
Capillary Electrophoresis-Laser Induced Fluorescence Method Development and Validation for Quantification of Nine Gangliosides—Application to Analysis of Cell Lines of CNS Origin


Abstract
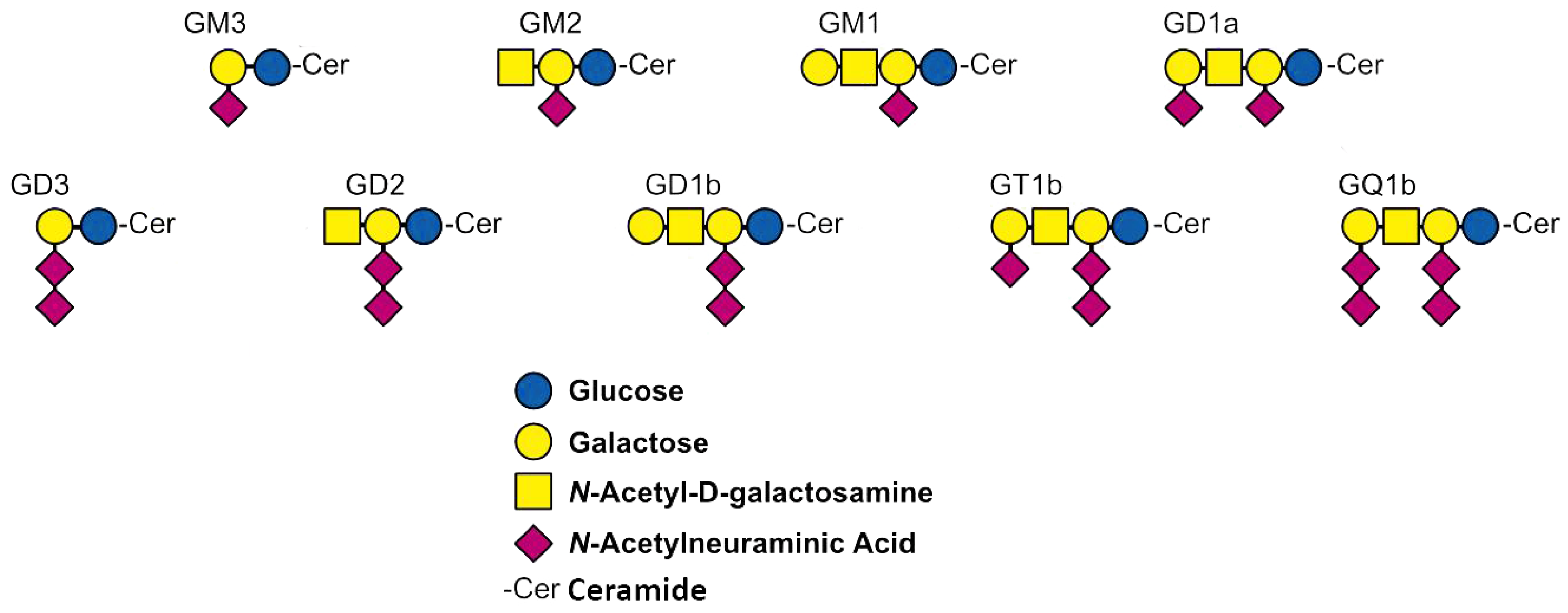
1. Introduction
2. Results and Discussion
2.1. Optimization of Separation Conditions
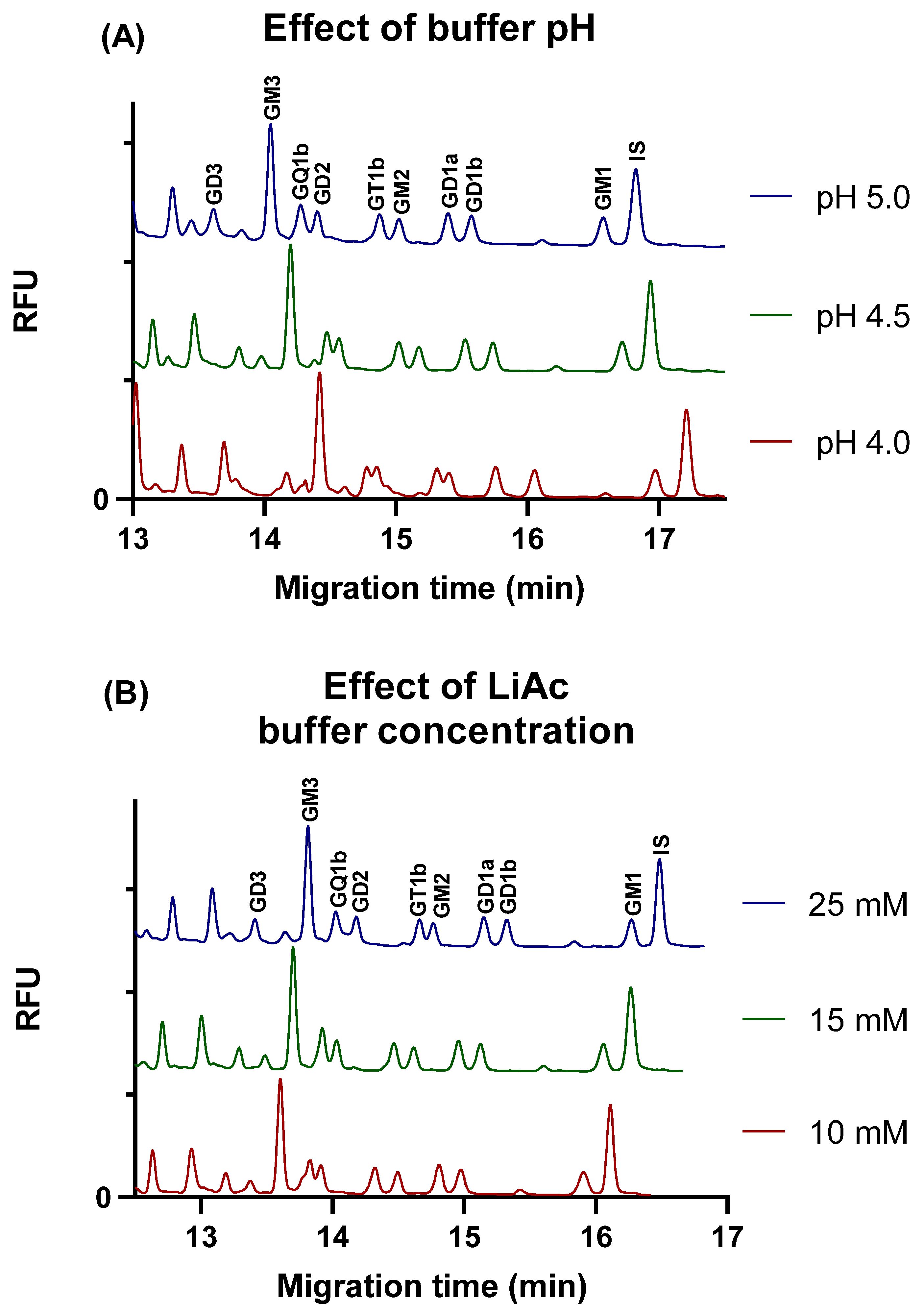
2.1.1. Effect of Buffer Concentration and Different Viscosity Enhancing Compounds on Separation
2.1.2. Effect of pH and Buffer Concentration on the Separation in the Presence of 5% w/v LPA
2.2. Optimization of the Digestion
2.3. Optimization of the Glycan Labeling
2.4. Method Validation
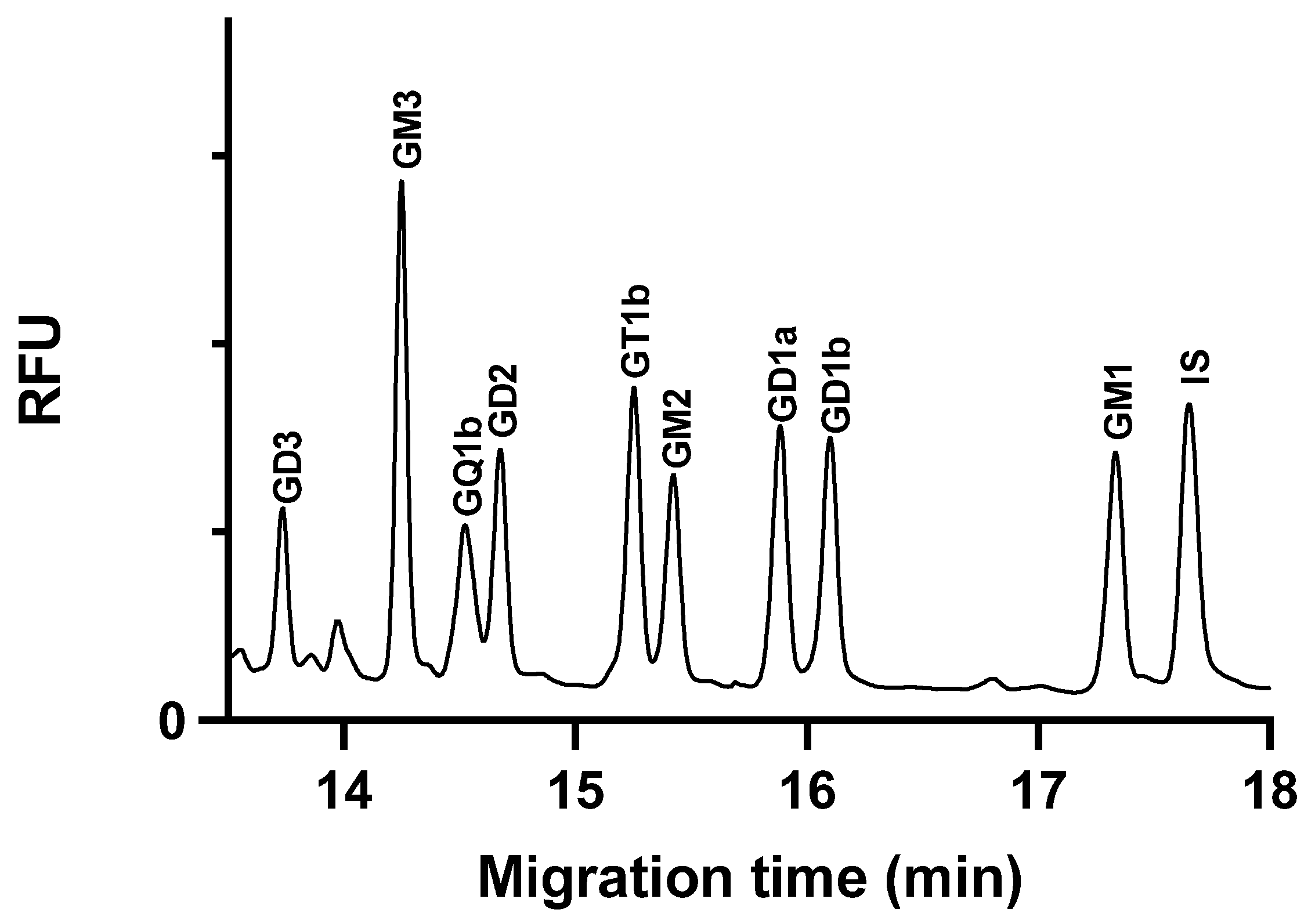
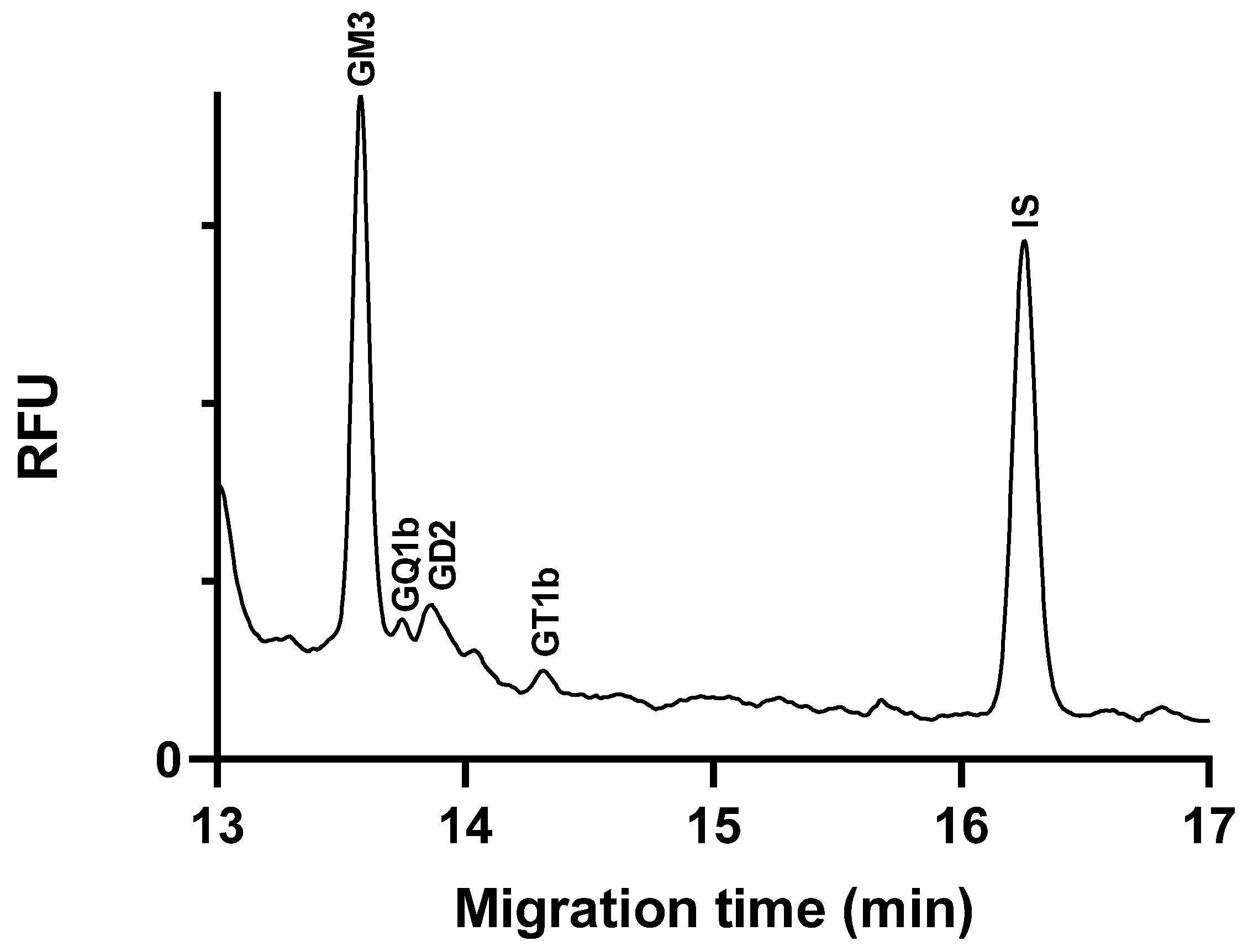
2.5. Application of the Method
3. Materials and Methods
3.1. Chemicals
3.2. Instrumentation
3.3. Sample Preparation
3.3.1. Preparation of Cells
3.3.2. Ganglioside Extraction
3.3.3. Digestion of Gangliosides by Endoglycoceramidase
3.3.4. Fluorescence Derivatization
3.4. Method Validation
3.4.1. Calibration Curves
3.4.2. Precision and Accuracy
3.4.3. Stability of the Extracted Sample
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klenk, E. Uber die Ganglioside, eine neue Gruppe von zuckerhaltigen Gehirnlipoiden. Biol. Chem. 1942, 273, 76–86. [Google Scholar] [CrossRef]
- Klenk, E. Beiträge zur Chemie der Lipoidosen [3. Mitteilung]. Niemann-Picksche Krankheit und amaurotische Idiotie. Biol. Chem. 1939, 262, 128–143. [Google Scholar] [CrossRef]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Sipione, S.; Monyror, J.; Galleguillos, D.; Steinberg, N.; Kadam, V. Gangliosides in the Brain: Physiology, Pathophysiology and Therapeutic Applications. Front. Neurosci. 2020, 14, 572965. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Yu, R.K. The expression and functions of glycoconjugates in neural stem cells. Glycobiology 2007, 17, 57R–74R. [Google Scholar] [CrossRef] [PubMed]
- Posse de Chaves, E.; Sipione, S. Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 2010, 584, 1748–1759. [Google Scholar] [CrossRef]
- Kolter, T. Ganglioside biochemistry. ISRN Biochem. 2012, 2012, 506160. [Google Scholar] [CrossRef] [PubMed]
- Tettamanti, G.; Bonali, F.; Marchesini, S.; Zambotti, V. A new procedure for the extraction, purification and fractionation of brain gangliosides. Biochim. Biophys. Acta 1973, 296, 160–170. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. Ganglioside Metabolism and Its Inherited Diseases. Methods Mol. Biol. 2018, 1804, 97–141. [Google Scholar]
- Segler-Stahl, K.; Webster, J.C.; Brunngraber, E.G. Changes in the concentration and composition of human brain gangliosides with aging. Gerontology 1983, 29, 161–168. [Google Scholar] [CrossRef]
- Yu, R.K.; Nakatani, Y.; Yanagisawa, M. The role of glycosphingolipid metabolism in the developing brain. J. Lipid Res. 2009, 50, S440–S445. [Google Scholar] [CrossRef]
- Regina Todeschini, A.; Hakomori, S.I. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim. Biophys. Acta 2008, 1780, 421–433. [Google Scholar] [CrossRef]
- Schengrund, C.L. The Ying and Yang of Ganglioside Function in Cancer. Cancers 2023, 15, 5362. [Google Scholar] [CrossRef]
- Sisu, E.; Flangea, C.; Serb, A.; Rizzi, A.; Zamfir, A.D. High-performance separation techniques hyphenated to mass spectrometry for ganglioside analysis. Electrophoresis 2011, 32, 1591–1609. [Google Scholar] [CrossRef]
- Muthing, J. TLC in structure and recognition studies of glycosphingolipids. Methods Mol. Biol. 1998, 76, 183–195. [Google Scholar]
- Vukelić, Z.; Kalanj-Bognar, S.; Froesch, M.; Bîndilă, L.; Radić, B.; Allen, M.; Peter-Katalinić, J.; Zamfir, A.D. Human gliosarcoma-associated ganglioside composition is complex and distinctive as evidenced by high-performance mass spectrometric determination and structural characterization. Glycobiology 2007, 17, 504–515. [Google Scholar] [CrossRef]
- Zamfir, A.D.; Vukelic, Z.; Schneider, A.; Sisu, E.; Dinca, N.; Ingendoh, A. A novel approach for ganglioside structural analysis based on electrospray multiple-stage mass spectrometry. J. Biomol. Tech. 2007, 18, 188–193. [Google Scholar]
- Zamfir, A.; Peter-Katalinic, J. Capillary electrophoresis-mass spectrometry for glycoscreening in biomedical research. Electrophoresis 2004, 25, 1949–1963. [Google Scholar] [CrossRef]
- Gobburi, A.L.P.; Kipruto, E.W.; Inman, D.M.; Anderson, D.J. A New LC-MS/MS Technique for Separation of Gangliosides Using a Phenyl-Hexyl Column: Systematic Separation According to Sialic Acid Class and Ceramide Subclass. J. Liq. Chromatogr. Relat. Technol. 2021, 44, 114–125. [Google Scholar] [CrossRef]
- Szabo, Z.; Guttman, A.; Rejtar, T.; Karger, B.L. Improved sample preparation method for glycan analysis of glycoproteins by CE-LIF and CE-MS. Electrophoresis 2010, 31, 1389–1395. [Google Scholar] [CrossRef]
- Guttman, A.; Kerekgyarto, M.; Jarvas, G. Effect of Separation Temperature on Structure Specific Glycan Migration in Capillary Electrophoresis. Anal. Chem. 2015, 87, 11630–11634. [Google Scholar] [CrossRef]
- Geda, O.; Tabi, T.; Szoko, E. Development and validation of capillary electrophoresis method for quantification of gangliosides in brain synaptosomes. J. Pharm. Biomed. Anal. 2021, 205, 114329. [Google Scholar] [CrossRef]
- Lienard--Mayor, T.; Yang, B.; Tran, N.T.; Bruneel, A.; Guttman, A.; Taverna, M.; Mai, T.D. High sensitivity capillary electrophoresis with fluorescent detection for glycan mapping. J. Chromatogr. A 2021, 1657, 462593. [Google Scholar] [CrossRef]
- Guttman, A.; Cooke, N.; Starr, C.M. Capillary electrophoresis separation of oligosaccharides: I. Effect of operational variables. Electrophoresis 1994, 15, 1518–1522. [Google Scholar] [CrossRef]
- Lu, G.; Crihfield, C.L.; Gattu, S.; Veltri, L.M.; Holland, L.A. Capillary Electrophoresis Separations of Glycans. Chem. Rev. 2018, 118, 7867–7885. [Google Scholar] [CrossRef]
- Chen, F.T.; Dobashi, T.S.; Evangelista, R.A. Quantitative analysis of sugar constituents of glycoproteins by capillary electrophoresis. Glycobiology 1998, 8, 1045–1052. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, L.; Thomas, S.; Hu, Y.; Li, S.; Cipollo, J.; Zhang, H. Modification of Sialic Acids on Solid Phase: Accurate Characterization of Protein Sialylation. Anal. Chem. 2017, 89, 6330–6335. [Google Scholar] [CrossRef]
- Busch, C.; Desai, A.; Moorthy, G.; Fox, E.; Balis, F. A validated HPLC-MS/MS method for estimating the concentration of the ganglioside, G(D2), in human plasma or serum. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1102–1103, 60–65. [Google Scholar] [CrossRef]
- Gu, J.; Tifft, C.J.; Soldin, S.J. Simultaneous quantification of GM1 and GM2 gangliosides by isotope dilution tandem mass spectrometry. Clin. Biochem. 2008, 41, 413–417. [Google Scholar] [CrossRef]
- Bassi, R.; Viani, P.; Giussani, P.; Riboni, L.; Tettamanti, G. GM3 ganglioside inhibits endothelin-1-mediated signal transduction in C6 glioma cells. FEBS Lett. 2001, 507, 101–104. [Google Scholar] [CrossRef]
- Zheng, C.; Terreni, M.; Sollogoub, M.; Zhang, Y. Ganglioside GM3 and Its Role in Cancer. Curr. Med. Chem. 2019, 26, 2933–2947. [Google Scholar] [CrossRef]
- Paret, C.; Ustjanzew, A.; Ersali, S.; Seidmann, L.; Jennemann, R.; Ziegler, N.; El Malki, K.; Russo, A.; Wingerter, A.; Ortmüller, F.; et al. GD2 Expression in Medulloblastoma and Neuroblastoma for Personalized Immunotherapy: A Matter of Subtype. Cancers 2022, 14, 6051. [Google Scholar] [CrossRef]
- Sabbih, G.O.; Danquah, M.K. Neuroblastoma GD2 Expression and Computational Analysis of Aptamer-Based Bioaffinity Targeting. Int. J. Mol. Sci. 2021, 22, 9101. [Google Scholar] [CrossRef]
- Mohd, A.B.; Mohd, O.B.; Alabdallat, Y.J.; Al Dwairy, S.Y.; Ghannam, R.A.; Hanaqtah, B.M.; Albakri, K.A. Safety and efficacy of dinutuximab in the treatment of neuroblastoma: A review. J. Res. Med. Sci. 2023, 28, 71. [Google Scholar]
- Svennerholm, L.; Fredman, P. A procedure for the quantitative isolation of brain gangliosides. Biochim. Biophys. Acta 1980, 617, 97–109. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- International Conference on Harmonization. ICH Guideline M10 on Bioanalytical Method Validation and Study Sample Analysis; International Conference on Harmonization: Geneva, Switzerland, 2022. [Google Scholar]
- International Conference on Harmonization. ICH Guidline Q2(R1) Validation of Analytical Procedures: Text and Methodology; International Conference on Harmonization: Geneva, Switzerland, 2005. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linear Range (nM) | Slope | Y-Intercept | Std. Error of Slope | Std. Error of Y-Intercept | Correlation Coefficient (R2) | LOQ (nM) | LOD (nM) | |
|---|---|---|---|---|---|---|---|---|
| GM1 | 125–1250 | 7.58 × 10−4 | −2.31 × 10−2 | 3.30 × 10−5 | 6.72 × 10−3 | 0.9944 | 88.7 | 29.2 |
| GM2 | 125–1250 | 5.84 × 10−4 | −1.78 × 10−2 | 9.81 × 10−6 | 2.00 × 10−3 | 0.9992 | 34.2 | 11.3 |
| GM3 | 500–2500 | 7.18 × 10−4 | −1.23 × 10−1 | 2.51 × 10−5 | 1.50 × 10−2 | 0.9963 | 208.5 | 68.8 |
| GD1a | 125–1250 | 7.36 × 10−4 | −3.66 × 10−2 | 3.09 × 10−5 | 1.82 × 10−3 | 0.9930 | 24.7 | 8.1 |
| GD1b | 125–1250 | 8.10 × 10−4 | −6.80 × 10−2 | 1.96 × 10−5 | 6.08 × 10−3 | 0.9982 | 75.1 | 24.7 |
| GD2 | 125–1250 | 5.59 × 10−4 | −8.34 × 10−2 | 2.04 × 10−5 | 5.12 × 10−3 | 0.9960 | 91.7 | 30.2 |
| GD3 | 125–1250 | 3.36 × 10−4 | 7.01 × 10−3 | 4.59 × 10−6 | 1.40 × 10−3 | 0.9993 | 41.6 | 13.7 |
| GT1b | 125–1250 | 7.48 × 10−4 | −5.01 × 10−2 | 4.02 × 10−5 | 7.76 × 10−3 | 0.9914 | 103.7 | 34.2 |
| GQ1b | 62.5–625 | 4.00 × 10−4 | −3.44 × 10−3 | 1.56 × 10−5 | 1.91 × 10−3 | 0.9940 | 47.7 | 15.7 |
| Intradays | n = 4 | Interdays | n = 4 | ||||
|---|---|---|---|---|---|---|---|
| Added Concentration (nM) | Mean (nM) | Accuracy (%) | Precision (RSD%) | Mean (nM) | Accuracy (%) | Precision (RSD%) | |
| 1000 | 1088.7 | 108.8 | 6.6 | 1038.2 | 103.8 | 9.2 | |
| GM1 | 500 | 498.02 | 99.6 | 1.5 | 489.7 | 97.9 | 2.8 |
| 125 | 132.9 | 106.3 | 6.0 | 112.1 | 89.7 | 9.8 | |
| 1000 | 1002.5 | 100.2 | 4.2 | 1066.9 | 106.6 | 12.7 | |
| GM2 | 500 | 497.3 | 99.4 | 1.5 | 555.9 | 111.2 | 5.6 |
| 125 | 107.2 | 85.7 | 7.8 | 112.3 | 89.8 | 13.3 | |
| 2000 | 2221.2 | 111.0 | 2.1 | 2295.8 | 114.7 | 5.9 | |
| GM3 | 1250 | 1070.9 | 85.6 | 8.1 | 1275.1 | 102.0 | 4.1 |
| 500 | 545.3 | 109.0 | 4.6 | 562.8 | 112.5 | 14.6 | |
| 1000 | 1041.3 | 104.1 | 7.6 | 1131.9 | 113.2 | 7.9 | |
| GD1a | 500 | 493.7 | 98.7 | 4.1 | 564.6 | 112.9 | 3.2 |
| 125 | 134.0 | 107.2 | 3.5 | 135.7 | 108.6 | 7.9 | |
| 1000 | 1063.8 | 106.3 | 9.7 | 1062.1 | 106.2 | 4.4 | |
| GD1b | 500 | 450.4 | 90.0 | 4.8 | 501.5 | 100.3 | 3.9 |
| 125 | 122.5 | 98.0 | 14.8 | 119.7 | 95.7 | 10.3 | |
| 1000 | 996.1 | 99.6 | 8.6 | 1098.7 | 109.8 | 14.1 | |
| GD2 | 500 | 464.6 | 92.9 | 5.6 | 474.0 | 94.8 | 11.7 |
| 125 | 117.6 | 94.0 | 11.7 | 130.6 | 104.5 | 10.3 | |
| 1000 | 1054.7 | 105.4 | 8.1 | 1123.0 | 112.3 | 4.2 | |
| GD3 | 500 | 495.1 | 99.0 | 7.4 | 480.1 | 96.0 | 5.5 |
| 125 | 119.8 | 95.9 | 5.5 | 121.4 | 97.1 | 13.3 | |
| 1000 | 1088.0 | 108.8 | 1.1 | 1093.6 | 109.3 | 8.6 | |
| GT1b | 500 | 443.9 | 88.7 | 9.6 | 528.2 | 105.6 | 10.2 |
| 125 | 111.4 | 89.1 | 8.8 | 130.2 | 104.2 | 8.1 | |
| 500 | 496.0 | 99.2 | 8.1 | 537.4 | 107.5 | 12.8 | |
| GQ1b | 250 | 268.7 | 107.5 | 4.6 | 276.9 | 110.7 | 8.9 |
| 62.5 | 58.4 | 93.5 | 7.2 | 73.7 | 117.9 | 13.2 |
| Ganglioside Content (nmol/mg Protein) | |
| C6 Glioblastoma | |
| GM3 | 20.20 ± 1.97 |
| GQ1b | 1.27 ± 0.32 |
| GD2 | 3.92 ± 0.04 |
| GT1b | 1.88 ± 0.14 |
| SH-SY5Y Neuroblastoma | |
| GM3 | 2.93 ± 0.28 |
| GQ1b | 3.42 ± 0.19 |
| GD2 | 24.60 ± 3.60 |
| GT1b | 2.15 ± 0.38 |
| GM2 | 6.77 ± 1.37 |
| GD1a | 3.60 ± 0.54 |
| GM1 | 1.30 ± 0.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarnóczi, K.; Geda, O.; Tábi, T.; Szökő, É. Capillary Electrophoresis-Laser Induced Fluorescence Method Development and Validation for Quantification of Nine Gangliosides—Application to Analysis of Cell Lines of CNS Origin. Molecules 2024, 29, 3769. https://doi.org/10.3390/molecules29163769
Tarnóczi K, Geda O, Tábi T, Szökő É. Capillary Electrophoresis-Laser Induced Fluorescence Method Development and Validation for Quantification of Nine Gangliosides—Application to Analysis of Cell Lines of CNS Origin. Molecules. 2024; 29(16):3769. https://doi.org/10.3390/molecules29163769
Chicago/Turabian StyleTarnóczi, Katinka, Orsolya Geda, Tamás Tábi, and Éva Szökő. 2024. "Capillary Electrophoresis-Laser Induced Fluorescence Method Development and Validation for Quantification of Nine Gangliosides—Application to Analysis of Cell Lines of CNS Origin" Molecules 29, no. 16: 3769. https://doi.org/10.3390/molecules29163769
APA StyleTarnóczi, K., Geda, O., Tábi, T., & Szökő, É. (2024). Capillary Electrophoresis-Laser Induced Fluorescence Method Development and Validation for Quantification of Nine Gangliosides—Application to Analysis of Cell Lines of CNS Origin. Molecules, 29(16), 3769. https://doi.org/10.3390/molecules29163769