
Designing C9N10 Anchored Single Mo Atom as an Efficient Electrocatalyst for Nitrogen Fixation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
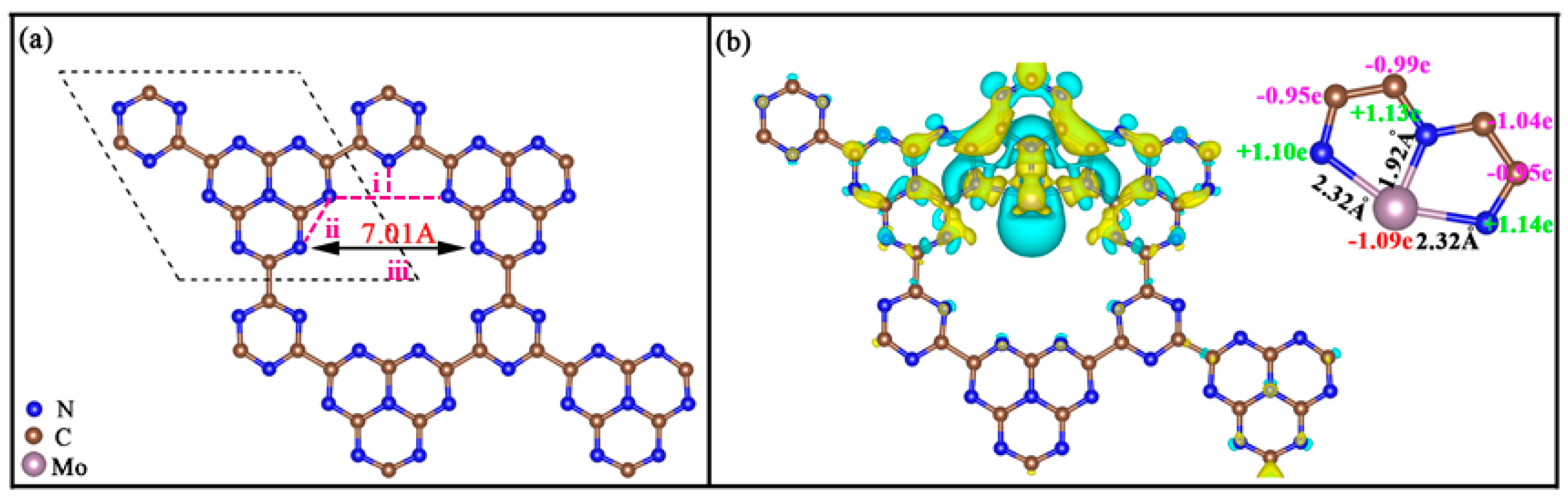
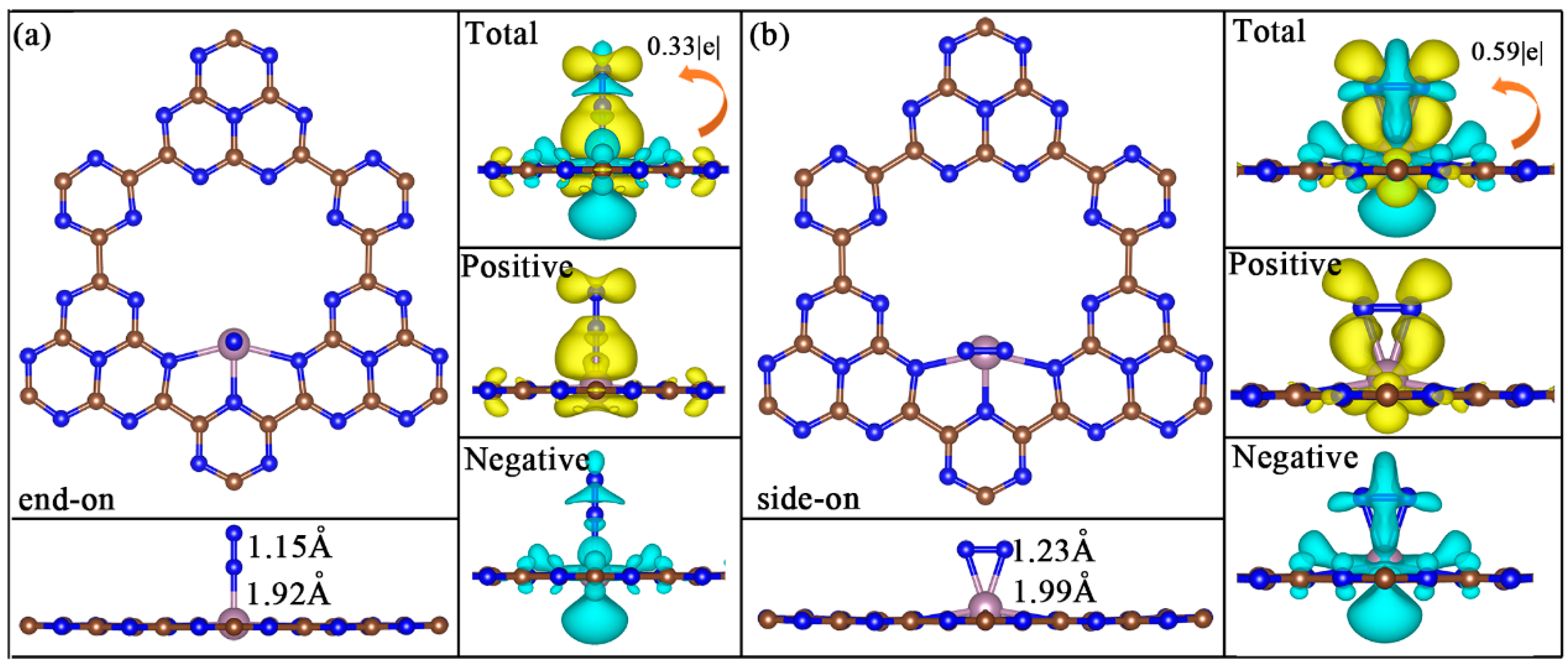
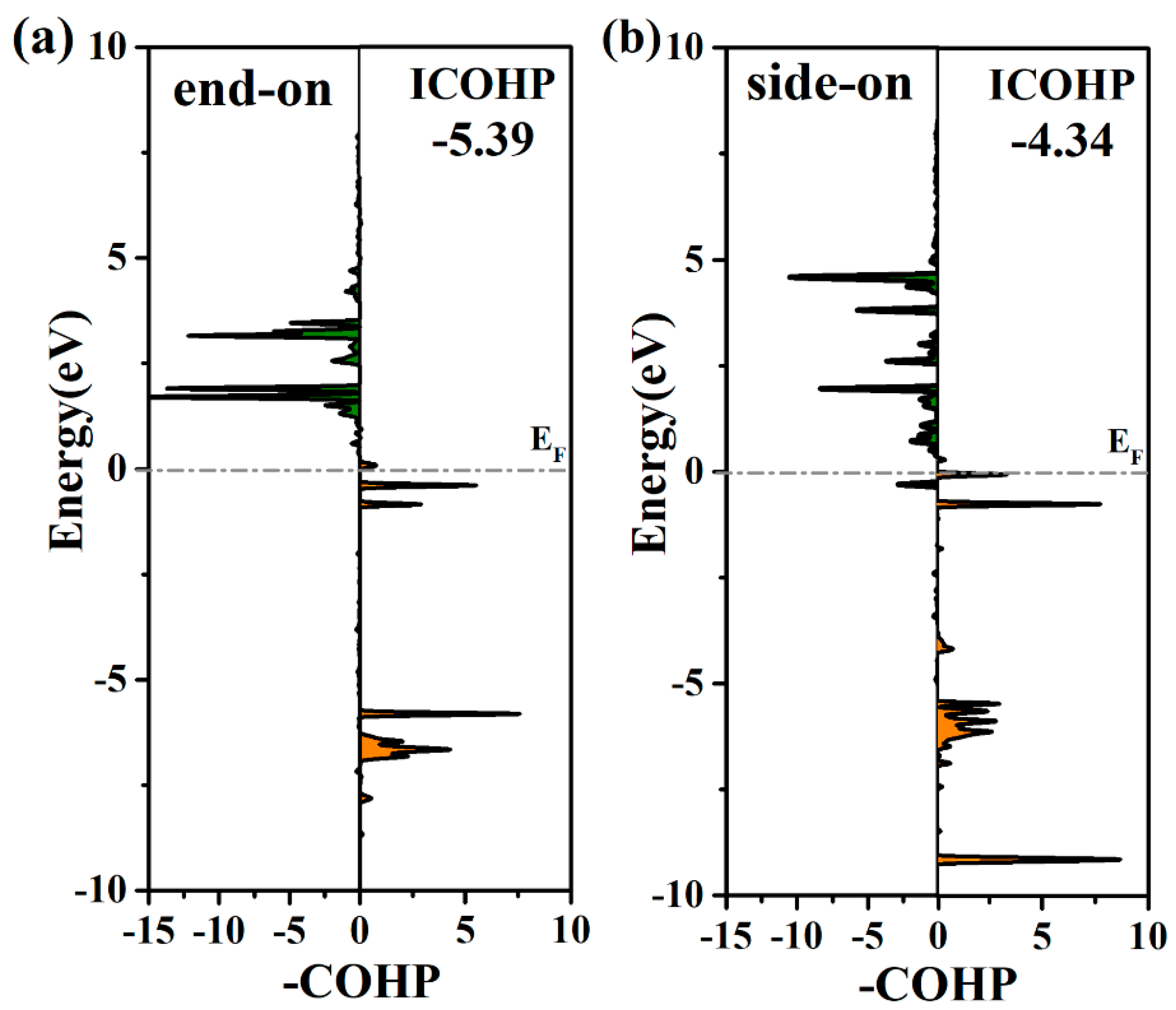
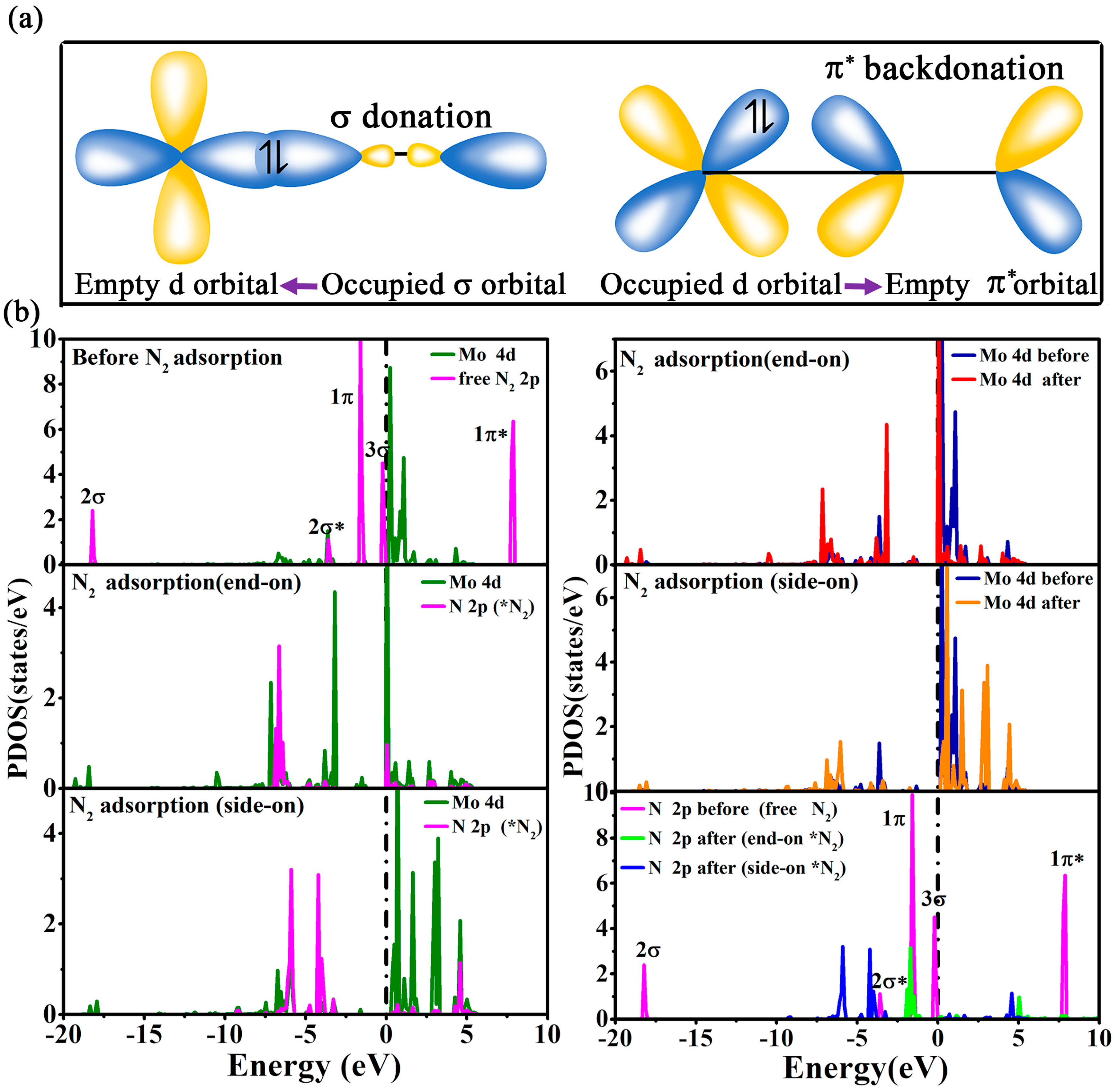
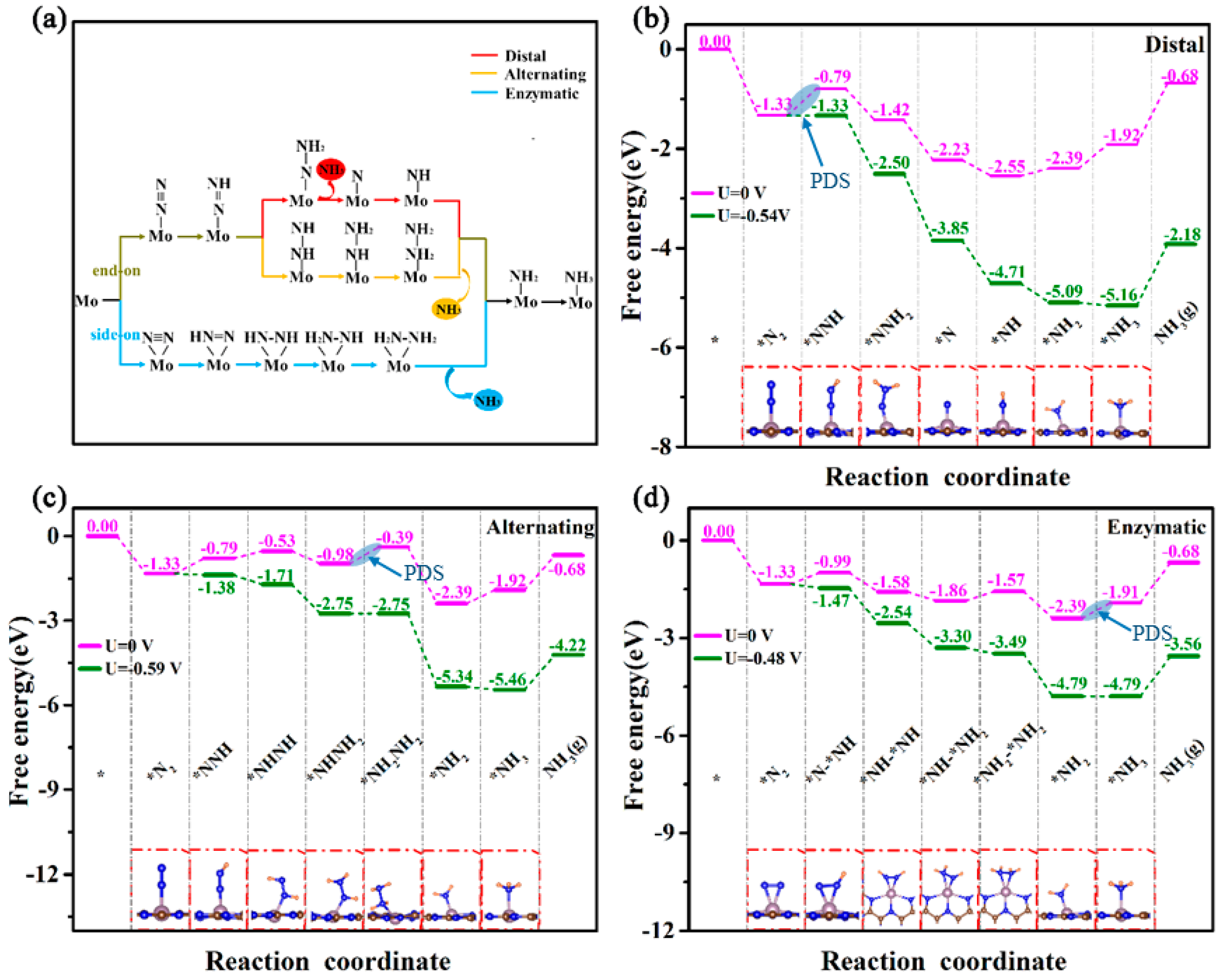
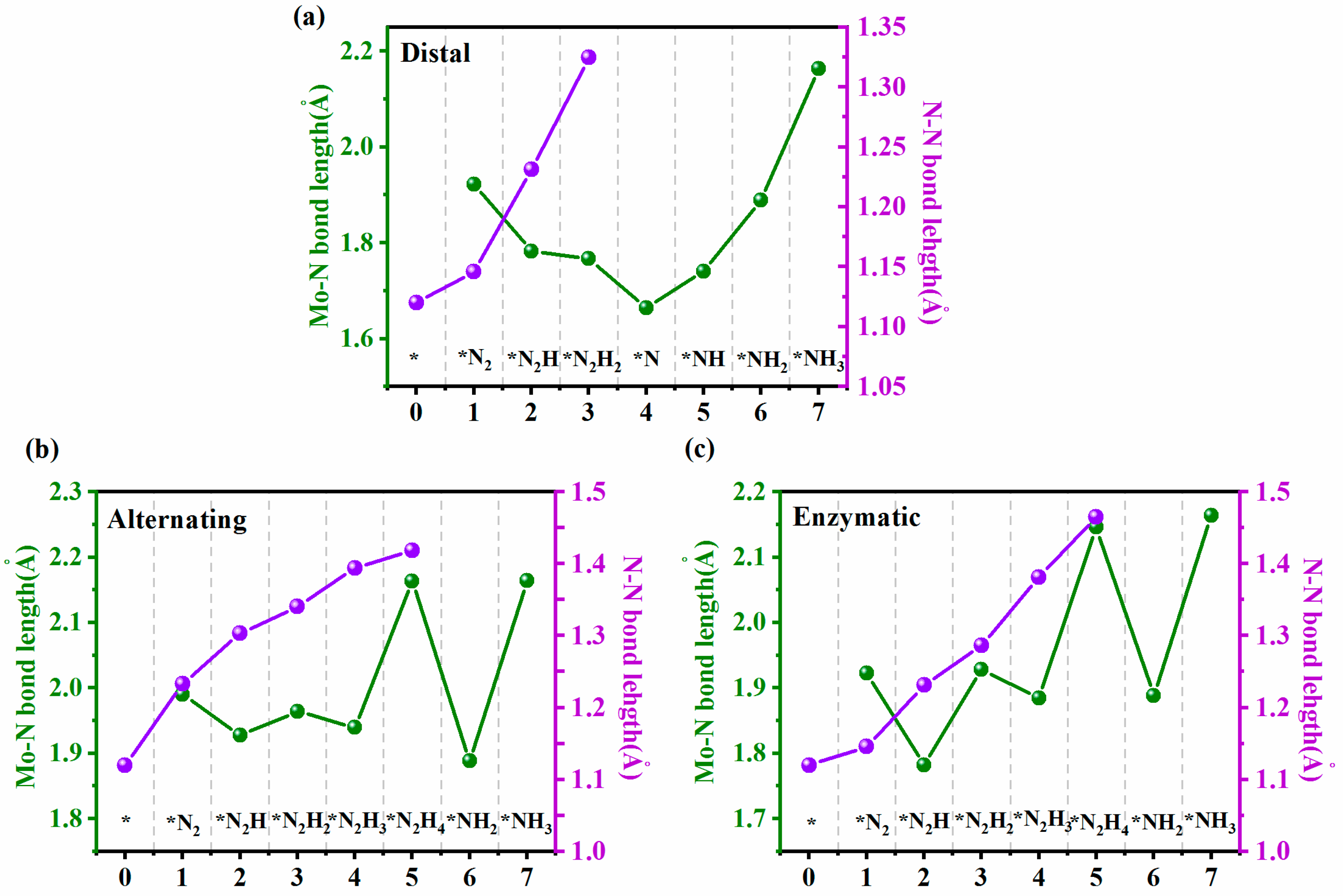
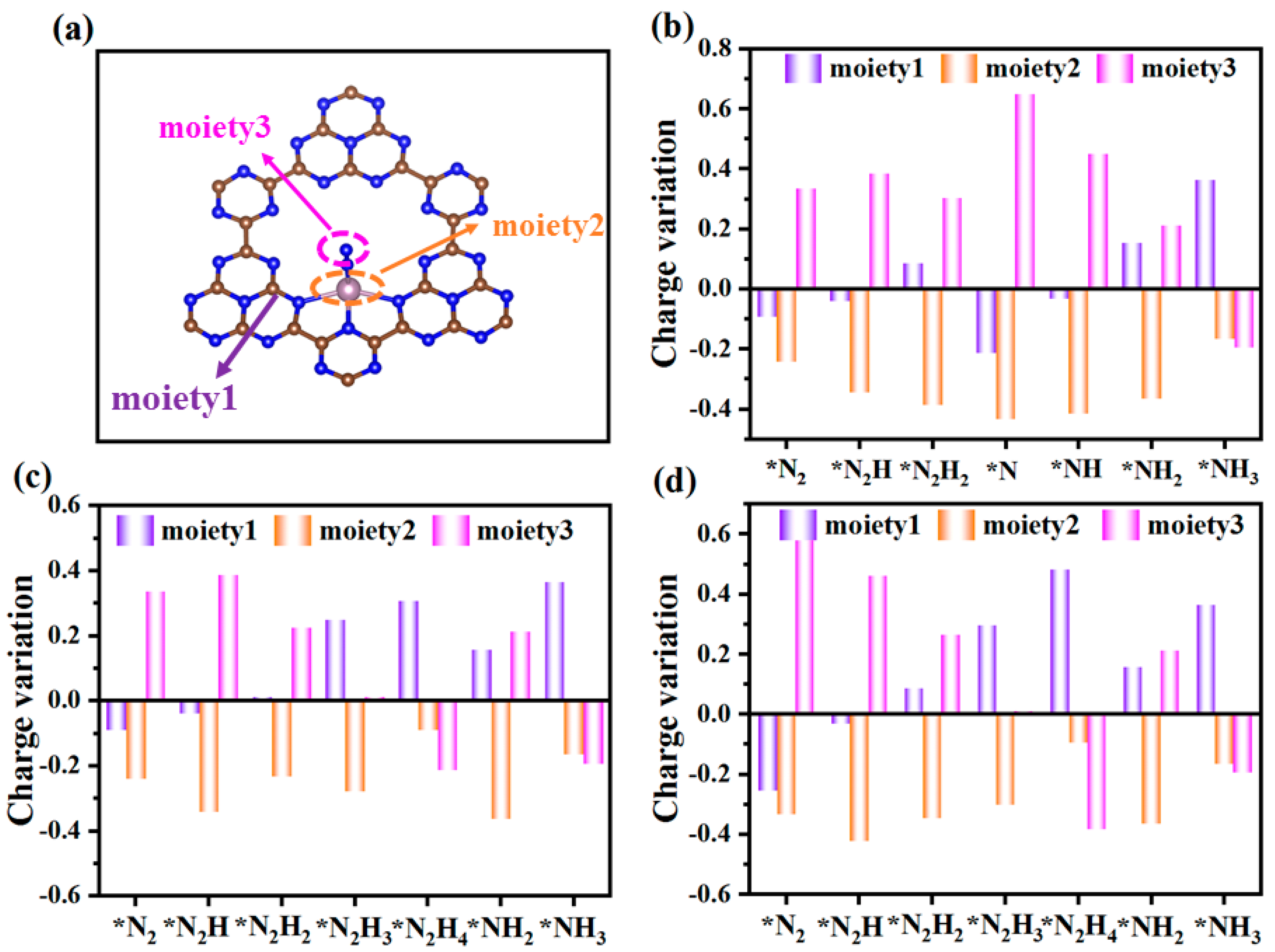
2. Results and Discussion
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, J.G.; Crooks, R.M.; Seefeldt, L.C.; Bren, K.L.; Bullock, R.M.; Darensbourg, M.Y.; Holland, P.L.; Hoffman, B.; Janik, M.J.; Jones, A.K.; et al. Beyond fossil fuel–driven nitrogen transformations. Science 2018, 360, eaar6611. [Google Scholar] [CrossRef]
- Li, L.; Wu, Z.; Zhu, H.; Robinson, G.H.; Xie, Y.; Schaefer, H.F. Reduction of Dinitrogen via 2,3′-Bipyridine-Mediated Tetraboration. J. Am. Chem. Soc. 2020, 142, 6244–6250. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, Y.; Du, Y.; Zhu, X.-D.; Gao, J.; Zhang, Y.-C.; Wu, G. P-Block Metal-Based Electrocatalysts for Nitrogen Reduction to Ammonia: A Minireview. Small 2023, 19, 2206776. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, X.; Guo, M.; Hu, W.; Huang, B.; Yuan, D. Enhanced Catalytic Activity of Bimetallic Ordered Catalysts for Nitrogen Reduction Reaction by Perturbation of Scaling Relations. ACS Catal. 2023, 13, 2190–2201. [Google Scholar] [CrossRef]
- Wang, M.; Ma, J.; Shang, Z.; Fu, L.; Zhang, H.; Li, M.-B.; Lu, K. Advances in ambient selective electrohydrogenation of nitrogen to ammonia: Strategies to strengthen nitrogen chemisorption. J. Mater. Chem. A 2023, 11, 3871–3887. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Dong, J.-H.; Tan, Z.; Wang, X.-F.; Song, X.-Z. The journey of iron-based electrocatalytic materials for nitrogen reduction reaction: From current status to future prospects. J. Mater. Chem. A 2023, 11, 11048–11077. [Google Scholar] [CrossRef]
- Feng, X.; Liu, J.; Chen, L.; Kong, Y.; Zhang, Z.; Zhang, Z.; Wang, D.; Liu, W.; Li, S.; Tong, L.; et al. Hydrogen Radical-Induced Electrocatalytic N2 Reduction at a Low Potential. J. Am. Chem. Soc. 2023, 145, 10259–10267. [Google Scholar] [CrossRef]
- Wu, X.; Nazemi, M.; Gupta, S.; Chismar, A.; Hong, K.; Jacobs, H.; Zhang, W.; Rigby, K.; Hedtke, T.; Wang, Q.; et al. Contrasting Capability of Single Atom Palladium for Thermocatalytic versus Electrocatalytic Nitrate Reduction Reaction. ACS Catal. 2023, 13, 6804–6812. [Google Scholar] [CrossRef]
- Du, C.; Qiu, C.; Fang, Z.; Li, P.; Gao, Y.; Wang, J.; Chen, W. Interface hydrophobic tunnel engineering: A general strategy to boost electrochemical conversion of N2 to NH3. Nano Energy 2022, 92, 106784. [Google Scholar] [CrossRef]
- Zhu, S.; Qin, M.; Chen, L.; Jiang, S.; Zhou, Y.; Jiang, J.; Zhang, W. Theoretical Investigation of Electrocatalytic Reduction of Nitrates to Ammonia on Highly Efficient and Selective g-C2N Monolayer-Supported Single Transition-Metal Atoms. J. Phys. Chem. Lett. 2023, 14, 4185–4191. [Google Scholar] [CrossRef]
- Cipriano, L.A.; Di Liberto, G.; Pacchioni, G. Superoxo and Peroxo Complexes on Single-Atom Catalysts: Impact on the Oxygen Evolution Reaction. ACS Catal. 2022, 12, 11682–11691. [Google Scholar] [CrossRef]
- Wei, X.; Cao, S.; Xu, H.; Jiang, C.; Wang, Z.; Ouyang, Y.; Lu, X.; Dai, F.; Sun, D. Novel Two-Dimensional Metal Organic Frameworks: High-Performance Bifunctional Electrocatalysts for OER/ORR. ACS Mater. Lett. 2022, 4, 1991–1998. [Google Scholar] [CrossRef]
- Cao, S.; Liu, Y.; Hu, Y.; Li, J.; Yang, C.; Chen, Z.; Wang, Z.; Wei, S.; Liu, S.; Lu, X. Precise electronic structure modulation on MXene-based single atom catalysts for high-performance electrocatalytic CO2 reduction reaction: A first-principle study. J. Colloid Interface Sci. 2023, 642, 273–282. [Google Scholar] [CrossRef]
- Ren, Y.; Sun, X.; Qi, K.; Zhao, Z. Single atom supported on MoS2 as efficient electrocatalysts for the CO2 reduction reaction: A DFT study. Appl. Surf. Sci. 2022, 602, 154211. [Google Scholar] [CrossRef]
- Choi, C.; Back, S.; Kim, N.-Y.; Lim, J.; Kim, Y.-H.; Jung, Y. Suppression of Hydrogen Evolution Reaction in Electrochemical N2 Reduction Using Single-Atom Catalysts: A Computational Guideline. ACS Catal. 2018, 8, 7517–7525. [Google Scholar] [CrossRef]
- Zhu, J.; Cai, L.; Tu, Y.; Zhang, L.; Zhang, W. Emerging ruthenium single-atom catalysts for the electrocatalytic hydrogen evolution reaction. J. Mater. Chem. A 2022, 10, 15370–15389. [Google Scholar] [CrossRef]
- Sathishkumar, N.; Chen, H.-T. Regulating the Coordination Environment of Single-Atom Catalysts Anchored on Thiophene Linked Porphyrin for an Efficient Nitrogen Reduction Reaction. ACS Appl. Mater. Interfaces 2023, 15, 15545–15560. [Google Scholar] [CrossRef]
- Ma, D.; Zeng, Z.; Liu, L.; Huang, X.; Jia, Y. Computational Evaluation of Electrocatalytic Nitrogen Reduction on TM Single-, Double-, and Triple-Atom Catalysts (TM = Mn, Fe, Co, Ni) Based on Graphdiyne Monolayers. J. Phys. Chem. C 2019, 123, 19066–19076. [Google Scholar] [CrossRef]
- Ma, Z.; Lv, P.; Wu, D.; Li, X.; Chu, K.; Ma, D.; Jia, Y. V (Nb) Single Atoms Anchored by the Edge of a Graphene Armchair Nanoribbon for Efficient Electrocatalytic Nitrogen Reduction: A Theoretical Study. Inorg. Chem. 2022, 61, 17864–17872. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, X.; Qin, J.; Liu, R. Theoretical screening of highly efficient single-atom catalysts for nitrogen reduction based on a defective C3N monolayer. Int. J. Hydrogen Energy 2022, 47, 5292–5306. [Google Scholar] [CrossRef]
- Wu, J.; Li, J.-H.; Yu, Y.-X. Single Nb or W Atom-Embedded BP Monolayers as Highly Selective and Stable Electrocatalysts for Nitrogen Fixation with Low-Onset Potentials. ACS Appl. Mater. Interfaces 2021, 13, 10026–10036. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Q.; Gong, H.; Xue, M. Single Mo atom supported on defective BC2N monolayers as promising electrochemical catalysts for nitrogen reduction reaction. Appl. Surf. Sci. 2021, 546, 149131. [Google Scholar] [CrossRef]
- Li, L.; Martirez, J.M.P.; Carter, E.A. Prediction of Highly Selective Electrocatalytic Nitrogen Reduction at Low Overpotential on a Mo-Doped g-GaN Monolayer. ACS Catal. 2020, 10, 12841–12857. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, T.; Yang, L.; Liu, R.; Zhang, G.; Jiang, J.; Luo, Y.; Lian, P.; Tang, S. Graphene–boron nitride hybrid-supported single Mo atom electrocatalysts for efficient nitrogen reduction reaction. J. Mater. Chem. A 2019, 7, 15173–15180. [Google Scholar] [CrossRef]
- Xue, Z.; Zhang, X.; Qin, J.; Liu, R. Anchoring Mo on C9N4 monolayers as an efficient single atom catalyst for nitrogen fixation. J. Energy Chem. 2021, 57, 443–450. [Google Scholar] [CrossRef]
- Baby, A.; Trovato, L.; Di Valentin, C. Single Atom Catalysts (SAC) trapped in defective and nitrogen-doped graphene supported on metal substrates. Carbon 2021, 174, 772–788. [Google Scholar] [CrossRef]
- Gao, S.; Ma, Z.; Xiao, C.; Du, W.; Sun, X.; Li, Q.; Sa, R.; Sun, C. High-Throughput computational screening of Single-atom embedded in defective BN nanotube for electrocatalytic nitrogen fixation. Appl. Surf. Sci. 2022, 591, 153130. [Google Scholar] [CrossRef]
- Lv, X.; Wei, W.; Wang, H.; Huang, B.; Dai, Y. Holey graphitic carbon nitride (g-CN) supported bifunctional single atom electrocatalysts for highly efficient overall water splitting. Appl. Catal. B Environ. 2020, 264, 118521. [Google Scholar] [CrossRef]
- Detz, H.; Butera, V. Insights into the mechanistic CO2 conversion to methanol on single Ru atom anchored on MoS2 monolayer. Mol. Catal. 2023, 535, 112878. [Google Scholar] [CrossRef]
- Wang, S.; Wei, W.; Lv, X.; Huang, B.; Dai, Y. W supported on g-CN manifests high activity and selectivity for N2 electroreduction to NH3. J. Mater. Chem. A 2020, 8, 1378–1385. [Google Scholar] [CrossRef]
- Niu, H.; Wan, X.; Wang, X.; Shao, C.; Robertson, J.; Zhang, Z.; Guo, Y. Single-Atom Rhodium on Defective g-C3N4: A Promising Bifunctional Oxygen Electrocatalyst. ACS Sustain. Chem. Eng. 2021, 9, 3590–3599. [Google Scholar] [CrossRef]
- Liu, S.; Liu, J.-Y. Rational design of highly efficient electrocatalytic single-atom catalysts for nitrogen reduction on nitrogen-doped graphene and g-C2N supports. J. Power Sources 2022, 535, 231449. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, X.; Zhang, F.; Fang, C.; Liu, D.; Zhou, Q. A High-Throughput Screening toward Efficient Nitrogen Fixation: Transition Metal Single-Atom Catalysts Anchored on an Emerging π–π Conjugated Graphitic Carbon Nitride (g-C10N3) Substrate with Dirac Dispersion. ACS Appl. Mater. Interfaces 2023, 15, 11812–11826. [Google Scholar] [CrossRef]
- Schwarzer, A.; Saplinova, T.; Kroke, E. Tri-s-triazines (s-heptazines)—From a “mystery molecule” to industrially relevant carbon nitride materials. Coord. Chem. Rev. 2013, 257, 2032–2062. [Google Scholar] [CrossRef]
- Li, H.; Hu, H.; Bao, C.; Guo, F.; Zhang, X.; Liu, X.; Hua, J.; Tan, J.; Wang, A.; Zhou, H.; et al. Forming heterojunction: An effective strategy to enhance the photocatalytic efficiency of a new metal-free organic photocatalyst for water splitting. Sci. Rep. 2016, 6, 29327. [Google Scholar] [CrossRef]
- Xia, L.; Wang, H.; Zhao, Y. Novel graphitic carbon nitride g-C9N10 as a promising platform to design efficient photocatalysts for dinitrogen reduction to ammonia: The first-principles investigation. J. Mater. Chem. A 2021, 9, 20615–20625. [Google Scholar] [CrossRef]
- Wang, M.; Huang, Y.; Ma, F.; Zhu, G.; Zhang, J.; Wei, X.; Hou, P.; Du, R.; Liu, J. Theoretical insights into the mechanism of nitrogen-to-ammonia electroreduction on TM/g-C9N10. Mol. Catal. 2023, 547, 113391. [Google Scholar] [CrossRef]
- Gao, D.; Yi, D.; Sun, C.; Yang, Y.; Wang, X. Breaking the Volcano-Shaped Relationship for Highly Efficient Electrocatalytic Nitrogen Reduction: A Computational Guideline. ACS Appl. Mater. Interfaces 2022, 14, 52806–52814. [Google Scholar] [CrossRef]
- Hu, X.; Xiong, L.; Fang, W.-H.; Su, N.Q. Computational Insight into Metallated Graphynes as Single Atom Electrocatalysts for Nitrogen Fixation. ACS Appl. Mater. Interfaces 2022, 14, 27861–27872. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Chen, L.; Zhang, X.; Zhang, P. Designing C9N10 Anchored Single Mo Atom as an Efficient Electrocatalyst for Nitrogen Fixation. Molecules 2024, 29, 4768. https://doi.org/10.3390/molecules29194768
Chen Y, Chen L, Zhang X, Zhang P. Designing C9N10 Anchored Single Mo Atom as an Efficient Electrocatalyst for Nitrogen Fixation. Molecules. 2024; 29(19):4768. https://doi.org/10.3390/molecules29194768
Chicago/Turabian StyleChen, Yibo, Liang Chen, Xinyu Zhang, and Pengyue Zhang. 2024. "Designing C9N10 Anchored Single Mo Atom as an Efficient Electrocatalyst for Nitrogen Fixation" Molecules 29, no. 19: 4768. https://doi.org/10.3390/molecules29194768
APA StyleChen, Y., Chen, L., Zhang, X., & Zhang, P. (2024). Designing C9N10 Anchored Single Mo Atom as an Efficient Electrocatalyst for Nitrogen Fixation. Molecules, 29(19), 4768. https://doi.org/10.3390/molecules29194768