin influences DNA synthesis in P388 leukemia cells in vivo. Pharm. Chem. J. 2004, 38, 651–653. [Google Scholar] [CrossRef]
- Ghantous, A.; Gali-Muhtasib, H.; Vuorela, H.; Saliba, N.A.; Darwiche, N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discovery Today 2010, 15, 668–678. [Google Scholar] [CrossRef]
- Li, J.; Li, S.S.; Guo, J.S.; Li, Q.Y.; Long, J.; Ma, C.; Ding, Y.H.; Yan, C.L.; Li, L.W.; Wu, Z.G.; et al. Natural product micheliolide (MCL) irreversibly activates pyruvate kinase M2 and suppresses leukemia. J. Med. Chem. 2018, 61, 4155–4164. [Google Scholar] [CrossRef]
- Wong, Y.K.; Xu, C.; Kalesh, K.A.; He, Y.; Lin, Q.; Wong, W.S.F.; Shen, H.M.; Wang, J. Artemisinin as an anticancer drug: Recent advances in target profiling and mechanisms of action. Med. Res. Rev. 2017, 37, 1492–1517. [Google Scholar] [CrossRef]
- Dai, Y.F.; Zhou, W.W.; Meng, J.; Du, X.L.; Sui, Y.P.; Dai, L.; Wang, P.Q.; Huo, H.R.; Sui, F. The pharmacological activities and mechanisms of artemisinin and its derivatives: A systematic review. Med. Chem. Res. 2017, 26, 867–880. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, X.; Liu, J.; Yuan, X.; He, Q. Therapeutic potentials and mechanisms of artemisinin and its derivatives for tumorigenesis and metastasis. Anti-Cancer Agents Med. Chem. 2020, 20, 520–535. [Google Scholar] [CrossRef] [PubMed]
- Augustin, Y.; Staines, H.M.; Krishna, S. Artemisinins as a novel anti-cancer therapy: Targeting a global cancer pandemic through drug repurposing. Pharm. Ther. 2020, 216, 107706. [Google Scholar] [CrossRef]
- Lesiak, K.; Koprowska, K.; Zalesna, I.; Nejc, D.; Düchler, M.; Czyz, M. Parthenolide, a sesquiterpene lactone from the medical herb feverfew, shows anticancer activity against human melanoma cells in vitro. Melanoma Res. 2010, 20, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Nasim, S.; Crooks, P.A. Antileukemic activity of aminoparthenolide analogs. Bioorg. Med. Chem. Lett. 2008, 18, 3870–3873. [Google Scholar] [CrossRef]
- Guzman, M.L.; Rossi, R.M.; Neelakantan, S.; Li, X.J.; Corbett, C.A.; Hassane, D.C.; Becker, M.W.; Bennett, J.M.; Sullivan, E.; Lachowicz, J.L.; et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood 2007, 110, 4427–4435. [Google Scholar] [CrossRef]
- Zhang, Q.; Lu, Y.X.; Ding, Y.H.; Zhai, J.D.; Ji, Q.; Ma, W.W.; Yang, M.; Fan, H.G.; Long, J.; Tong, Z.H.; et al. Guaianolide sesquiterpene lactones, a source to discover agents that selectively inhibit acute myelogenous leukemia stem and progenitor cells. J. Med. Chem. 2012, 55, 8757–8769. [Google Scholar] [CrossRef] [PubMed]
- Rozalski, M.; Krajewska, U.; Panczyk, M.; Mirowski, M.; Rozalska, B.; Wasek, T.; Janecki, T. Synthesis and biological evaluation of 4-methylideneisoxazolidin-5-ones—A new class of highly cytotoxic α-methylidene-γ-lactones. Eur. J. Med. Chem. 2007, 42, 248–255. [Google Scholar] [CrossRef]
- Polo, L.M.; Castro, C.M.; Cruzado, M.C.; Collino, C.J.G.; Cuello-Carrión, F.D.; Ciocca, D.R.; Giordano, O.S.; Ferrari, M.; López, L.A. 11,13-dihydro-dehydroleucodine, a derivative of dehydroleucodine with an inactivated alkylating function conserves the anti-proliferative activity in G2 but does not cause cytotoxicity. Eur. J. Pharmacol. 2007, 556, 19–26. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Eakin, M.A.; Thomas, A.M. Tumor inhibitors. 69. Structure-cytotoxicity relations among the sesquiterpene lactones. J. Med. Chem. 1971, 14, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Q.; Mao, X.M.; Fan, R.; Li, S.Y.; Shang, J.; Zhang, T.; Li, R.H.; Li, H.Q.; Hui, Y.; Chen, W.H.; et al. Trilobolide-6-O-isobutyrate suppresses hepatocellular carcinoma tumorigenesis through inhibition of IL-6/STAT3 signaling pathway. Phytother. Res. 2021, 35, 5741–5753. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Q.; Yi, J.L.; Chen, G.Y.; Liu, Z.X.; Hui, Y.; Chen, W.H. Structure modification and anti-tumor activity evaluation on sesquiterpene lactones TBA and TBB from Sphagneticola trilobata. Nat. Prod. Res. 2023, 37, 3766–3771. [Google Scholar]
- Hui, Y.; Cao, J.; Lin, J.; Yang, J.N.; Liu, Y.J.; Han, C.R.; Song, X.P.; Dai, C.Y.; Zhang, X.P.; Chen, W.H.; et al. Eudesmanolides and other constituents from the flowers of Wedelia trilobata. Chem. Biodivers. 2018, 15, 1700411. [Google Scholar] [CrossRef]
- Wu, M.Y.; Han, Z.B.; Ni, H.Z.; Wang, N.Z.; Ding, K.L.; Lu, Y.X. Phosphine-catalyzed divergent domino processes between γ-substituted allenoates and carbonyl-activated alkenes. Chem. Sci. 2022, 13, 3161–3168. [Google Scholar] [CrossRef]
- Guo, T.L.; Mo, K.L.; Zhang, N.N.; Xiao, L.P.C.; Liu, W.L.; Wen, L.L. Embedded homogeneous ultra-fine Pd nanoparticles within MOF ultra-thin nanosheets for heterogeneous catalysis. Dalton Trans. 2021, 50, 1774–1779. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, J.Z.; Soto-Acosta, R.; Mao, L.L.; Lian, J.H.; Chen, K.; Pillon, G.; Zhang, G.Q.; Geraghty, R.J.; Zheng, S.P. One-pot synthesis of 1-hydroxyacridones from para-quinols and ortho-methoxycarbonylaryl isocyanates. J. Org. Chem. 2020, 85, 4515–4524. [Google Scholar] [CrossRef]
- Wang, X.J.; Zhang, X.M.; Zheng, B.B.; Hu, N.; Xie, W.D.; Row, K. Synthesis of 13-amino telekin derivatives and their cytotoxic activity. Nat. Prod. Res. 2015, 29, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Kardile, R.A.; Sarkate, A.P.; Lokwani, D.K.; Tiwari, S.V.; Azad, R.; Thopate, S.R. Design, synthesis, and biological evaluation of novel quinoline derivatives as small molecule mutant EGFR inhibitors targeting resistance in NSCLC: In vitro screening and ADME predictions. Eur. J. Med. Chem. 2023, 245, 114889. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.P.; Zhang, Z.; Lv, J.Y.; Zhang, X.Q.; Zhang, Z.Y.; Han, S.T.; Liu, Y.W.; Liu, W.W.; Ji, J.; Shi, D.H. Design, synthesis and anti-cancer evaluation of genistein-1,3,5-triazine derivatives. Tetrahedron 2023, 134, 133293. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| HepG2 | Huh7 | Compound | HepG2 | Huh7 | |
| 1 | 27.77 ± 0.92 | 28.15 ± 0.87 | 11 | 47.98 ± 2.53 | 56.69 ± 1.09 |
| 2 | 22.12 ± 1.13 | 14.04 ± 2.37 | 12 | >70 | >70 |
| 3 | 14.42 ± 1.69 | 24.17 ± 0.57 | 13 | >70 | >70 |
| 4 | 39.43 ± 2.84 | 44.07 ± 3.30 | 14 | >70 | >70 |
| 5 | 15.00 ± 1.07 | 49.69 ± 2.46 | 15 | >70 | >70 |
| 6 | >70 | >70 | 16 | >70 | >70 |
| 7 | >70 | >70 | 17 | >70 | >70 |
| 8 | >70 | >70 | 18 | >70 | >70 |
| 9 | >70 | >70 | adriamycin | 26.80 ± 1.33 | 23.87 ± 1.62 |
| 10 | 9.73 ± 1.72 | 18.86 ± 0.88 | |||
| Physicochemical Properties | Water Solubility | ||
| Formula | C21H28O7 | Log S (ESOL) | −3.35 |
| Molecular weight | 392.44 g/mol | Solubility | 1.76 × 10−1 mg/mL; 4.49 × 10−4 mol/L |
| Num. heavy atoms | 28 | Class | Soluble |
| Num. arom. heavy atoms | 0 | Log S (Ali) | −3.95 |
| Fraction Csp3 | 0.67 | Solubility | 4.43 × 10−2 mg/mL; 1.13 × 10−4 mol/L |
| Num. rotatable bonds | 5 | Class | Soluble |
| Num. H-bond acceptors | 7 | Log S (SILICOS-IT) | −2.30 |
| Num. H-bond donors | 1 | Solubility | 1.99 × 100 mg/mL; 5.06 × 10−3 mol/L |
| Molar Refractivity | 100.53 | Class | Soluble |
| TPSA | 99.13 Å2 | Pharmacokinetics | |
| Lipophilicity | GI absorption | High | |
| Log Po/w (iLOGP) | 2.71 | BBB permeant | No |
| Log Po/w (XLOGP3) | 2.23 | P-gp substrate | No |
| Log Po/w (WLOGP) | 1.93 | CYP1A2 inhibitor | No |
| Log Po/w (MLOGP) | 2.04 | CYP2C19 inhibitor | No |
| Log Po/w (SILICOS-IT) | 2.12 | CYP2C9 inhibitor | No |
| Consensus Log Po/w | 2.20 | CYP2D6 inhibitor | No |
| Druglikeness | CYP3A4 inhibitor | No | |
| Lipinski | Yes; 0 violation | Log Kp (skin permeation) | −7.11 cm/s |
| Ghose | Yes | Medicinal Chemistry | |
| Veber | Yes | PAINS | 0 alert |
| Egan | Yes | Brenk | 3 alerts: isolated_alkene, michael_acceptor_1, more_than_2_esters |
| Muegge | Yes | Lead-likeness | No; 1 violation: MW > 350 |
| Bioavailability Score | 0.55 | Synthetic accessibility | 5.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Yi, G.; Qian, Y.; Yang, X.; Chen, G.; Hui, Y.; Chen, W. Design, Synthesis, and Anti-Hepatocellular Carcinoma Evaluation of Sesquiterpene Lactone Epimers Trilobolide-6-O-isobutyrate Analogs. Molecules 2024, 29, 393. https://doi.org/10.3390/molecules29020393
Zhou X, Yi G, Qian Y, Yang X, Chen G, Hui Y, Chen W. Design, Synthesis, and Anti-Hepatocellular Carcinoma Evaluation of Sesquiterpene Lactone Epimers Trilobolide-6-O-isobutyrate Analogs. Molecules. 2024; 29(2):393. https://doi.org/10.3390/molecules29020393
Chicago/Turabian StyleZhou, Xiuqiao, Guohui Yi, Yiming Qian, Xiaorong Yang, Guangying Chen, Yang Hui, and Wenhao Chen. 2024. "Design, Synthesis, and Anti-Hepatocellular Carcinoma Evaluation of Sesquiterpene Lactone Epimers Trilobolide-6-O-isobutyrate Analogs" Molecules 29, no. 2: 393. https://doi.org/10.3390/molecules29020393
APA StyleZhou, X., Yi, G., Qian, Y., Yang, X., Chen, G., Hui, Y., & Chen, W. (2024). Design, Synthesis, and Anti-Hepatocellular Carcinoma Evaluation of Sesquiterpene Lactone Epimers Trilobolide-6-O-isobutyrate Analogs. Molecules, 29(2), 393. https://doi.org/10.3390/molecules29020393