
The Effect of Solvents on the Crystal Morphology of Isosorbide Mononitrate and Its Molecular Mechanisms

Abstract
1. Introduction
2. Results and Discussion
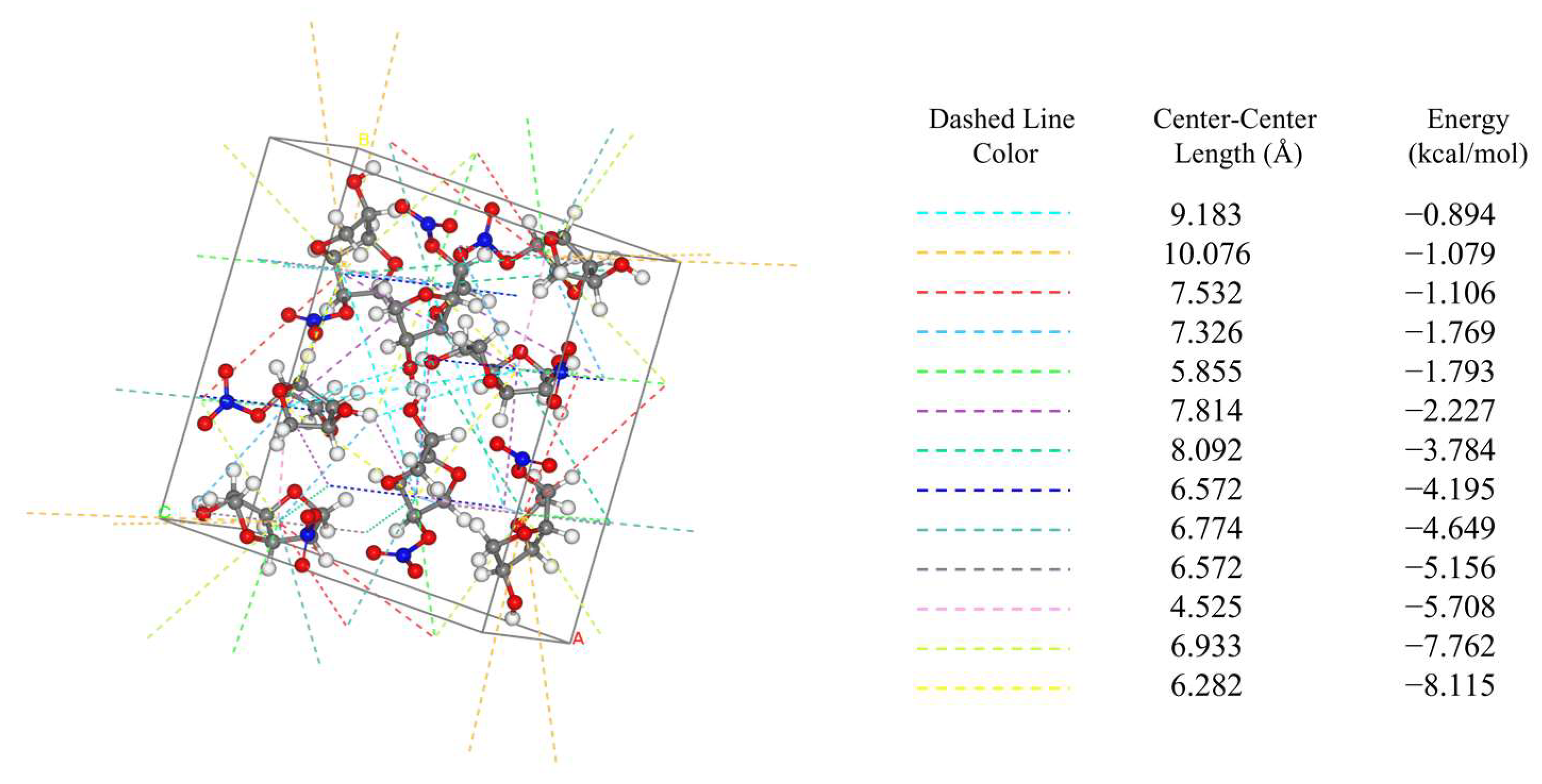
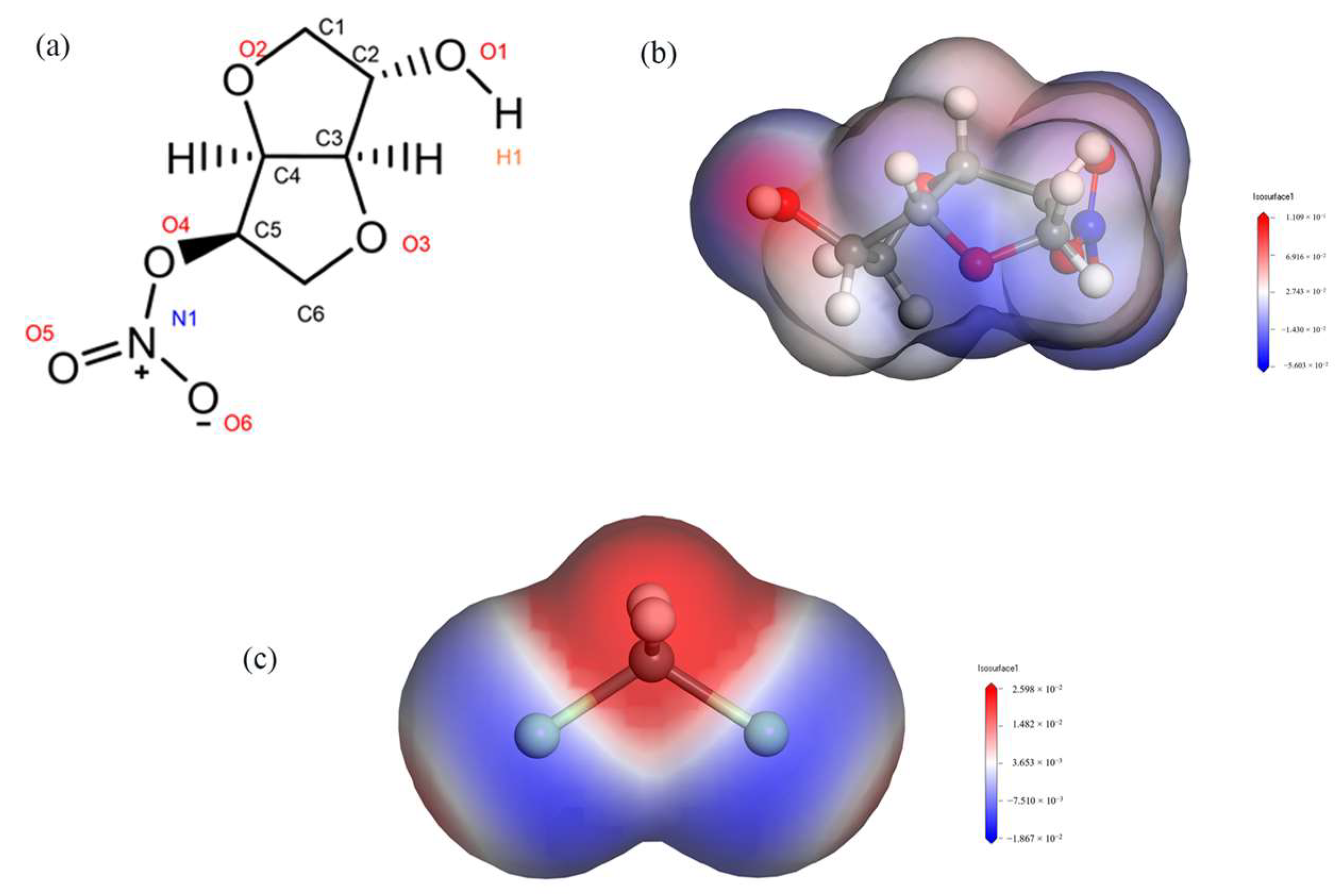
2.1. Intermolecular Interactions within the Crystal Cell
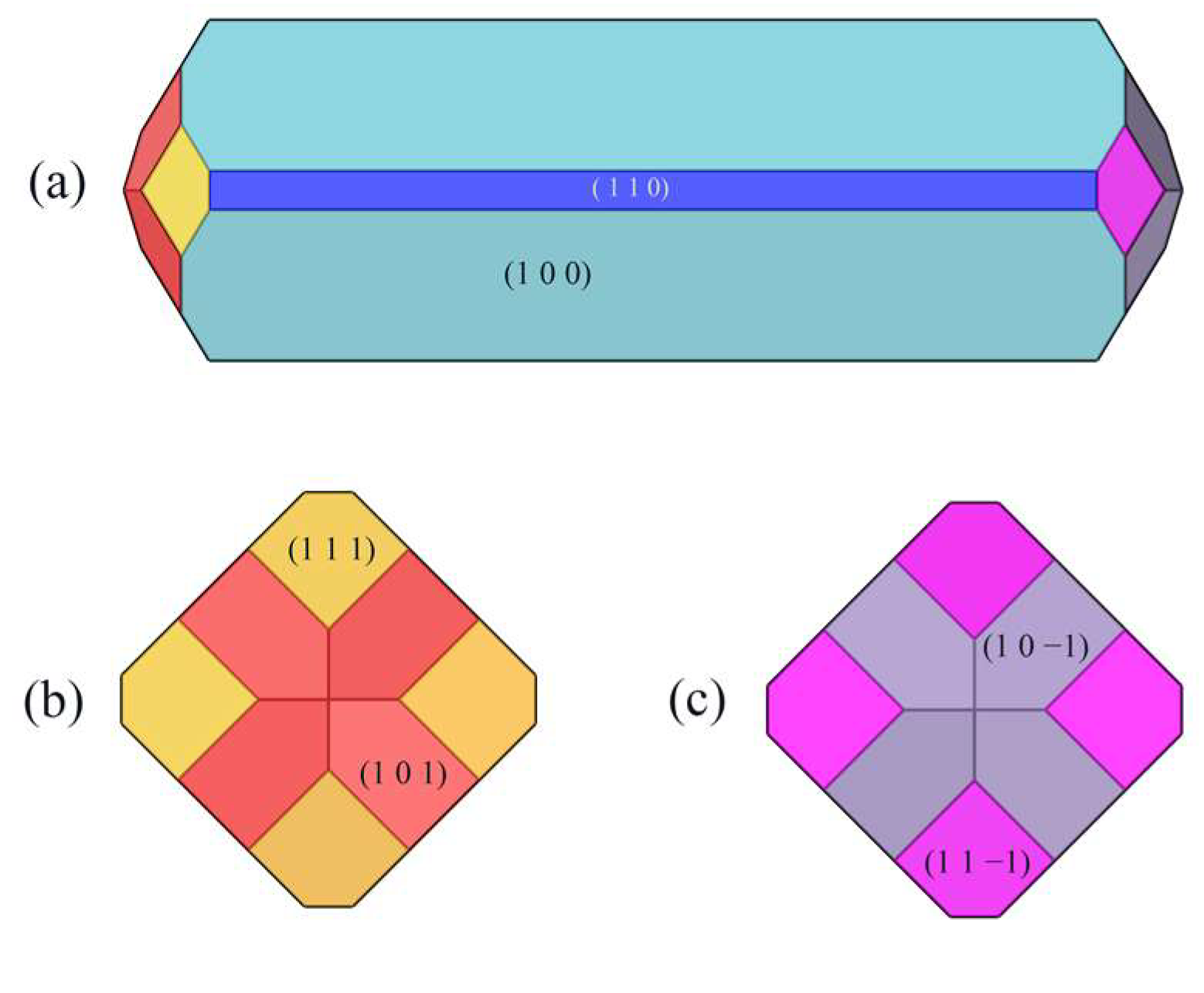
2.2. Crystal Morphology in Vacuum
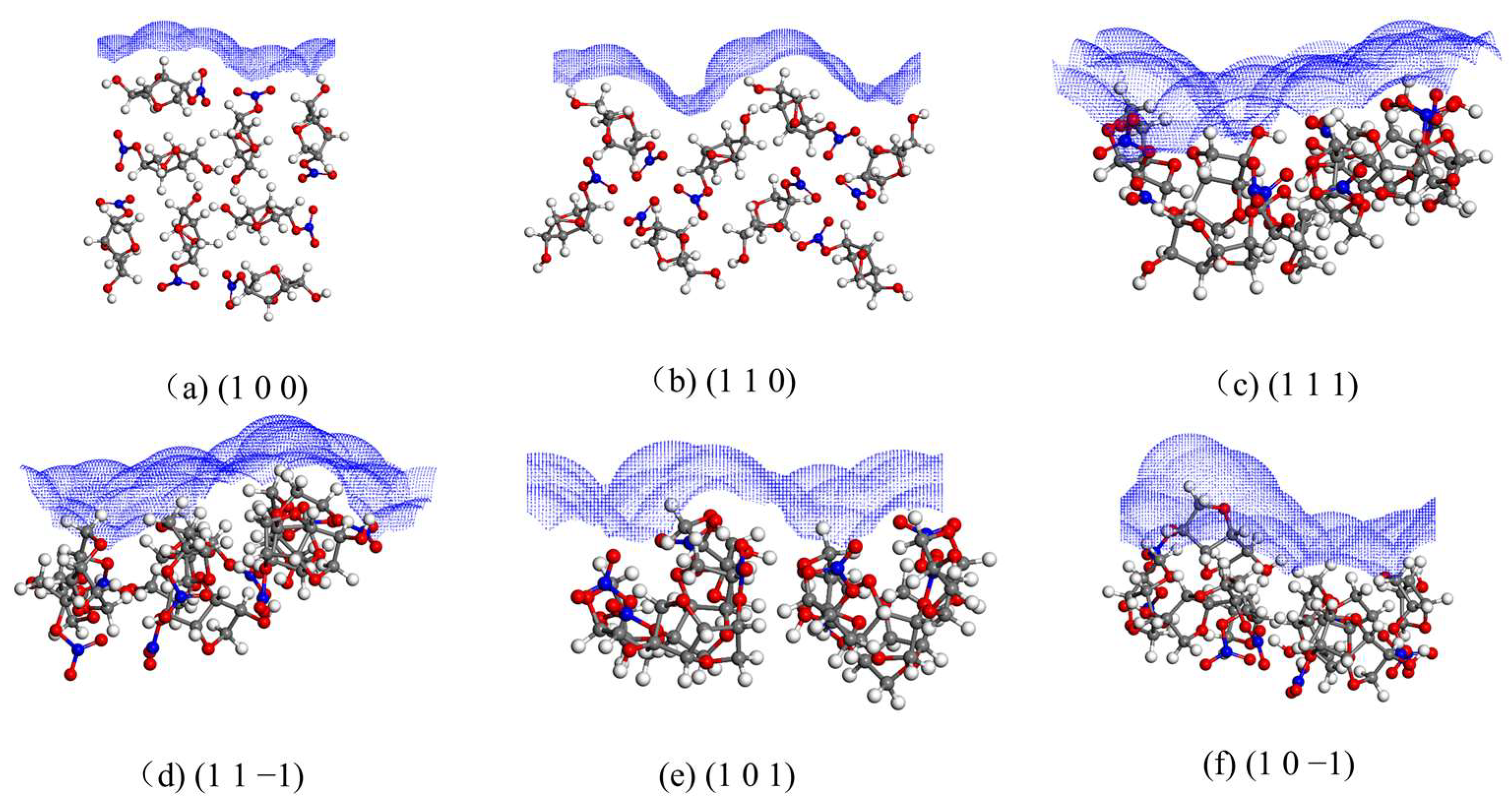
2.3. Structural Properties of Crystal Faces
2.4. Effect of Solvent CH2Cl2 on the Morphology of ISMN
2.4.1. Crystal Morphology in the CH2Cl2 Solvent System
2.4.2. Diffusion Coefficient
2.4.3. Radial Distribution Function Analysis
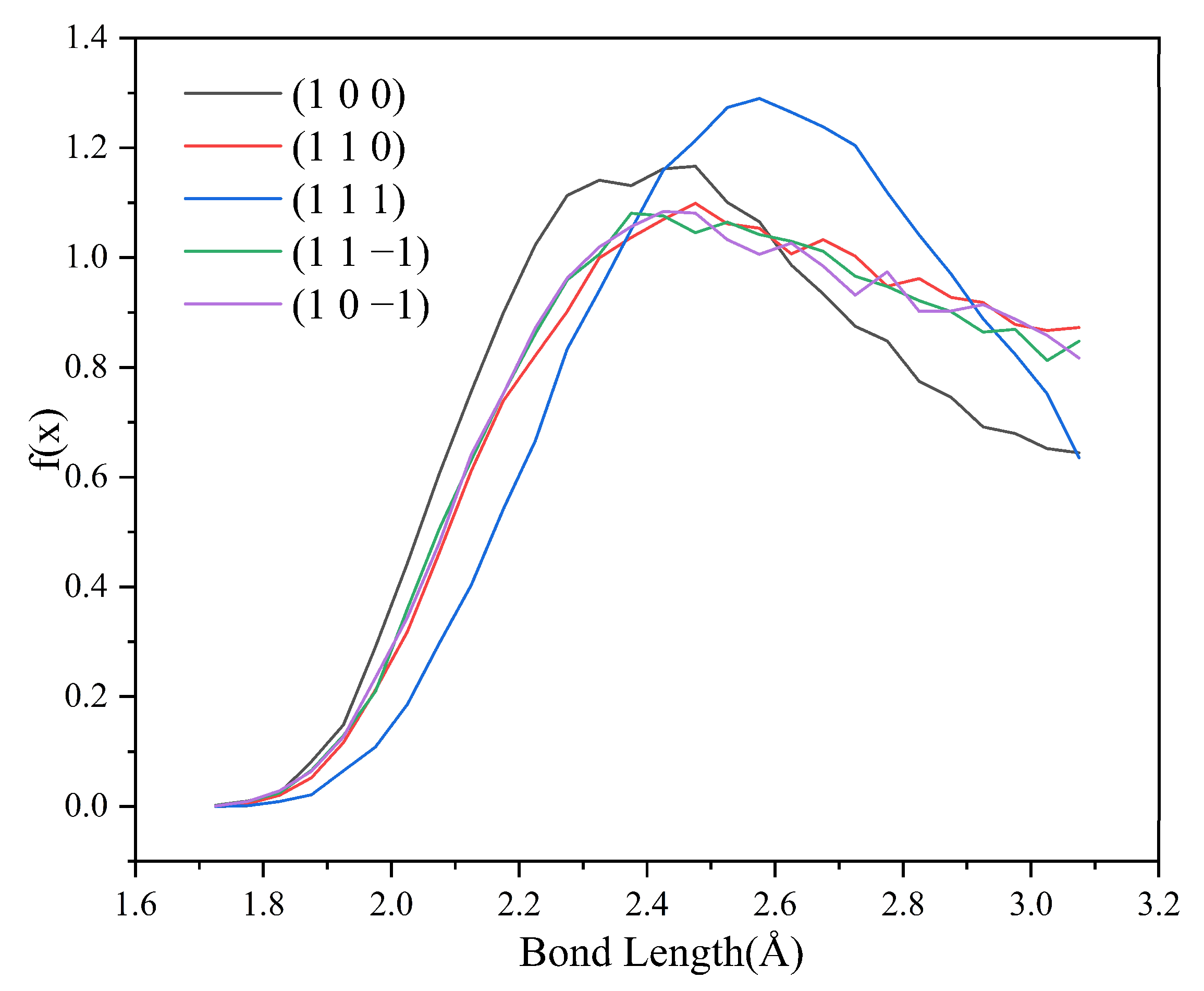
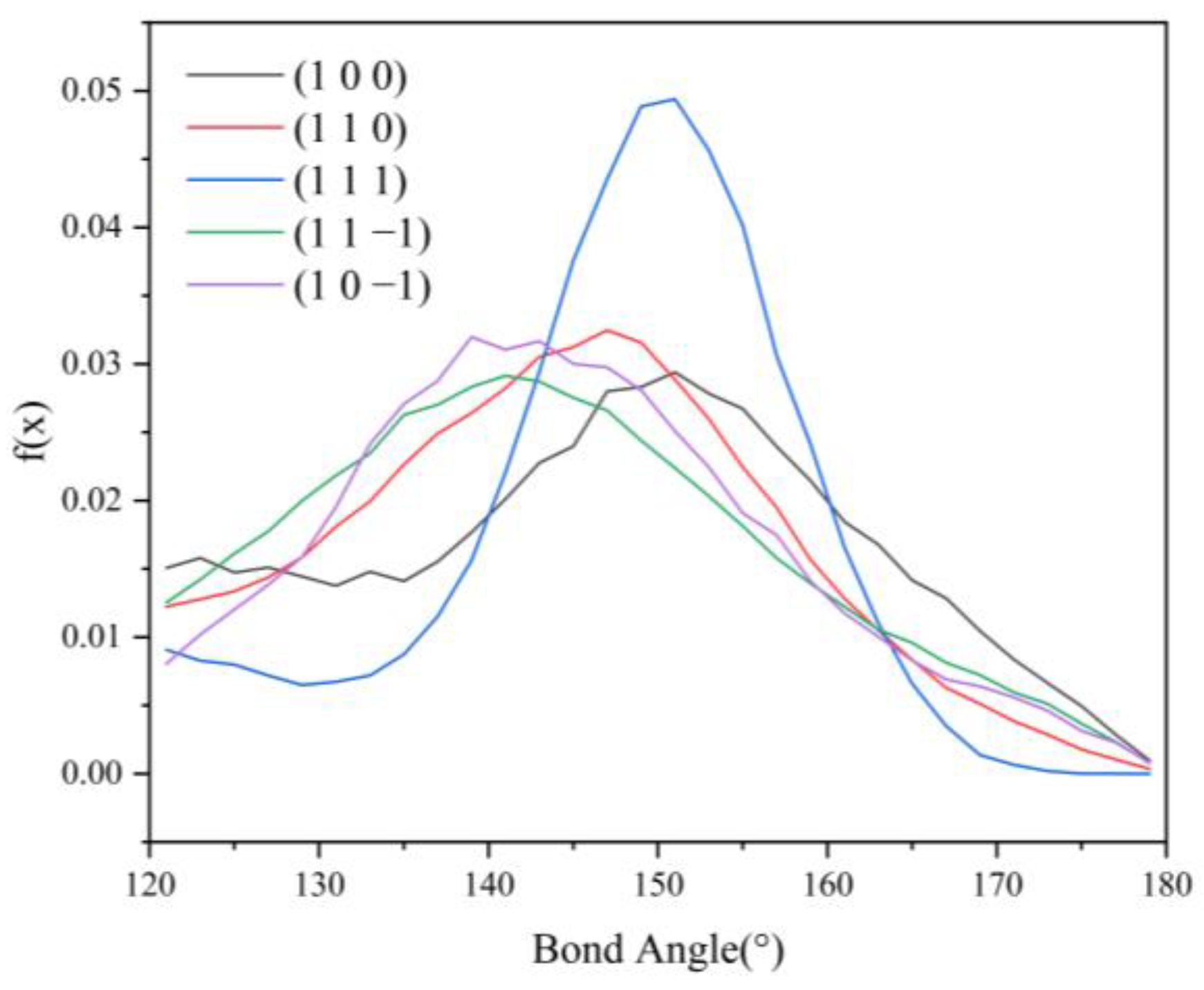
2.4.4. Analysis of Hydrogen Bonding Interactions
2.4.5. Effect of Hydrogen Bonds on Changes in Crystal Morphology
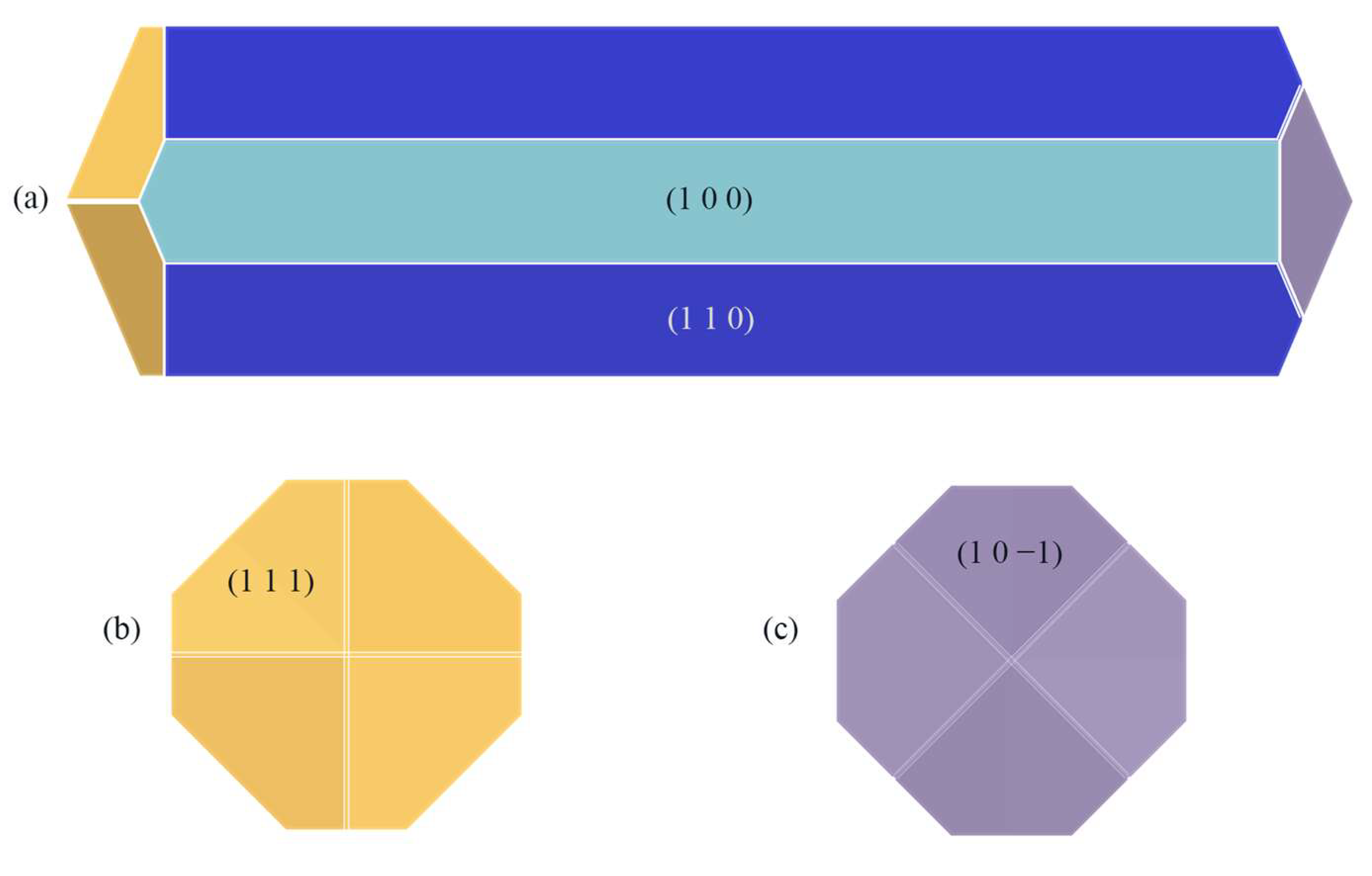
2.5. Prediction of Crystal Morphology in the CH2Cl2-C6H14 Solvent System and Analysis of Results
2.5.1. Crystal Morphology in the CH2Cl2-C6H14 Solvent System
2.5.2. Diffusion Coefficient of CH2Cl2 in the CH2Cl2-C6H14 Solvent System
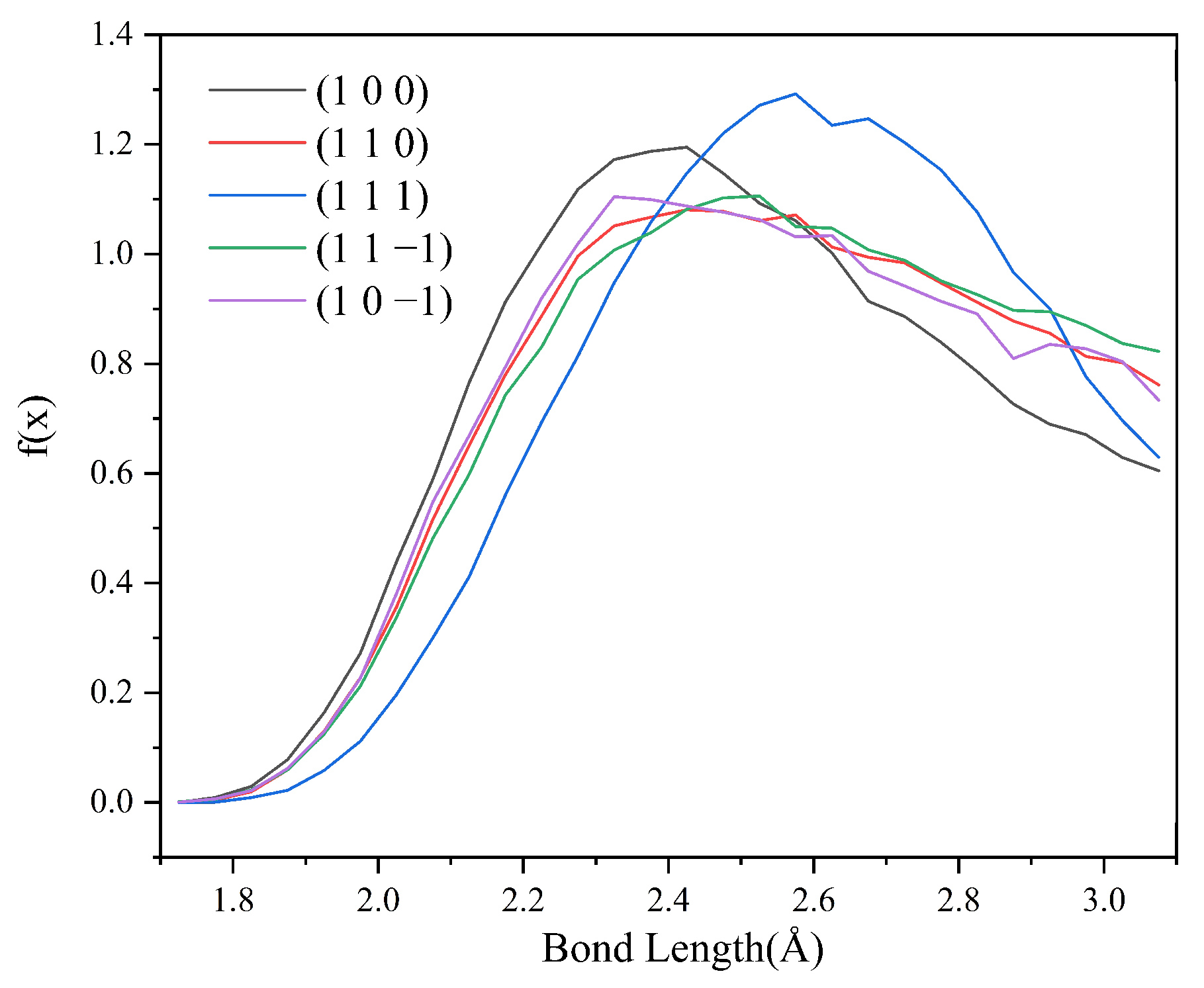
2.5.3. Analysis of Hydrogen Bond Interactions in the CH2Cl2-C6H14 Solvent System
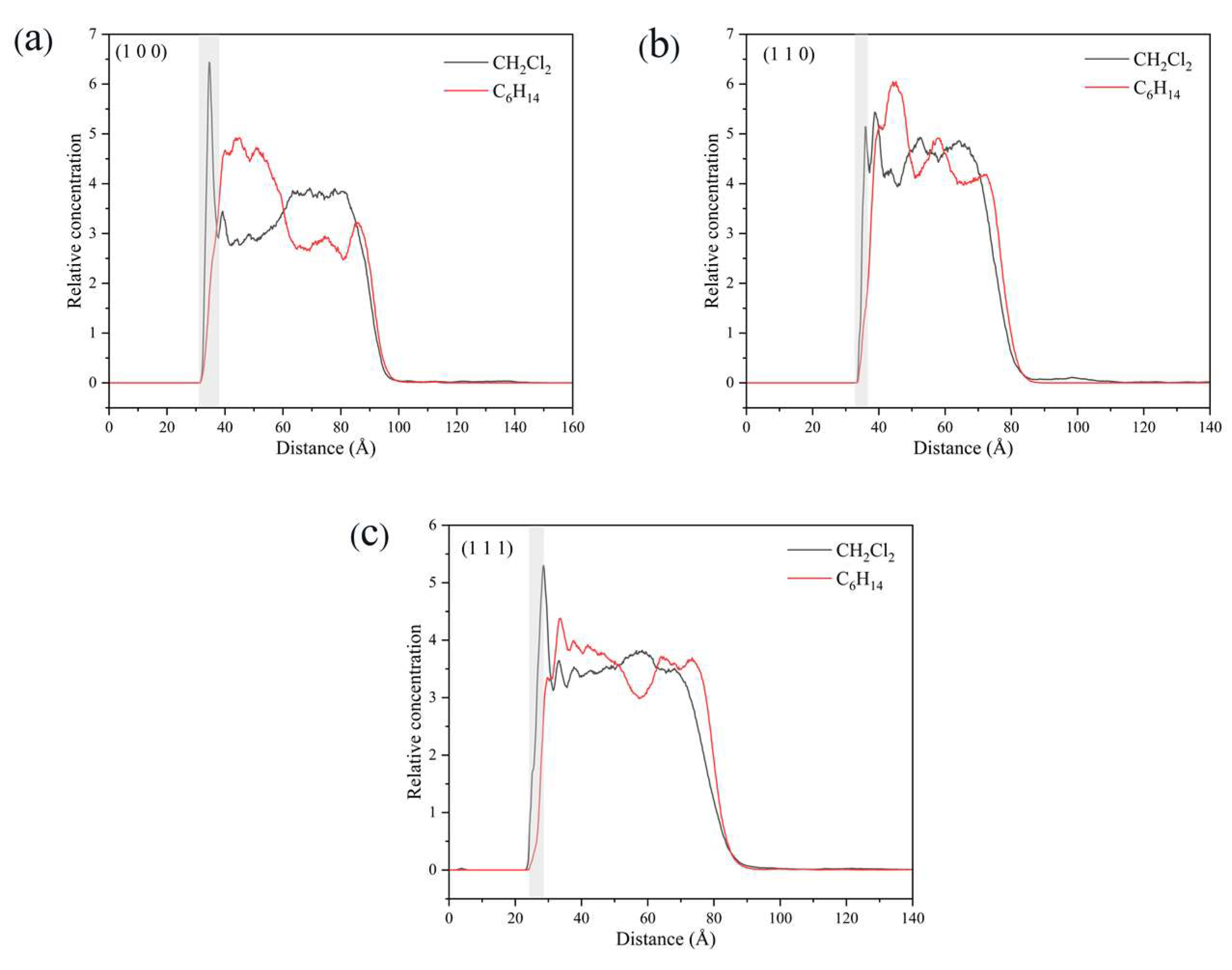
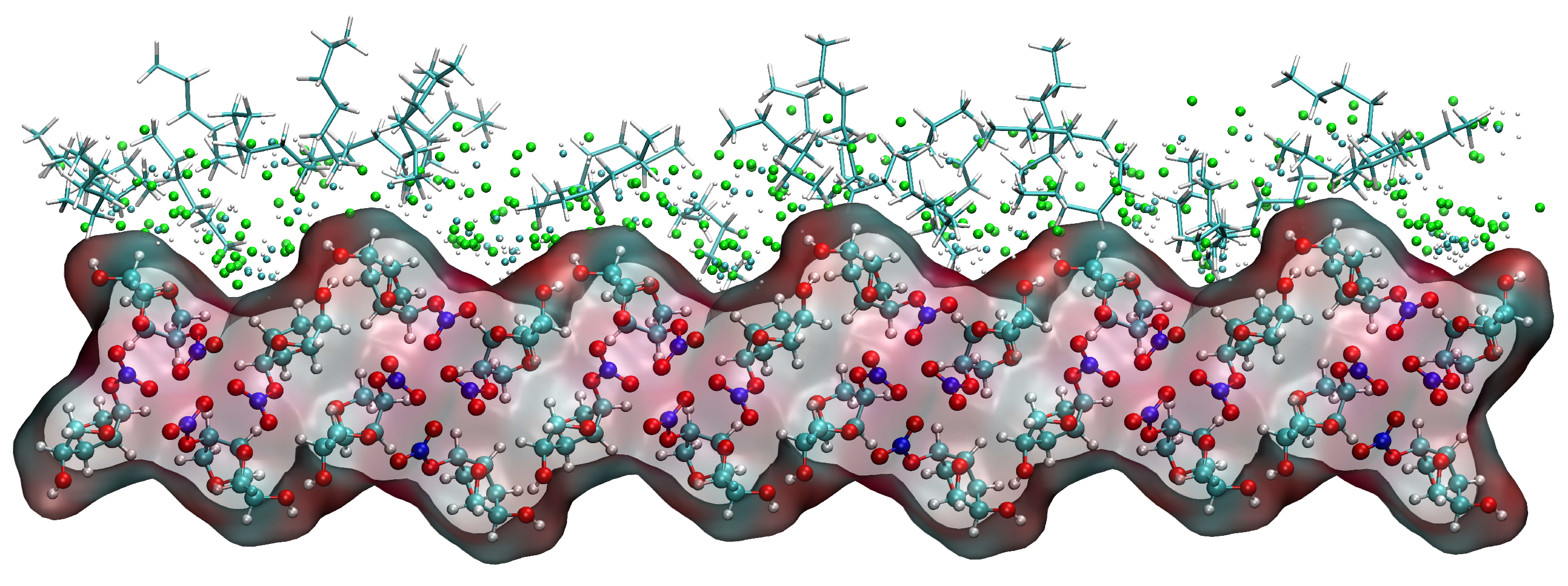
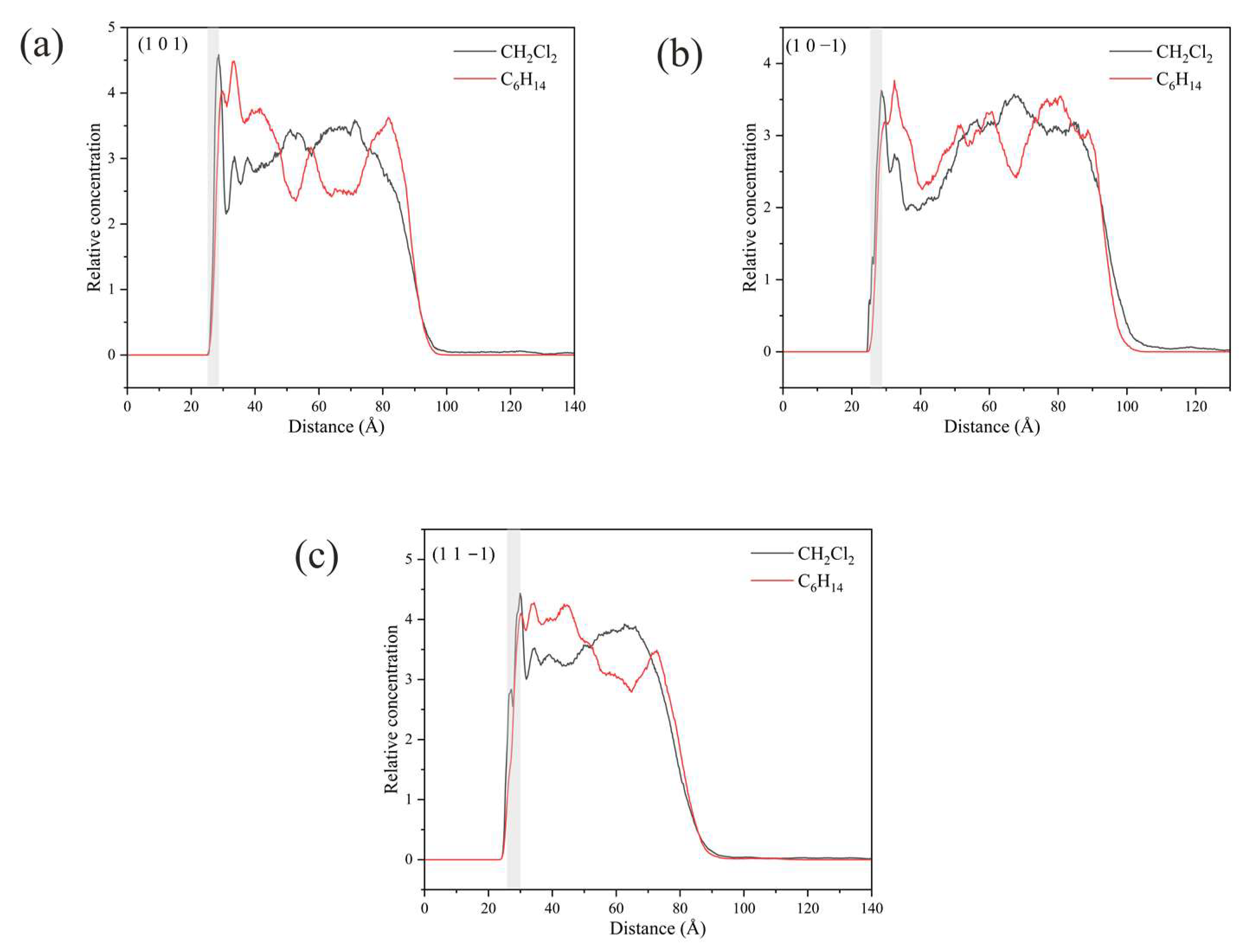
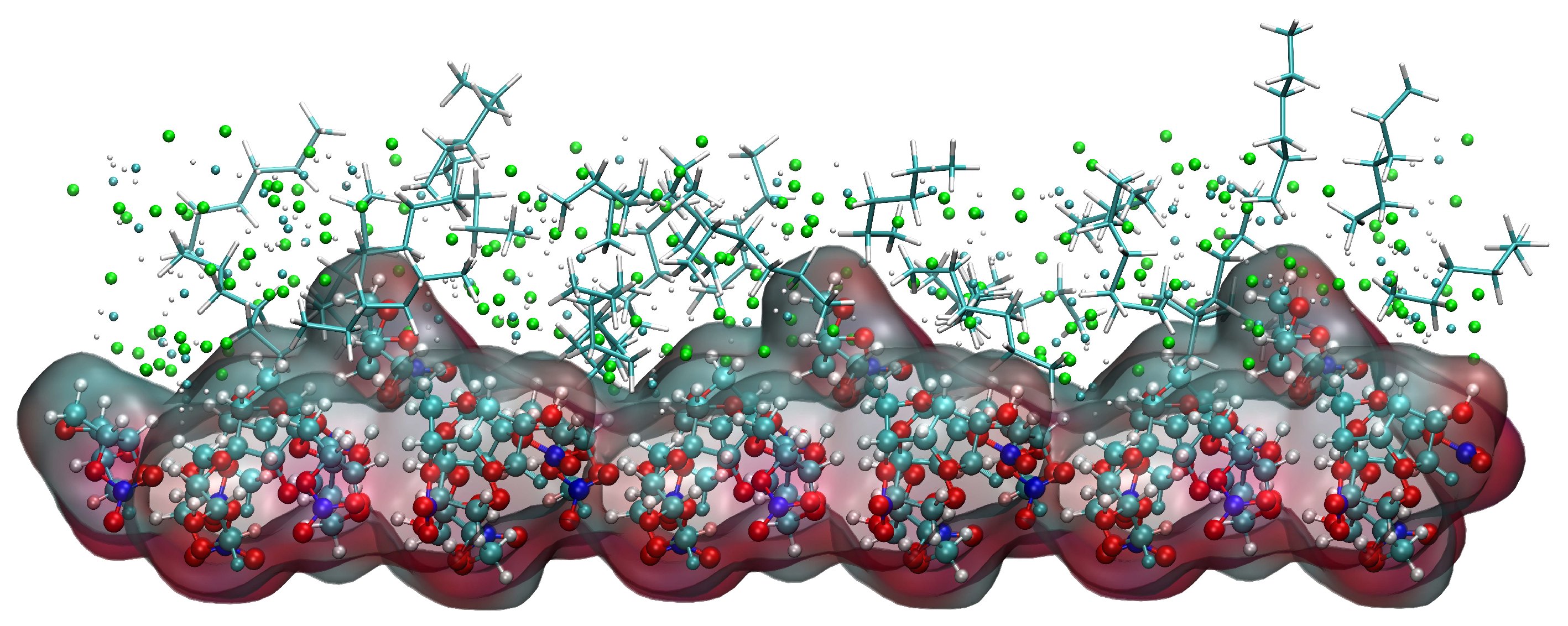
2.5.4. Relative Concentration Distributions of CH2Cl2 and C6H14
2.5.5. Analysis of Changes in the Crystal Morphology of ISMN in the CH2Cl2-C6H14 Solvent System
3. Simulation
3.1. Calculation Methodology
3.2. Simulation Methods and Details
3.2.1. Optimization of Lattice Parameters
3.2.2. Construction of the Simulation System
3.2.3. Molecular Dynamics Simulation Details
4. Conclusions
- In the CH2Cl2 solvent system, the type of interaction present at each crystal face was determined using RDF analysis. Hydrogen bond interactions determine the crystal morphology to a certain extent, with the bond length, bond angle, and number of hydrogen bonds affecting the crystal morphology.
- The use of C6H14 as a volatile agent accelerates crystal growth and increases the diffusion rate of CH2Cl2 molecules; the addition of C6H14 affects the interaction between the crystal face layer and the solution layer mainly by influencing the number of hydrogen bonds in the vicinity of the crystal face.
- The addition of C6H14 did not have a major effect on the overall morphology of the crystals, mainly because the three morphologically important growth faces, (1 0 0), (1 1 0), and (1 1 1), are still dominated by CH2Cl2 solvent molecules in the vicinity of the crystal faces.
- In the CH2Cl2-C6H14 solvent system, the difference in density distribution near the (1 0 −1) crystal face may account for the reappearance of the (1 1 −1) crystal face as a morphologically important growth surface.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bolourchian, N.; Nili, M.; Foroutan, S.M.; Mahboubi, A.; Nokhodchi, A. The use of cooling and anti-solvent precipitation technique to tailor dissolution and physicochemical properties of meloxicam for better performance. J. Drug Deliv. Sci. Technol. 2020, 55, 101485. [Google Scholar] [CrossRef]
- Kumar, D.; Thipparaboina, R.; Shastri, N.R. Impact of Nisoldipine Crystal Morphology on Its Biopharmaceutical Properties: A Layer Docking Assisted Study. Org. Process Res. Dev. 2015, 19, 1912–1917. [Google Scholar] [CrossRef]
- Modi, S.R.; Dantuluri, A.K.R.; Puri, V.; Pawar, Y.B.; Nandekar, P.; Sangamwar, A.T.; Perumalla, S.R.; Sun, C.C.; Bansal, A.K. Impact of Crystal Habit on Biopharmaceutical Performance of Celecoxib. Cryst. Growth Des. 2013, 13, 2824–2832. [Google Scholar] [CrossRef]
- Gouthami, K.S.; Kumar, D.; Thipparaboina, R.; Chavan, R.B.; Shastri, N.R. Can crystal engineering be as beneficial as micronisation and overcome its pitfalls?: A case study with cilostazol. Int. J. Pharm. 2015, 491, 26–34. [Google Scholar] [CrossRef]
- Wu, P.; Huang, G.; Liu, W.; Gao, Q.; Gong, S.; Jin, M.; Yu, K. Study on the Characterization and Stability of Different Crystal Habits of Tacrolimus Monohydrate. Chin. J. Mod. Appl. Pharm. 2022, 39, 1856–1862. [Google Scholar]
- Dhondale, M.R.; Thakor, P.; Nambiar, A.G.; Singh, M.; Agrawal, A.K.; Shastri, N.R.; Kumar, D. Co-Crystallization Approach to Enhance the Stability of Moisture-Sensitive Drugs. Pharmaceutics 2023, 15, 189. [Google Scholar] [CrossRef]
- Maghsoodi, M.; Kiafar, F. Co-precipitation with PVP and Agar to Improve Physicomechanical Properties of Ibuprofen. Iran. J. Basic Med. Sci. 2013, 16, 635–642. [Google Scholar] [PubMed]
- Zhao, Z.; Liu, G.; Lin, Q.; Jiang, Y. Co-Crystal of Paracetamol and Trimethylglycine Prepared by a Supercritical CO2 Anti-Solvent Process. Chem. Eng. Technol. 2018, 41, 1122–1131. [Google Scholar] [CrossRef]
- Ren, Y.; Shen, J.; Yu, K.X.; Phan, C.U.; Chen, G.X.; Liu, J.Y.; Hu, X.R.; Feng, J.Y. Impact of Crystal Habit on Solubility of Ticagrelor. Crystals 2019, 9, 556. [Google Scholar] [CrossRef]
- Phan, C.U.; Shen, J.; Yu, K.X.; Mao, J.M.; Tang, G.P. Impact of Crystal Habit on the Dissolution Rate and In Vivo Pharmacokinetics of Sorafenib Tosylate. Molecules 2021, 26, 3469. [Google Scholar] [CrossRef]
- Pu, S.Y.; Hadinoto, K. Salting-Out crystallization of glycopeptide Vancomycin: Phase behavior study to control the crystal habit. Chem. Eng. Sci. 2022, 262, 118057. [Google Scholar] [CrossRef]
- Cao, Y.C.; Yao, T.; Zhang, G.M.; Zhou, Z.Y.; Zhai, L.H.; Wu, S.G. Nucleation behavior of isosorbide 5-mononitrate revealed from metastable zone widths by combining nucleation theory model and molecular simulation. J. Mol. Liq. 2022, 363, 119846. [Google Scholar] [CrossRef]
- Zhu, S.G.; Yang, J.T.; Zhang, G.M.; Chen, C.F.; Zhang, F.L. An Improved Process for Industrial Production of Isosorbide-5-mononitrate: Recycling of Wastes. Org. Process Res. Dev. 2018, 22, 991–995. [Google Scholar] [CrossRef]
- Yan, X.Y.; Wang, N.; Ji, X.T.; Feng, Y.G.; Li, J.; Wang, T.; Huang, X.; Hao, H.X. The Effect of Solvents on the Crystal Morphology of Pyriproxyfen. Crystals 2023, 13, 195. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, X.G.; Cai, L.; Lu, W.; Gao, K.P.; Shi, Z.Q.; Zhang, Q.M. Pharmacokinetics of three organic nitrates in Chinese healthy male volunteers. Arzneim.-Drug Res. 2004, 54, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; Marston, R.W.; Quigley, P.F.; Roberts, S.M. New preparative routes to isosorbide 5-mononitrate. J. Chem. Soc.-Perkin Trans. 2000, 1, 1809–1810. [Google Scholar] [CrossRef]
- Song, L.C.; Lv, S.; Guo, H.; Cui, Y.B.; Zhang, X.; Zhang, S.G.; Tian, Y.; Yang, C.H. Crystal morphology regulation of pronamide through solvent selection: Prediction and implementation. J. Chem. Thermodyn. 2022, 169, 106743. [Google Scholar] [CrossRef]
- Chen, X.; He, L.; Li, X.; Zhou, Z.; Ren, Z. Molecular Simulation Studies on the Growth Process and Properties of Ammonium Dinitramide Crystal. J. Phys. Chem. C 2019, 123, 10940–10948. [Google Scholar] [CrossRef]
- Smith, L.A.; Duncan, A.; Thomson, G.B.; Roberts, K.J.; Machin, D.; McLeod, G. Crystallisation of sodium dodecyl sulphate from aqueous solution: Phase identification, crystal morphology, surface chemistry and kinetic interface roughening. J. Cryst. Growth 2004, 263, 480–490. [Google Scholar] [CrossRef]
- Clydesdale, G.; Roberts, K.J.; Docherty, R. HABIT95—A program for predicting the morphology of molecular crystals as a function of the growth environment. J. Cryst. Growth 1996, 166, 78–83. [Google Scholar] [CrossRef]
- Ma, C.Y.; Moldovan, A.A.; Maloney, A.G.P.; Roberts, K.J. Exploring the CSD Drug Subset: An Analysis of Lattice Energies and Constituent Intermolecular Interactions for the Crystal Structures of Pharmaceuticals. J. Pharm. Sci. 2023, 112, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Childs, S.L.; Wood, P.A.; Rodríguez-Hornedo, N.; Reddy, L.S.; Hardcastle, K.I. Analysis of 50 Crystal Structures Containing Carbamazepine Using the Materials Module of Mercury CSD. Cryst. Growth Des. 2009, 9, 1869–1888. [Google Scholar] [CrossRef]
- Wang, K.; Huang, S.L.; Zhu, W.H. Solvent effects on the crystal morphology of β-HMX studied by modified attachment energy and occupancy models. J. Cryst. Growth 2022, 585, 126605. [Google Scholar] [CrossRef]
- Lan, G.C.; Jin, S.H.; Li, J.; Wang, J.Y.; Li, J.X.; Chen, S.S.; Li, L.J. The study of external growth environments on the crystal morphology of ε-HNIW by molecular dynamics simulation. J. Mater. Sci. 2018, 53, 12921–12936. [Google Scholar] [CrossRef]
- Liu, N.; Li, Y.N.; Zeman, S.; Shu, Y.J.; Wang, B.Z.; Zhou, Y.S.; Zhao, Q.L.; Wang, W.L. Crystal morphology of 3,4-bis(3-nitrofurazan-4-yl)furoxan (DNTF) in a solvent system: Molecular dynamics simulation and sensitivity study. Crystengcomm 2016, 18, 2843–2851. [Google Scholar] [CrossRef]
- Li, L.; Ji, X.T.; Cheng, X.W.; Li, D.N.; Wang, T.; Huang, X.; Wang, N.; Yin, Q.X.; Hao, H.X. Effect of the solvent on the morphology of sulfamerazine crystals and its molecular mechanism. Crystengcomm 2022, 24, 5497–5506. [Google Scholar] [CrossRef]
- Lutz, D.; Rasper, J.; Gielsdorf, W.; Settlage, J.A.; Jaeger, H. Improved automated simultaneous determination of isosorbide dinitrate and its metabolites in plasma by capillary column gas chromatography. J. High Resolut. Chromatogr. Chromatogr. Commun. 1984, 7, 58–65. [Google Scholar] [CrossRef]
- Chen, G.; Chen, C.Y.; Xia, M.Z.; Lei, W.; Wang, F.Y.; Gong, X.D. A study of the solvent effect on the crystal morphology of hexogen by means of molecular dynamics simulations. RSC Adv. 2015, 5, 25581–25589. [Google Scholar] [CrossRef]
- Chen, F.; Zhou, T.; Li, J.; Wang, X.L.; Cao, D.L.; Wang, J.L.; Yang, Z.J. Crystal morphology of dihydroxylammonium 5,5′-bistetrazole-1,1′-diolate (TKX-50) under solvents system with different polarity using molecular dynamics. Comput. Mater. Sci. 2019, 168, 48–57. [Google Scholar] [CrossRef]
- Liu, N.; Zhou, C.; Wu, Z.K.; Lu, X.M.; Wang, B.Z.; Wang, W.L. Theoretical study on crystal morphologies of 1,1-diamino-2,2-dinitroethene in solvents: Modified attachment energy model and occupancy model. J. Mol. Graph. Model. 2018, 85, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Li, Q.F.; Gou, R.J.; Zhang, S.H.; Ren, F.D.; Wang, L.; Guan, R. Growth morphology of CL-20/HMX cocrystal explosive: Insights from solvent behavior under different temperatures. J. Mol. Model. 2017, 23, 360. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Xu, J.P.; Han, Y.; Zhang, J.L.; Li, W. Solvent-Antisolvent Competitive Interactions Mediate Imidacloprid Polymorphs in Antisolvent Crystallization. Cryst. Growth Des. 2021, 21, 4318–4328. [Google Scholar] [CrossRef]
- Zhu, D.; Zhang, S.H.; Cui, P.P.; Wang, C.; Dai, J.Y.; Zhou, L.; Huang, Y.H.; Hou, B.H.; Hao, H.X.; Zhou, L.N.; et al. Solvent Effects on Catechol Crystal Habits and Aspect Ratios: A Combination of Experiments and Molecular Dynamics Simulation Study. Crystals 2020, 10, 316. [Google Scholar] [CrossRef]
- Sun, T.; Liu, Q.; Xiao, J.J.; Zhao, F.; Xiao, H.M. Molecular Dynamics Simulation of Interface Interactions and Mechanical Properties of CL-20/HMX Cocrystal and Its Based PBXs. Acta Chim. Sin. 2014, 72, 1036–1042. [Google Scholar] [CrossRef]
- Li, J.; Jin, S.H.; Lan, G.C.; Ma, X.; Ruan, J.; Zhang, B.; Chen, S.S.; Li, L.J. Morphology control of 3-nitro-1,2,4-triazole-5-one (NTO) by molecular dynamics simulation. Crystengcomm 2018, 20, 6252–6260. [Google Scholar] [CrossRef]
- Rozas, I. On the nature of hydrogen bonds: An overview on computational studies and a word about patterns. Phys. Chem. Chem. Phys. 2007, 9, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Winn, D.; Doherty, M.F. Modeling crystal shapes of organic materials grown from solution. Aiche J. 2000, 46, 1348–1367. [Google Scholar] [CrossRef]
- Deij, M.A.; van Eupen, J.; Meekes, H.; Verwer, P.; Bennema, P.; Vlieg, E. Experimental and computational morphology of three polymorphs of the free base of Venlafaxine: A comparison of morphology prediction methods. Int. J. Pharm. 2008, 353, 113–123. [Google Scholar] [CrossRef]
- Zhao, Y.P.; Su, G.W.; Liu, G.Z.; Wei, H.Y.; Dang, L.P. Effect of modification of binary solvent molecules on ε-CL-20 crystal morphology: A molecular dynamics study. Crystengcomm 2021, 23, 3524–3536. [Google Scholar] [CrossRef]
- Chen, H.; Duan, S.J.; Sun, Y.Z.; Song, X.F.; Yu, J.G. Molecular dynamics simulations of solvent effects on the crystal morphology of lithium carbonate. RSC Adv. 2020, 10, 5604–5609. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.L.; Dong, Y.M. Crystal morphology study of N,N′-diacetylchitobiose by molecular dynamics simulation. Carbohydr. Res. 2011, 346, 2457–2462. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.Z.; Zhao, X.; Rui, J.H.; Qiu, S.Q.; Li, Y.S. Molecular Dynamics Simulation of β-HMX Crystal Morphology Induced by Polymer Additives. Crystals 2022, 12, 164. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, Z.Z. Solvent effects on the crystal growth structure and morphology of the pharmaceutical dirithromycin. J. Cryst. Growth 2017, 480, 18–27. [Google Scholar] [CrossRef]
- Cheng, X.; Huang, X.; Zheng, Z.; Dong, Y.; Wang, T.; Hao, H. Toward Understanding the Growth of Cefradine in Aqueous Solution. Cryst. Growth Des. 2021, 21, 1993–2004. [Google Scholar] [CrossRef]
- Liang, C.J.; Zhuang, J.H.; Zhuang, C.H.; Zhang, Z.X.; Lv, G.L.; Zhang, G.Q. Crystal morphology prediction and experimental verification of venlafaxine hydrochloride. Powder Diffr. 2022, 37, 133–142. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Niu, S.Y.; Lai, W.P.; Yu, T.; Ma, Y.D.; Gao, H.X.; Zhao, F.Q.; Ge, Z.X. Crystal morphology prediction of energetic materials grown from solution: Insights into the accurate calculation of attachment energies. Crystengcomm 2019, 21, 4910–4917. [Google Scholar] [CrossRef]
- Santschi, N.; Wagner, S.; Daniliuc, C.; Hermann, S.; Schäfers, M.; Gilmour, R. Synthesis of 2-[18F]Fluoro-2-deoxyisosorbide 5-mononitrate and Assessment of Its in vivo Biodistribution as Determined by Dynamic Positron Emission Tomography (PET). Chemmedchem 2015, 10, 1724–1732. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applications—Overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Mudalip, S.K.A.; Adam, F.; Abu Bakar, M.R. Evaluation of the intermolecular interactions and polymorphism of mefenamic acid crystals in N,N-dimethyl formamide solution: A molecular dynamics simulation and experimental study. Comptes Rendus Chim. 2019, 22, 771–778. [Google Scholar] [CrossRef]
- Cao, Z.; Wu, Y.; Niu, M.; Li, Y.; Li, T.; Ren, B. Exploring the solvent effect on risperidone (form I) crystal morphology: A combination of molecular dynamics simulation and experimental study. J. Mol. Liq. 2023, 376, 121358. [Google Scholar] [CrossRef]
- Lan, G.C.; Jin, S.H.; Li, J.; Lu, Z.Y.; Chen, M.L.; Wu, N.N.; Chen, S.S.; Li, L.J. Molecular dynamics investigation on the morphology of HNIW affected by the growth condition. J. Energetic Mater. 2019, 37, 44–56. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (h k l) | Multiplicity | dhkl (Å) | Eatt (kcal/mol/Unit Cell) | Total Habit Facet Area (Å2) | Total Habit Facet Area Percentage |
|---|---|---|---|---|---|
| (1 0 0) | 4 | 15.42 | −48.81 | 26,086.60 | 72.56% |
| (1 1 0) | 4 | 10.90 | −61.87 | 4621.26 | 12.85% |
| (1 0 1) | 4 | 6.05 | −177.07 | 1339.40 | 3.73% |
| (1 0 −1) | 4 | 6.05 | −177.07 | 1339.40 | 3.73% |
| (1 1 1) | 4 | 5.63 | −170.28 | 1282.82 | 3.57% |
| (1 1 −1) | 4 | 5.63 | −170.28 | 1282.82 | 3.57% |
| (h k l) | Ahkl (Å2) | Aacc (Å2) | S |
|---|---|---|---|
| (1 0 0) | 101.342 | 131.615 | 1.299 |
| (1 1 0) | 143.319 | 202.505 | 1.413 |
| (1 1 1) | 277.607 | 392.680 | 1.415 |
| (1 1 −1) | 277.607 | 391.540 | 1.410 |
| (1 0 1) | 258.448 | 340.865 | 1.319 |
| (1 0 −1) | 258.448 | 356.375 | 1.379 |
| (h k l) | dhkl (Å) | Eatt (kcal/mol/Unit Cell) | Zcry | Zhkl | Ahkl (Å2) | Abox (Å2) | Eint (kcal/mol) | E’att (kcal/mol/Unit Cell) |
|---|---|---|---|---|---|---|---|---|
| (1 0 0) | 15.42 | −48.81 | 8 | 8 | 101.34 | 2432.21 | −577.44 | −24.75 |
| (1 1 0) | 10.90 | −61.87 | 8 | 8 | 143.32 | 3439.66 | −919.91 | −23.54 |
| (1 1 1) | 5.63 | −170.28 | 8 | 8 | 277.61 | 6663.03 | −2211.43 | −78.15 |
| (1 1 −1) | 5.63 | −170.28 | 8 | 8 | 277.61 | 6663.03 | −2074.29 | −83.86 |
| (1 0 1) | 6.05 | −177.07 | 8 | 8 | 258.45 | 2326.03 | −805.57 | −87.56 |
| (1 0 −1) | 6.05 | −177.07 | 8 | 8 | 258.45 | 2326.03 | −840.60 | −83.67 |
| (h k l) | HB Length a (Å) | HB Angle b (°) | S1% c (HB Length < 2.5 Å) | S2% d (HB Angle > 150°) | NHB e | NH1 f | NHB-per H1 g | Abox h(Å2) | NHB-unit area i | Ahkl j (Å2) | NHB-hkl k |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (1 0 0) | 2.52 | 147.13 | 50.00% | 44.67% | 24.80 | 18 | 1.38 | 2432.21 | 0.0102 | 101.342 | 1.03 |
| (1 1 0) | 2.57 | 144.13 | 42.34% | 31.99% | 47.19 | 48 | 0.98 | 3439.66 | 0.0137 | 143.319 | 1.97 |
| (1 1 1) | 2.59 | 147.53 | 37.49% | 45.55% | 20.92 | 24 | 0.87 | 6663.03 | 0.0031 | 277.607 | 0.87 |
| (1 1 −1) | 2.56 | 144.21 | 43.60% | 31.93% | 49.50 | 48 | 1.03 | 6663.03 | 0.0074 | 277.607 | 2.06 |
| (1 0 −1) | 2.56 | 144.28 | 43.80% | 30.99% | 17.68 | 18 | 0.98 | 2326.03 | 0.0076 | 258.448 | 1.96 |
| (h k l) | dhkl (Å) | Eatt (kcal/mol/Unit Cell) | Zcry | Zhkl | Ahkl (Å2) | Abox (Å2) | Eint (kcal/mol) | E’att (kcal/mol/Unit Cell) |
|---|---|---|---|---|---|---|---|---|
| (1 0 0) | 15.42 | −48.81 | 8 | 8 | 101.34 | 2432.21 | −556.13 | −25.63 |
| (1 1 0) | 10.90 | −61.87 | 8 | 8 | 143.32 | 3439.66 | −903.67 | −24.22 |
| (1 1 1) | 5.63 | −170.28 | 8 | 8 | 277.61 | 6663.03 | −2126.07 | −81.70 |
| (1 1 −1) | 5.63 | −170.28 | 8 | 8 | 277.61 | 6663.03 | −1991.14 | −87.33 |
| (1 0 1) | 6.05 | −177.07 | 8 | 8 | 258.45 | 2326.03 | −766.75 | −91.87 |
| (1 0 −1) | 6.05 | −177.07 | 8 | 8 | 258.45 | 2326.03 | −786.70 | −89.66 |
| (h k l) | Dα1 a (10−8 m2/s) | Dα2 b (10−8 m2/s) |
|---|---|---|
| (1 0 0) | 0.2661 | 0.3192 |
| (1 1 0) | 0.2650 | 0.3657 |
| (1 1 1) | 0.3526 | 0.4465 |
| (1 1 −1) | 0.3773 | 0.4369 |
| (1 0 1) | 0.2670 | 0.3475 |
| (1 0 −1) | 0.3038 | 0.3673 |
| (h k l) | HB Length a (Å) | HB Angle b (°) | S1% c (HB Length < 2.5 Å) | S2% d (HB Angle > 150°) | NHB e | NH1 f | NHB-per H1 g | Abox h (Å2) | NHB-unit area i | Ahkl j (Å2) | NHB-hkl k |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (1 0 0) | 2.51 | 147.27 | 50.49% | 45.16% | 22.87 | 18 | 1.27 | 2432.21 | 0.0094 | 101.342 | 0.95 |
| (1 1 0) | 2.55 | 144.49 | 44.53% | 33.08% | 40.29 | 48 | 0.84 | 3439.66 | 0.0117 | 143.319 | 1.68 |
| (1 1 1) | 2.59 | 147.76 | 37.75% | 45.95% | 20.72 | 24 | 0.86 | 6663.03 | 0.0031 | 277.607 | 0.86 |
| (1 1 −1) | 2.57 | 143.93 | 43.01% | 31.22% | 39.87 | 48 | 0.83 | 6663.03 | 0.0060 | 277.607 | 1.66 |
| (1 0 −1) | 2.55 | 144.61 | 45.73% | 31.60% | 15.85 | 18 | 0.88 | 2326.03 | 0.0068 | 258.448 | 1.76 |
| Lattice Parameter | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) |
|---|---|---|---|---|---|---|
| Primitive unit cell | 15.992 | 15.992 | 6.523 | 90 | 90 | 90 |
| Cambridge Structural Database | 15.926 | 15.926 | 6.509 | 90 | 90 | 90 |
| COMPASS | 15.419 | 15.419 | 6.572 | 90 | 90 | 90 |
| Relative error | 3.58% | 3.58% | 0.75% | 0.00% | 0.00% | 0.00% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Zhang, G.; Zhou, Z.; Sun, Y.; Wang, Y.; Yang, Y.; Zhang, X. The Effect of Solvents on the Crystal Morphology of Isosorbide Mononitrate and Its Molecular Mechanisms. Molecules 2024, 29, 367. https://doi.org/10.3390/molecules29020367
Li P, Zhang G, Zhou Z, Sun Y, Wang Y, Yang Y, Zhang X. The Effect of Solvents on the Crystal Morphology of Isosorbide Mononitrate and Its Molecular Mechanisms. Molecules. 2024; 29(2):367. https://doi.org/10.3390/molecules29020367
Chicago/Turabian StyleLi, Penghui, Guimin Zhang, Zongyi Zhou, Ying Sun, Yan Wang, Yu Yang, and Xiaolai Zhang. 2024. "The Effect of Solvents on the Crystal Morphology of Isosorbide Mononitrate and Its Molecular Mechanisms" Molecules 29, no. 2: 367. https://doi.org/10.3390/molecules29020367
APA StyleLi, P., Zhang, G., Zhou, Z., Sun, Y., Wang, Y., Yang, Y., & Zhang, X. (2024). The Effect of Solvents on the Crystal Morphology of Isosorbide Mononitrate and Its Molecular Mechanisms. Molecules, 29(2), 367. https://doi.org/10.3390/molecules29020367