
Cl©Li5Cl5−: A Star-like Superhalogen Anion Featuring a Planar Pentacoordinate Chlorine at the Center



Abstract

1. Introduction
2. Computational Details
3. Result and Discussion
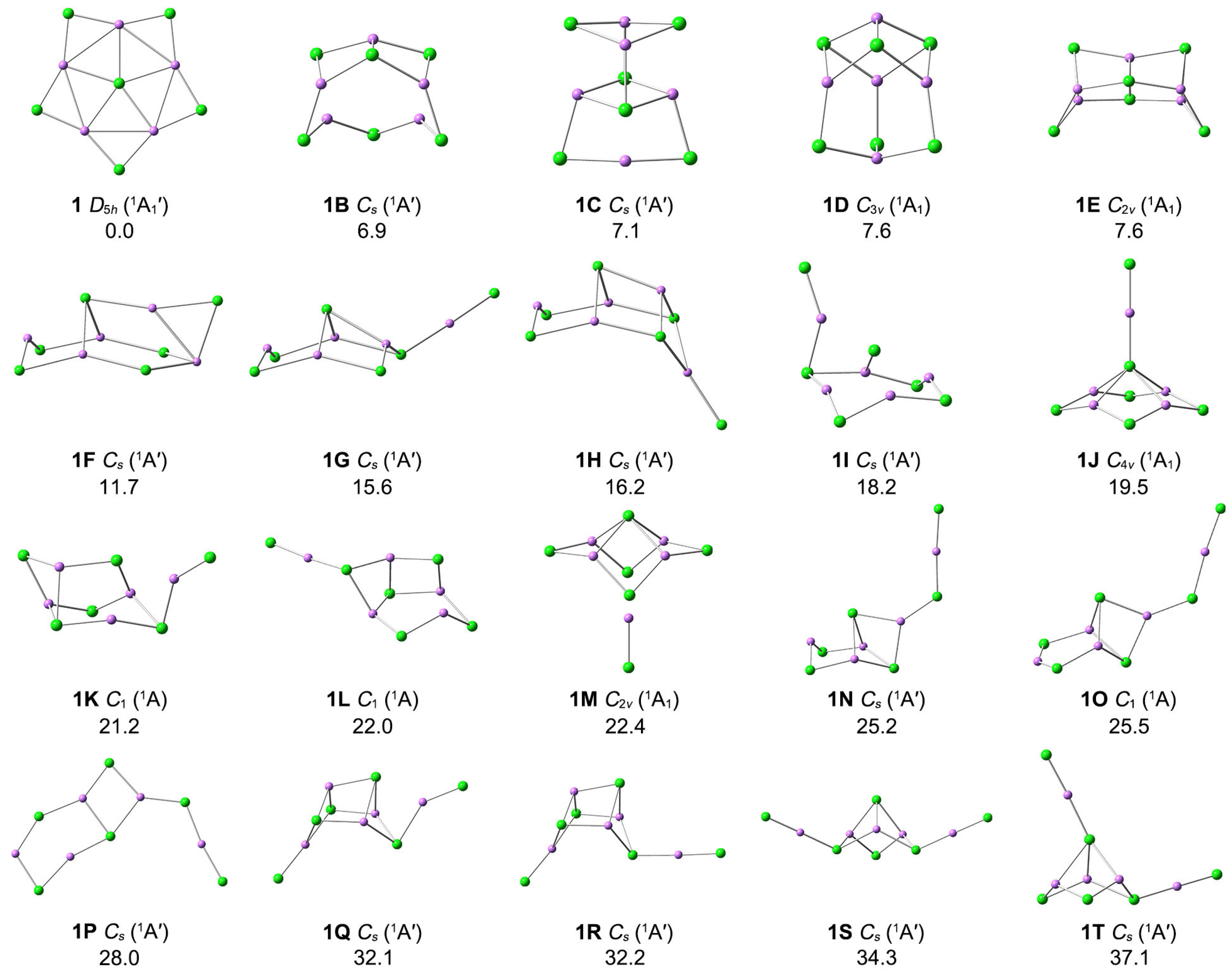
3.1. Structures and Stability
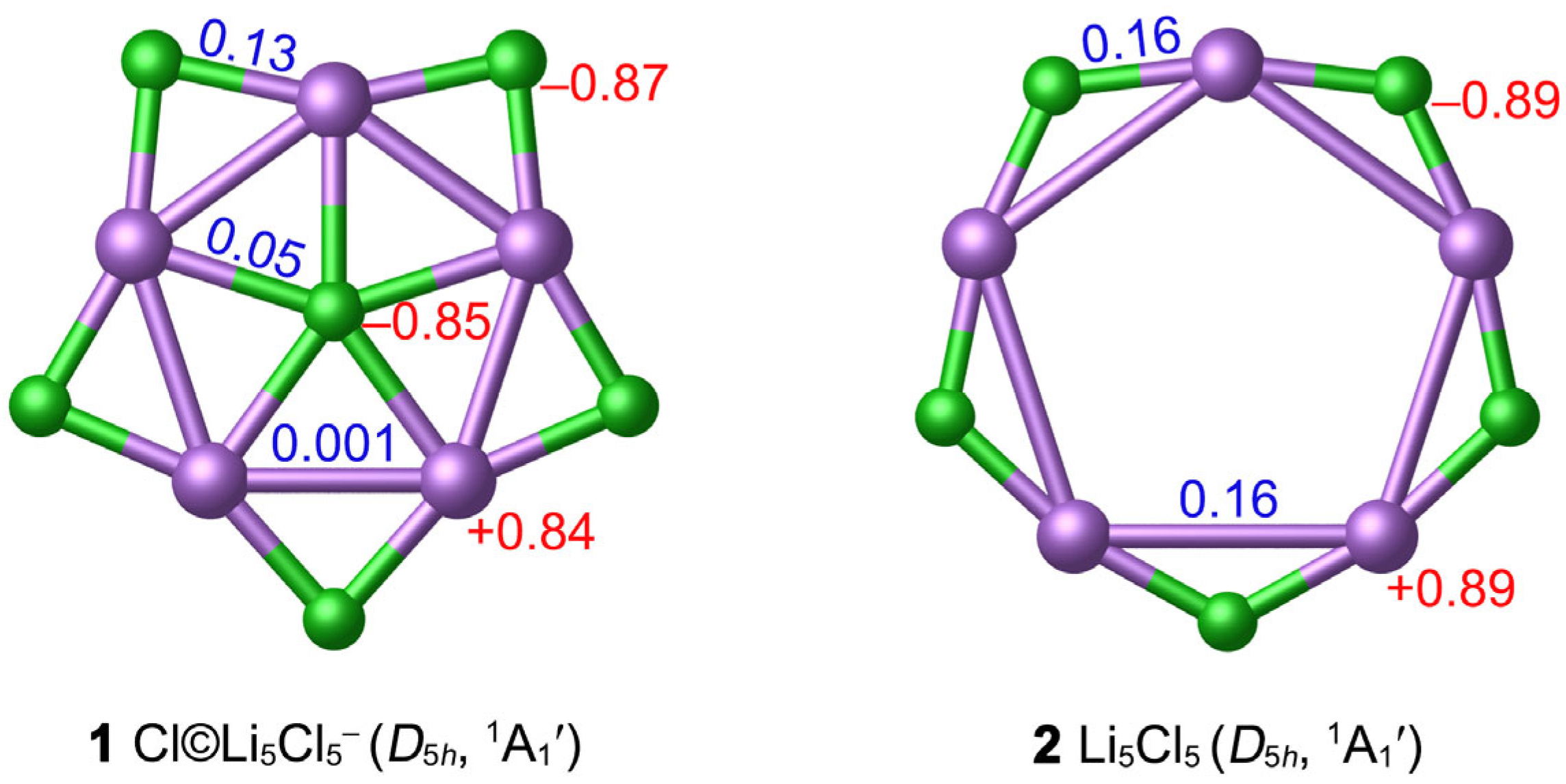
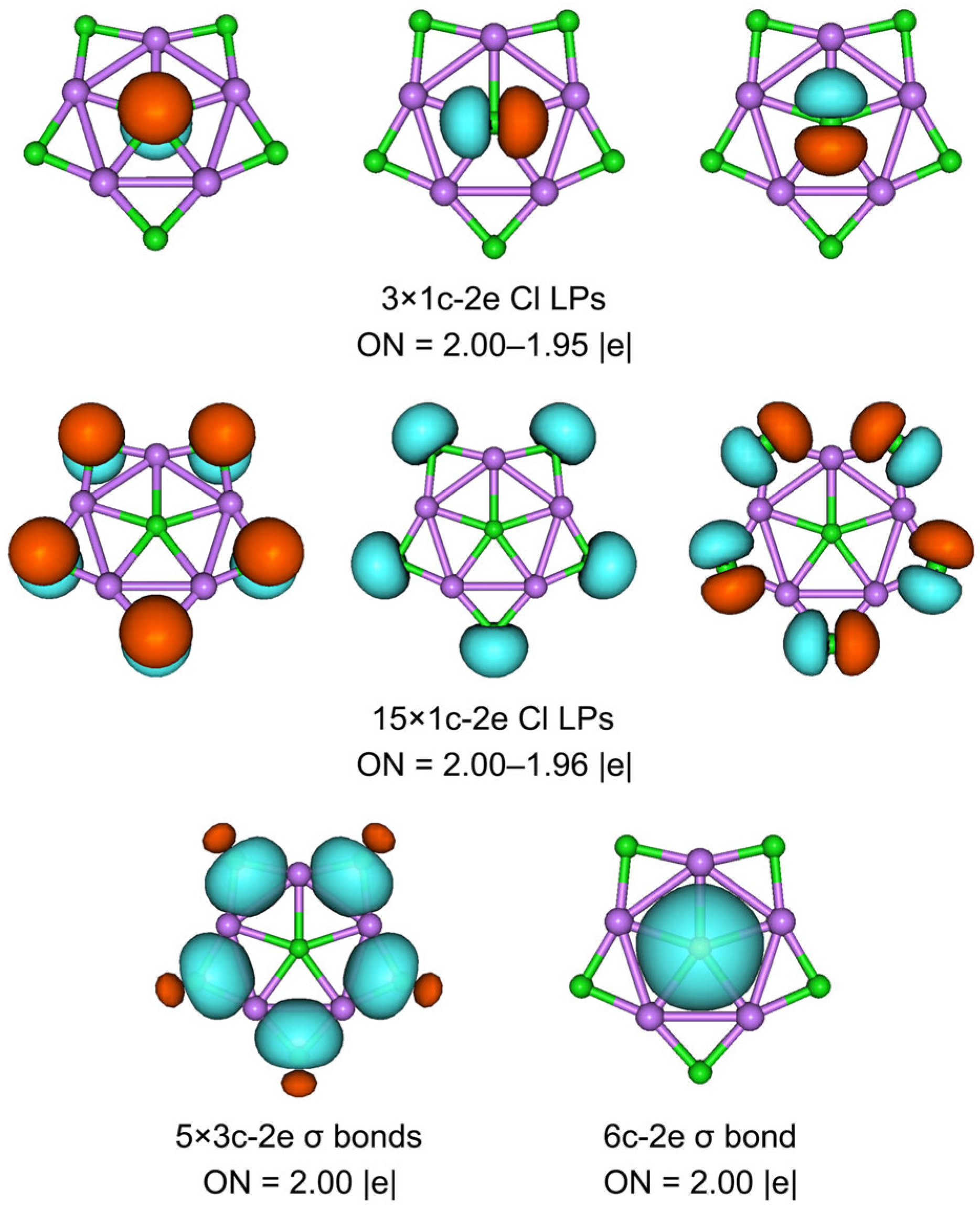
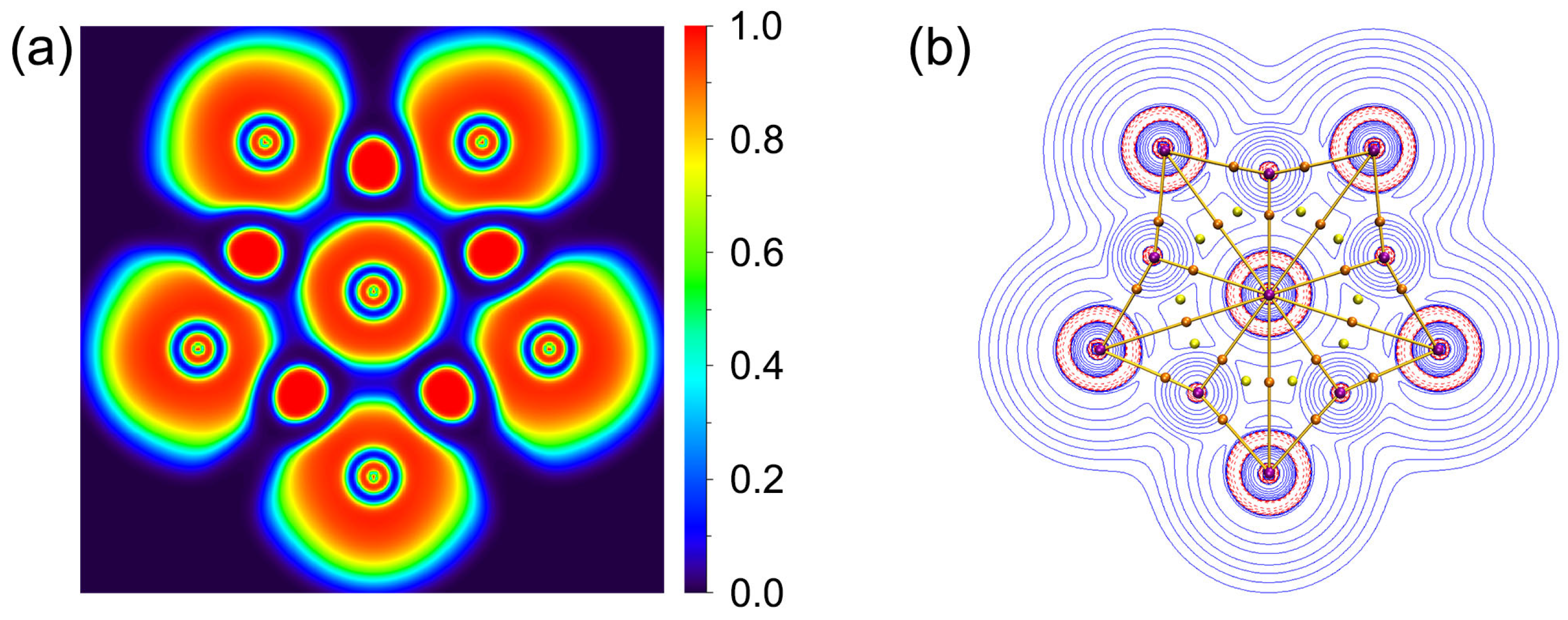
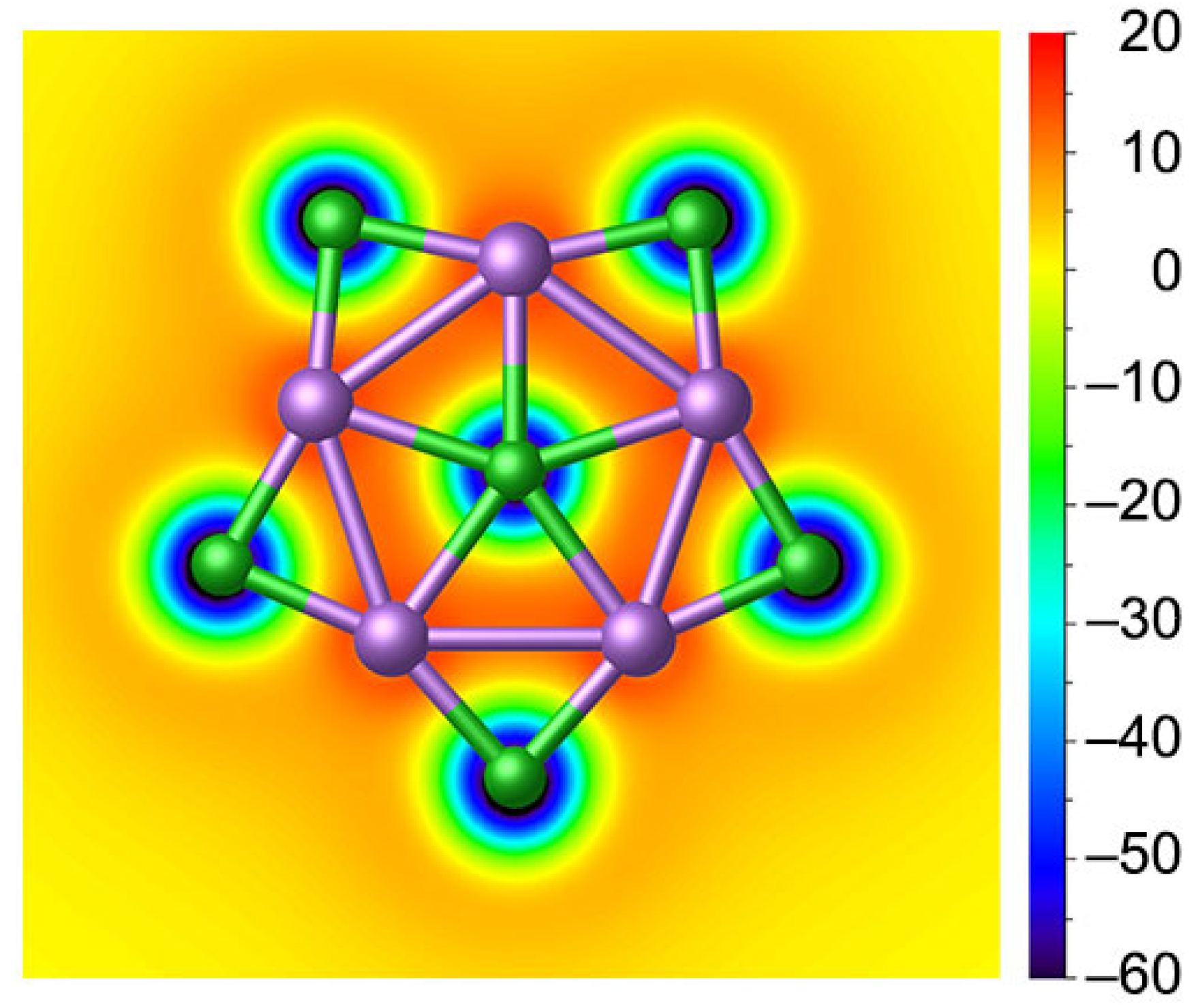
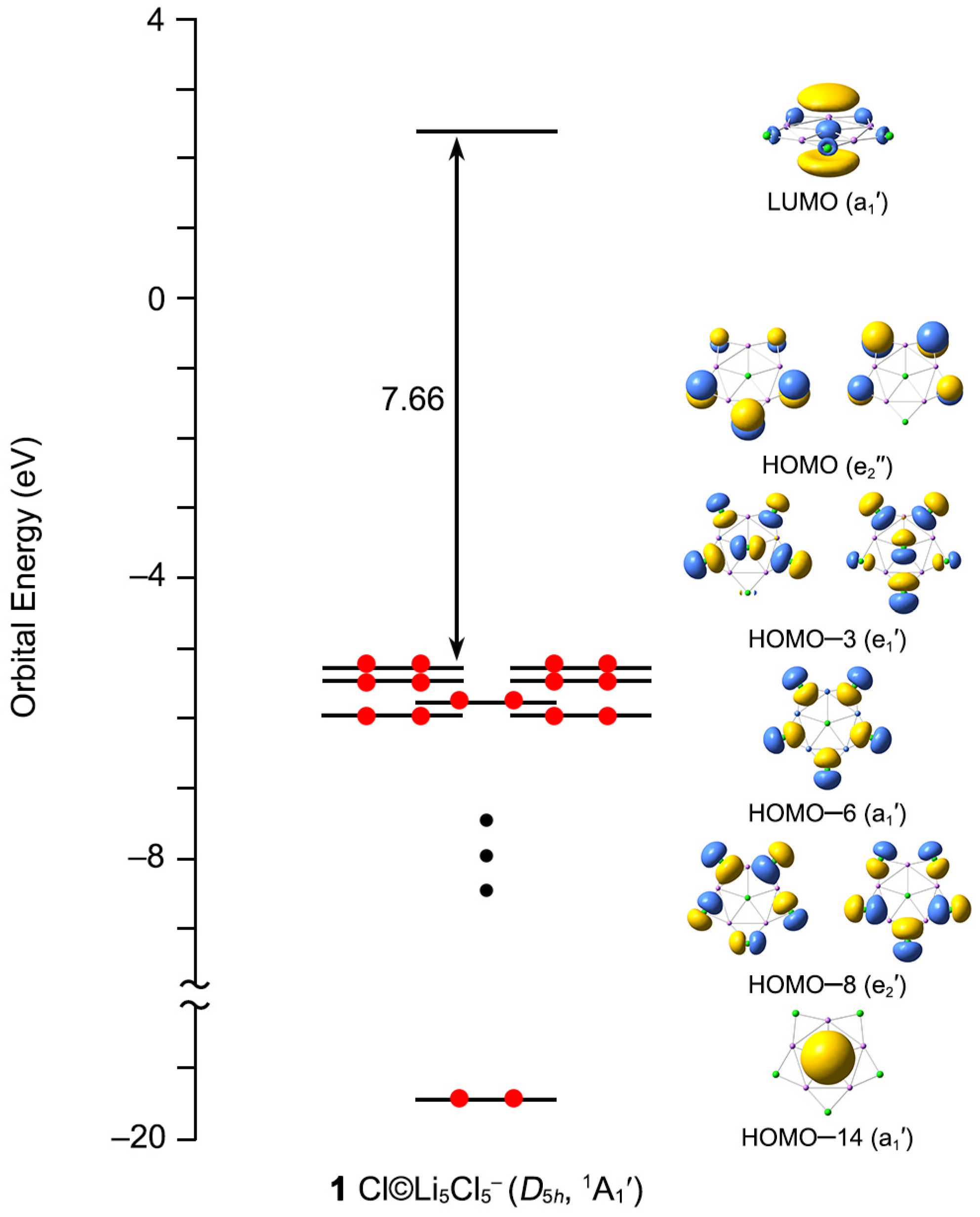
3.2. Chemical Bonding
3.3. Superhalogen Anion Characteristic
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Monkhorst, H.J. Activation energy for interconversion of enantiomers containing an asymmetric carbon atom without breaking bonds. Chem. Commun. 1968, 1111–1112. [Google Scholar] [CrossRef]
- Hoffmann, R.; Alder, R.W.; Wilcox, C.F. Planar tetracoordinate carbon. J. Am. Chem. Soc. 1970, 92, 4992–4993. [Google Scholar] [CrossRef]
- Collins, J.B.; Dill, J.D.; Jemmis, E.D.; Apeloig, Y.; Schleyer, P.v.R.; Seeger, R.; Pople, J.A. Stabilization of planar tetracoordinate carbon. J. Am. Chem. Soc. 1976, 98, 5419–5427. [Google Scholar] [CrossRef]
- Yang, L.M.; Ganz, E.; Chen, Z.F.; Wang, Z.X.; Schleyer, P.v.R. Four decades of the chemistry of planar hypercoordinate compounds. Angew. Chem. Int. Ed. 2015, 54, 9468–9501. [Google Scholar] [CrossRef]
- Vassilev-Galindo, V.; Pan, S.; Donald, K.J.; Merino, G. Planar pentacoordinate carbons. Nat. Rev. Chem. 2018, 2, 114. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.F.; Chen, Z.F. Planar hypercoordinate motifs in two-dimensional materials. Acc. Chem. Res. 2020, 53, 887–895. [Google Scholar] [CrossRef]
- Exner, K.; Schleyer, P.v.R. Planar hexacoordinate carbon: A viable possibility. Science 2000, 290, 1937–1940. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.S.; Boldyrev, A.I.; Simons, J. Tetracoordinated planar carbon in the Al4C− anion. A combined photoelectron spectroscopy and ab initio study. J. Am. Chem. Soc. 1999, 121, 6033–6038. [Google Scholar] [CrossRef]
- Li, X.; Zhang, H.F.; Wang, L.S.; Geske, G.D.; Boldyrev, A.I. Pentaatomic tetracoordinate planar carbon, [CAl4]2−: A new structural unit and its salt complexes. Angew. Chem. Int. Ed. 2000, 39, 3630–3632. [Google Scholar] [CrossRef]
- Wang, L.S.; Boldyrev, A.I.; Li, X.; Simons, J. Experimental observation of pentaatomic tetracoordinate planar carbon-containing molecules. J. Am. Chem. Soc. 2000, 122, 7681–7687. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.X.; Yu, S.; Ding, Y.H.; Bowen, K.H. Identifying the hydrogenated planar tetracoordinate carbon: A combined experimental and theoretical study of CAl4H and CAl4H−. J. Phys. Chem. Lett. 2017, 8, 2263–2267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.J.; Dai, W.S.; Xu, H.G.; Xu, X.L.; Zheng, W.J. Structural evolution of carbon-doped aluminum clusters AlnC− (n = 6–15): Anion photoelectron spectroscopy and theoretical calculations. J. Phys. Chem. A 2022, 126, 5621–5631. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.X.; Barroso, J.; Orozco-Ic, M.; Ortiz-Chi, F.; Guo, J.C.; Merino, G. CAl11−: A molecular rotor with a quasi-planar tetracoordinate carbon. Chem. Commun. 2023, 59, 4966–4969. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; An, W.; Ito, K.; Schleyer, P.v.R.; Zeng, X.C. Planar pentacoordinate carbon in CAl5+: A global minimum. J. Am. Chem. Soc. 2008, 130, 10394–10400. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Parra, L.; Diego, L.; Yañez, O.; Inostroza, D.; Barroso, J.; Vásquez-Espinal, A.; Merino, G.; Tiznado, W. Planar hexacoordinate carbons: Half covalent, half ionic. Angew. Chem. Int. Ed. 2021, 60, 8700–8704. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.v.R.; Boldyrev, A.I. A new, general strategy for achieving planar tetracoordinate geometries for carbon and other second row periodic elements. J. Chem. Soc. Chem. Commun. 1991, 1536–1538. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Li, X.; Wang, L.S. Experimental observation of pentatomic tetracoordinate planar Si- and Ge-containing molecules: MAl4− and MAl4. Angew. Chem. Int. Ed. 2000, 39, 3307–3310. [Google Scholar] [CrossRef]
- Wang, M.H.; Dong, X.; Cui, Z.H.; Orozco-Ic, M.; Ding, Y.H.; Barroso, J.; Merino, G. Planar pentacoordinate silicon and germanium atoms. Chem. Commun. 2020, 56, 13772–13775. [Google Scholar] [CrossRef]
- Chen, C.; Wang, M.H.; Feng, L.Y.; Zhao, L.Q.; Guo, J.C.; Zhai, H.J.; Cui, Z.H.; Pan, S.; Merino, G. Bare and ligand protected planar hexacoordinate silicon in SiSb3M3+ (M = Ca, Sr, Ba) clusters. Chem. Sci. 2022, 13, 8045–8051. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.Q.; Cui, Z.H. σ-Aromaticity planar pentacoordinate beryllium atoms. Inorg. Chem. 2021, 60, 16053–16058. [Google Scholar] [CrossRef]
- Jin, B.; Guan, X.L.; Yan, M.; Wang, Y.J.; Wu, Y.B. Planar hexacoordinate beryllium: Covalent bonding between s–block metals. Chem. Eur. J. 2023, 29, e202302672. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.H.; Chen, C.; Pan, S.; Cui, Z.H. Planar hexacoordinate gallium. Chem. Sci. 2021, 12, 15067–15076. [Google Scholar] [CrossRef]
- Wang, M.H.; Kalita, A.J.; Orozco-Ic, M.; Yan, G.R.; Chen, C.; Yan, B.; Castillo-Toraya, G.; Tiznado, W.; Guha, A.K.; Pan, S.; et al. Planar pentacoordinate s–block metals. Chem. Sci. 2023, 14, 8785–8791. [Google Scholar] [CrossRef] [PubMed]
- Kalita, A.J.; Rohman, S.S.; Sahu, P.P.; Guha, A.K. Planar tetracoordinate hydrogen: Pushing the limit of multicentre bonding. Angew. Chem. Int. Ed. 2024, 63, e202317312. [Google Scholar] [CrossRef] [PubMed]
- Sarmah, K.; Kalita, A.J.; Guha, A.K. Alkali metal decorated planar tetracoordinate hydrogen. Inorg. Chem. 2023, 62, 20919–20922. [Google Scholar] [CrossRef] [PubMed]
- Sarmah, K.; Kalita, A.J.; Purkayastha, S.K.; Guha, A.K. Pushing the extreme of multicentre bonding: Planar pentacoordinate hydride. Angew. Chem. Int. Ed. 2024, 63, e202318741. [Google Scholar] [CrossRef]
- Castillo-Toraya, G.; Orozco-Ic, M.; Dzib, E.; Zarate, X.; Ortíz-Chi, F.; Cui, Z.H.; Barroso, J.; Merino, G. Planar tetracoordinate fluorine atoms. Chem. Sci. 2021, 12, 6699–6704. [Google Scholar] [CrossRef] [PubMed]
- Sarmah, K.; Kalita, A.J.; Guha, A.K. Planar tetracoordinate fluorine atom: Global minimum with viable possibility. Phys. Chem. Chem. Phys. 2024, 26, 6678–6682. [Google Scholar] [CrossRef]
- Law, C.K.; Chien, S.H.; Li, W.K.; Cheung, Y.S. Thermochemistry of chlorine fluorides ClFn, n = 1–7, and their singly charged cations and anions: A Gaussian-3 and Gaussian-3X Study. J. Phys. Chem. A 2002, 106, 11271–11276. [Google Scholar] [CrossRef]
- Gutsev, G.L.; Boldyrev, A.I. DVM-Xα calculations on the ionization potentials of MXk+1−complex anions and the electron affinities of MXk+1 “superhalogens”. Chem. Phys. 1981, 56, 277–283. [Google Scholar] [CrossRef]
- Wang, X.B.; Ding, C.F.; Wang, L.S.; Boldyrev, A.I.; Simons, J. First experimental photoelectron spectra of superhalogens and their theoretical interpretations. J. Chem. Phys. 1999, 110, 4763–4771. [Google Scholar] [CrossRef]
- Anusiewicz, I. Electrophilic substituents as ligands in superhalogen anions. J. Phys. Chem. A 2009, 113, 6511–6516. [Google Scholar] [CrossRef] [PubMed]
- Anusiewicz, I. Superhalogen anions utilizing acidic functional groups as ligands. J. Phys. Chem. A 2009, 113, 11429–11434. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K. M(BO)k+1− anions: Novel superhalogens based on boronyl ligands. J. Phys. Chem. A 2022, 126, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K. Recent progress on the design and applications of superhalogens. Chem. Commun. 2023, 59, 5943–5960. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.X.; Jin, Y.X.; Guo, J.C. D4h H©K4H4−: A planar tetracoordinate hydrogen global minimum. Chem. Commun. 2024, 60, 6300–6303. [Google Scholar] [CrossRef]
- Saunders, M. Stochastic search for isomers on a quantum mechanical surface. J. Comput. Chem. 2004, 25, 621–626. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Purvis III, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Millam, J.M.; Bakken, V.; Chen, W.; Hase, W.L.; Schlegel, H.B. Ab initio classical trajectories on the Born–Oppenheimer surface: Hessian-based integrators using fifth-order polynomial and rational function fits. J. Chem. Phys. 1999, 111, 3800–3805. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef] [PubMed]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Fliegl, H.; Taubert, S.; Lehtonen, O.; Sundholm, D. The gauge including magnetically induced current method. Phys. Chem. Chem. Phys. 2011, 13, 20500–20518. [Google Scholar] [CrossRef] [PubMed]
- Sundholm, D.; Fliegl, H.; Berger, R.J.F. Calculations of magnetically induced current densities: Theory and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 639–678. [Google Scholar] [CrossRef]
- Jusélius, J.; Sundholm, D.; Gauss, J. Calculation of current densities using gauge-including atomic orbitals. J. Chem. Phys. 2004, 121, 3952–3963. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ziegler, T.; Atkins, A.J.; Autschbach, J.; Bashford, D.; Baseggio, O.; Brces, A.; Bickelhaupt, F.M.; Bo, C.; Boerritger, P.M.; et al. ADF2023; Vrije Universiteit: Amsterdam, The Netherlands, 2023; Available online: https://www.scm.com/amsterdam-modeling-suite/adf (accessed on 5 April 2023).
- Legault, C.Y. CYLview, 1.0b; Universite. de Sherbrooke: Sherbrooke, QC, USA, 2009; Available online: http://www.cylview.org (accessed on 16 November 2022).
- Varetto, U. Molekel 5.4.0.8; Swiss National Supercomputing Center: Manno, Switzerland, 2009. [Google Scholar]
- Pyykkö, P. Additive covalent radii for single-, double-, and triple-bonded molecules and tetrahedrally bonded crystals: A summary. J. Phys. Chem. A 2015, 119, 2326–2337. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.J.; van Eikema Hommes, N.J.R. Nucleus-independent chemical shifts: A simple and efficient aromaticity probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title 1 | 1 | 2 |
|---|---|---|
| (Cl–Li) | −110.7 | — |
| (Cl–Li) | −105.5 (95.3%) | — |
| (Cl–Li) | −5.2 (4.7%) | — |
| (Li–Li) | 89.2 | 69.3 |
| (Li–Li) | 89.2 (100.0%) | 69.3 (100.0%) |
| (Li–Li) | 0.0 (0.0%) | 0.0 (0.0%) |
| (Cl a–Li) | −139.4 | −136.0 |
| (Cl a–Li) | −124.9 (89.6%) | −120.1 (88.3%) |
| (Cl a−Li) | −14.5 (10.4%) | −15.9 (11.7%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, L.-X.; Gao, C.-Y.; Guo, J.-C.; Li, S.-D. Cl©Li5Cl5−: A Star-like Superhalogen Anion Featuring a Planar Pentacoordinate Chlorine at the Center. Molecules 2024, 29, 3831. https://doi.org/10.3390/molecules29163831
Bai L-X, Gao C-Y, Guo J-C, Li S-D. Cl©Li5Cl5−: A Star-like Superhalogen Anion Featuring a Planar Pentacoordinate Chlorine at the Center. Molecules. 2024; 29(16):3831. https://doi.org/10.3390/molecules29163831
Chicago/Turabian StyleBai, Li-Xia, Cai-Yue Gao, Jin-Chang Guo, and Si-Dian Li. 2024. "Cl©Li5Cl5−: A Star-like Superhalogen Anion Featuring a Planar Pentacoordinate Chlorine at the Center" Molecules 29, no. 16: 3831. https://doi.org/10.3390/molecules29163831
APA StyleBai, L.-X., Gao, C.-Y., Guo, J.-C., & Li, S.-D. (2024). Cl©Li5Cl5−: A Star-like Superhalogen Anion Featuring a Planar Pentacoordinate Chlorine at the Center. Molecules, 29(16), 3831. https://doi.org/10.3390/molecules29163831