
Insight into the Binding Interaction between PEDCs and hERRγ Utilizing Molecular Docking and Molecular Dynamics Simulations


Abstract

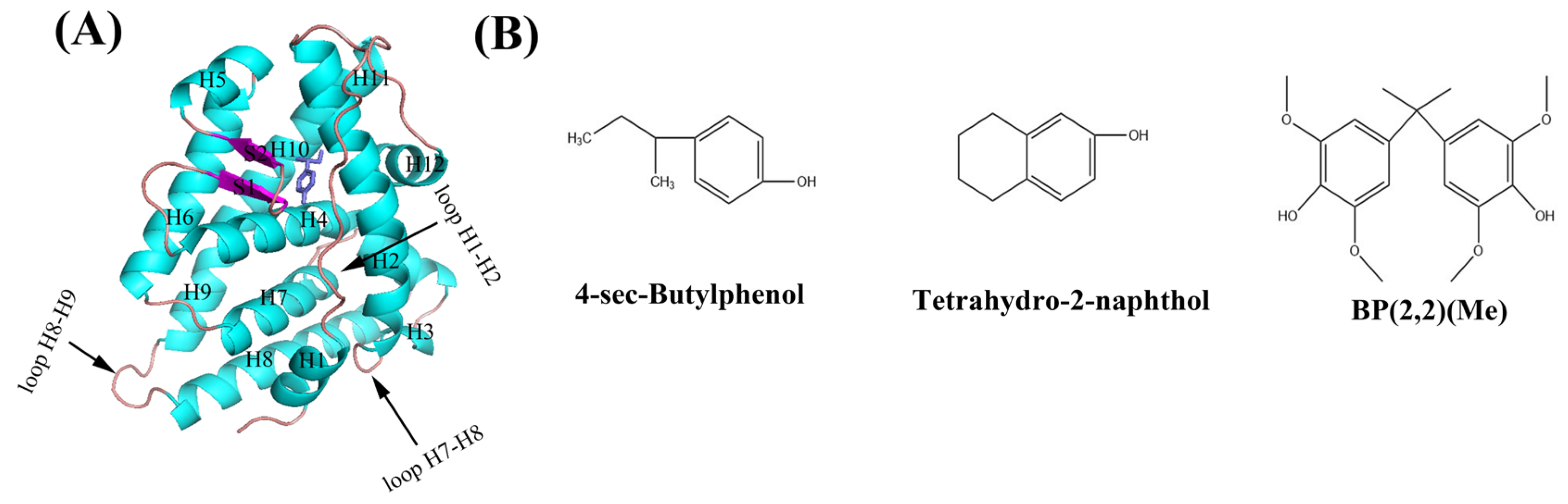
1. Introduction
2. Results and Discussion
2.1. Dynamics of hERRγ-PEDC Complexes
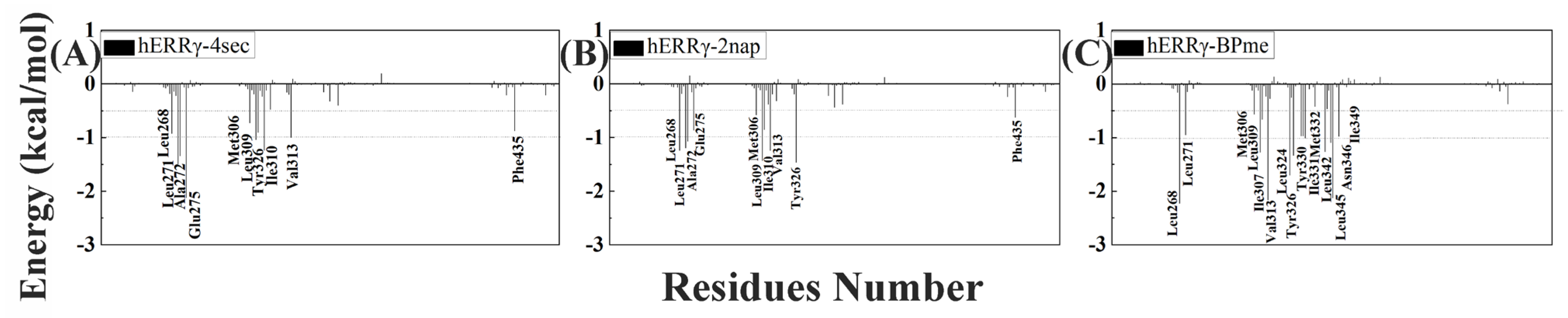
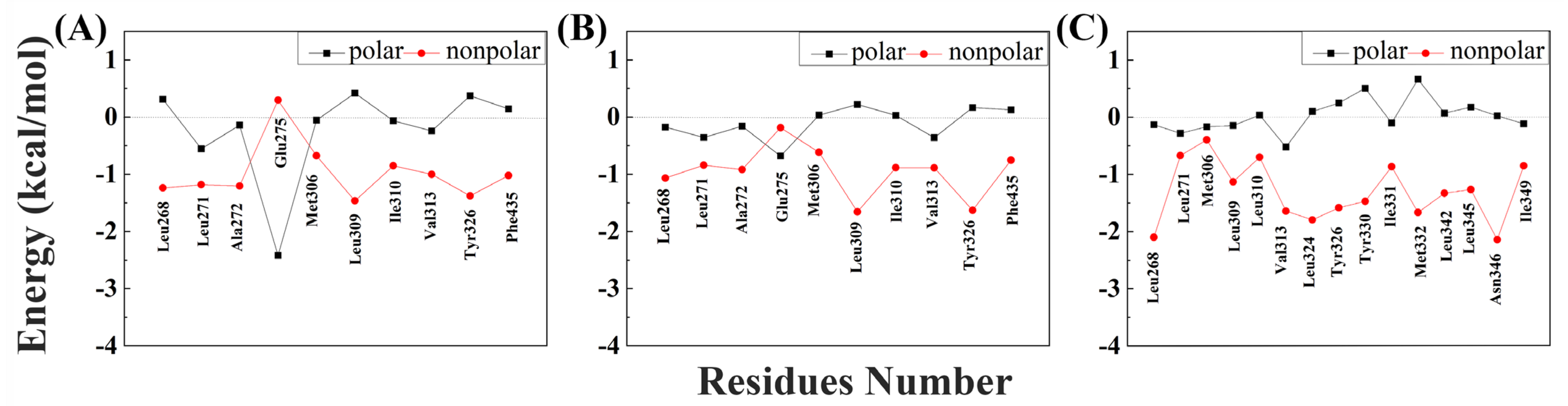
2.2. Analysis of Free Energy between hERRγ and PEDCs
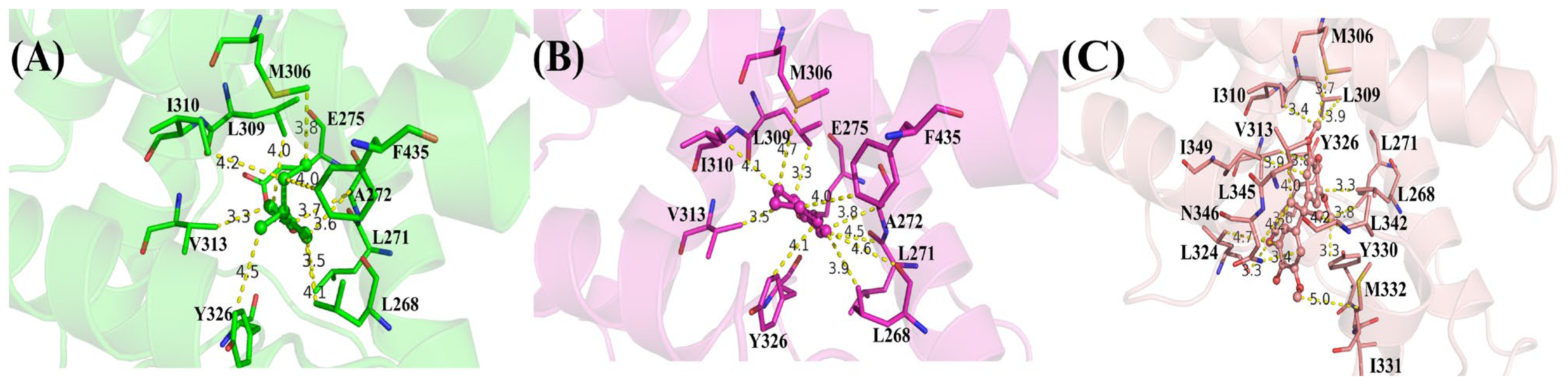
2.3. Structure and Energy Relationship between hERRγ and PEDCs
3. Materials and Methods
3.1. Molecular Docking
3.2. System Setup
3.3. MD Simulation
3.4. MM-PBSA Calculation
3.5. SIE Calculation
3.6. Conformational Dynamics Analysis
3.7. Principal Component Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wan, M.L.Y.; Co, V.A.; El-Nezami, H. Endocrine disrupting chemicals and breast cancer: A systematic review of epidemiological studies. Crit. Rev. Food Sci. Nutr. 2022, 62, 6549–6576. [Google Scholar] [CrossRef]
- Ismanto, A.; Hadibarata, T.; Kristanti, R.A.; Maslukah, L.; Safinatunnajah, N.; Kusumastuti, W. Endocrine disrupting chemicals (EDCs) in environmental matrices: Occurrence, fate, health impact, physio-chemical and bioremediation technology. Environ. Pollut. 2022, 302, 119061. [Google Scholar] [CrossRef]
- Kouiti, M.; Castillo-Hermoso, M.Á.; Youlyouz-Marfak, I.; Khan, K.S.; Thangaratinam, S.; Olmedo-Requena, R.; Zamora, J.; Jiménez-Moléon, J.J. Persistent organic pollutant exposure as a risk factor of gestational diabetes mellitus: A systematic review and meta-analysis. BJOG-Int. J. Obstet. Gy. 2024, 131, 579–588. [Google Scholar] [CrossRef]
- Sieck, N.E.; Bruening, M.; van Woerden, I.; Whisner, C.; Payne-Sturges, D.C. Effects of Behavioral, Clinical, and Policy Interventions in Reducing Human Exposure to Bisphenols and Phthalates: A Scoping Review. Environ. Health Perspect. 2024, 132, 36001. [Google Scholar] [CrossRef]
- Pan, Z.; Tang, C.; Cao, Y.; Xuan, Y.; Zhou, Q. Distribution and source apportionment of phenolic EDCs in rivers in the Pearl River Delta, South China. Environ. Sci. Pollut. Res. 2023, 30, 48248–48259. [Google Scholar] [CrossRef]
- Otitoju, O.B.; Alfred, M.O.; Ogunlaja, O.O.; Olorunnisola, C.G.; Olukanni, O.D.; Ogunlaja, A.; Omorogie, M.O.; Unuabonah, E.I. Pollution and risk assessment of phenolic compounds in drinking water sources from South-Western Nigeria. Environ. Sci. Pollut. Res. 2023, 30, 76798–76817. [Google Scholar] [CrossRef]
- Xu, R.; Liu, S.; Pan, Y.-F.; Wu, N.-N.; Huang, Q.-Y.; Li, H.-X.; Lin, L.; Hou, R.; Xu, X.-R.; Cheng, Y.-Y. Steroid metabolites as overlooked emerging contaminants: Insights from multimedia partitioning and source–sink simulation in an estuarine environment. J. Hazard. Mater. 2024, 461, 132673. [Google Scholar] [CrossRef]
- Ibor, O.R.; Nnadozie, P.; Ogarekpe, D.M.; Idogho, O.; Anyanti, J.; Aizobu, D.; Onyezobi, C.; Chukwuka, A.V.; Adeogun, A.O.; Arukwe, A. Public health implications of endocrine disrupting chemicals in drinking water and aquatic food resources in Nigeria: A state-of-the-science review. Sci. Total. Environ. 2023, 858, 159835. [Google Scholar] [CrossRef]
- Zhang, C.; Lu, J.; Wu, J. Enhanced removal of phenolic endocrine disrupting chemicals from coastal waters by intertidal macroalgae. J. Hazard. Mater. 2021, 411, 125105. [Google Scholar] [CrossRef]
- Alizada, S.; Eminli, F.; Vakhshouri, A.R. A review of novel electrode materials and techniques for electro-Fenton process applied to the degradation of phenolic compounds. Water Environ. J. 2023, 37, 390–401. [Google Scholar] [CrossRef]
- Cavallini, A.; Notarnicola, M.; Giannini, R.; Montemurro, S.; Lorusso, D.; Visconti, A.; Minervini, F.; Caruso, M.G. Oestrogen receptor-related receptor alpha (ERRα) and oestrogen receptors (ERα and ERβ) exhibit different gene expression in human colorectal tumour progression. Eur. J. Cancer 2005, 41, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Zhu, X.; Wang, L.; Zheng, X.; Wang, J. Effect of bisphenol A on the ovarian expressions of estrogen-related receptor gene and protein in queen honey bee (Apis mellifera). Apidologie 2023, 54, 60. [Google Scholar] [CrossRef]
- Chen, L.; Lin, X.; Shi, S.; Li, M.; Mortimer, M.; Fang, W.; Li, F.; Guo, L.-H. Activation of estrogen-related receptor: An alternative mechanism of hexafluoropropylene oxide homologs estrogenic effects. Sci. Total. Environ. 2023, 901, 166257. [Google Scholar] [CrossRef] [PubMed]
- Thouennon, E.; Delfosse, V.; Bailly, R.; Blanc, P.; Boulahtouf, A.; Grimaldi, M.; Barducci, A.; Bourguet, W.; Balaguer, P. Insights into the activation mechanism of human estrogen-related receptor γ by environmental endocrine disruptors. Cell. Mol. Life Sci. 2019, 76, 4769–4781. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, S.; Bhattacharyya, R.; Banerjee, D. Plastic compounds and liver diseases: Whether bisphenol A is the only culprit. Liver Int. 2024, 44, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, S.; Gusev, Y.; Singh, S.; Riggins, R.B. ERRγ target genes are poor prognostic factors in Tamoxifen-treated breast cancer. J. Exp. Clin. Cancer Res. 2015, 34, 45. [Google Scholar] [CrossRef] [PubMed]
- Lasheras, J.; Pardo, R.; Velilla, M.; Poncelas, M.; Salvatella, N.; Simó, R.; Ruiz-Meana, M.; Zamora, M.; Villena, J.A. Cardiac-Specific Overexpression of ERRγ in Mice Induces Severe Heart Dysfunction and Early Lethality. Int. J. Mol. Sci. 2021, 22, 8047. [Google Scholar] [CrossRef] [PubMed]
- Amitrano, A.; Mahajan, J.S.; Korley, L.T.J.; Epps, T.H. Estrogenic activity of lignin-derivable alternatives to bisphenol A assessed via molecular docking simulations. RSC Adv. 2021, 11, 22149–22158. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, M.T.; Aki-Yalcin, E. Molecular docking: Principles, advances, and its applications in drug discovery. Lett. Drug. Des. Discov. 2024, 21, 480–495. [Google Scholar] [CrossRef]
- Shanker, S.; Sanner, M.F. Predicting Protein–Peptide Interactions: Benchmarking Deep Learning Techniques and a Comparison with Focused Docking. J. Chem. Inf. Model. 2023, 63, 3158–3170. [Google Scholar] [CrossRef]
- Vitali, E.; Ficarelli, F.; Bisson, M.; Gadioli, D.; Accordi, G.; Fatica, M.; Beccari, A.R.; Palermo, G. GPU-optimized approaches to molecular docking-based virtual screening in drug discovery: A comparative analysis. J. Parallel Distrib. Comput. 2024, 186, 104819. [Google Scholar] [CrossRef]
- Yasir, M.; Park, J.; Han, E.-T.; Park, W.S.; Han, J.-H.; Kwon, Y.-S.; Lee, H.-J.; Chun, W. Machine Learning-Based Drug Repositioning of Novel Janus Kinase 2 Inhibitors Utilizing Molecular Docking and Molecular Dynamic Simulation. J. Chem. Inf. Model. 2023, 63, 6487–6500. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, K.; Fujie, T.; Takabatake, K.; Akiyama, Y. QUBO Problem Formulation of Fragment-Based Protein–Ligand Flexible Docking. Entropy 2024, 26, 397. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Liu, X.; Liu, X.-C.; Pan, W.-X.; Fu, J.-J.; Zhang, A.-Q. The Effect of Structural Diversity on Ligand Specificity and Resulting Signaling Differences of Estrogen Receptor α. Chem. Res. Toxicol. 2019, 32, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Q.; Zhang, Y.; Niu, Y.; Yao, X.; Liu, H. The Molecular Mechanism of Bisphenol A (BPA) as an Endocrine Disruptor by Interacting with Nuclear Receptors: Insights from Molecular Dynamics (MD) Simulations. PLoS ONE 2015, 10, e0120330. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Liu, H.; Yao, X. Molecular mechanism of hiv-1 integrase-vdna interactions and strand transfer inhibitor action: A molecular modeling perspective. J. Comput. Chem. 2012, 33, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Na, L.; Zhou, W.; Yue, G.; Wang, J.; Fu, W.; Sun, H.; Li, D.; Duan, M.; Hou, T. Molecular dynamics simulations revealed the regulation of ligands to the interactions between androgen receptor and its coactivator. J. Chem. Inf. Model. 2018, 58, 1652–1661. [Google Scholar]
- Auffinger, P.; Westhof, E. Rna hydration: Three nanoseconds of multiple molecular dynamics simulations of the solvated trnaasp anticodon hairpin. J. Mol. Biol. 1997, 269, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Caves, L.S.D.; Evanseck, J.D.; Karplus, M. Locally accessible conformations of proteins: Multiple molecular dynamics simulations of crambin. Protein Sci. 2010, 7, 7649–7666. [Google Scholar] [CrossRef]
- Chen, L.; Huang, X.; Li, Y.; Zhao, B.; Liang, M.; Wang, R. Structural and energetic basis of interaction between human estrogen-related receptor γ and environmental endocrine disruptors from multiple molecular dynamics simulations and free energy predictions. J. Hazard. Mater. 2023, 443, 130174. [Google Scholar] [CrossRef]
- Knapp, B.; Ospina, L.; Deane, C.M. Avoiding False Positive Conclusions in Molecular Simulation: The Importance of Replicas. J. Chem. Theory Comput. 2018, 14, 6127–6138. [Google Scholar] [CrossRef]
- Wang, L.; Lu, D.; Wang, Y.; Xu, X.; Zhong, P.; Yang, Z. Binding selectivity-dependent molecular mechanism of inhibitors towards CDK2 and CDK6 investigated by multiple short molecular dynamics and free energy landscapes. J. Enzym. Inhib. Med. Chem. 2023, 38, 84–99. [Google Scholar] [CrossRef]
- Wang, R.; Zheng, Q. Multiple Molecular Dynamics Simulations and Energy Analysis Unravel the Dynamic Properties and Binding Mechanism of Mutants HIV-1 Protease with DRV and CA-p2. Microbiol. Spectr. 2022, 10, e0074821. [Google Scholar] [CrossRef] [PubMed]
- Sulea, T.; Cui, Q.; Purisima, E.O. Solvated interaction energy (SIE) for scoring protein–ligand binding affinities. 2. Benchmark in the CSAR-2010 scoring exercise. J. Chem. Inf. Model. 2011, 51, 2066–2081. [Google Scholar] [CrossRef]
- Naïm, M.; Bhat, S.; Rankin, K.; Dennis, S.; Chowdhury, S.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.; Jakalian, A.; et al. Solvated interaction energy (SIE) for scoring protein−ligand binding affinities. 1. Exploring the parameter space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef]
- Huang, K.; Luo, S.; Cong, Y.; Zhong, S.; Zhang, J.Z.; Duan, L. An accurate free energy estimator: Based on MM/PBSA combined with interaction entropy for protein–ligand binding affinity. Nanoscale 2020, 12, 10737–10750. [Google Scholar] [CrossRef]
- Ravindranathan, K.; Tirado-Rives, J.; Jorgensen, W.L.; Guimarães, C.R.W. Improving MM-GB/SA Scoring through the Application of the Variable Dielectric Model. J. Chem. Theory Comput. 2011, 7, 3859–3865. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate—DNA Helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Gohlke, H.; Case, D.A. Converging free energy estimates: MM-PB (GB) SA studies on the protein–protein complex Ras–Raf. J. Comput. Chem. 2004, 25, 238–250. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuserica, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Case, D.A.; Cheatham, T.E., III; Darden, T.A.; Glhike, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.L.; Wang, B.; Woods, R.J. The Amber biomolecular simualtion programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N.log (N) method for Ewald sums in large systems. J. Comput. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- KrUtler, V.; Gunsteren, W.F.V.; Hünenberger, H.P. A fast shake algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2015, 22, 501–508. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr 2002, 40, 82–92. [Google Scholar]
- Hou, T.; Wang, J.; Wang, Y.; Li, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDB bind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef] [PubMed]
- Swanson, J.M.; Henchman, R.H.; McCammon, J.A. Revisiting Free Energy Calculations: A Theoretical Connection to MM/PBSA and Direct Calculation of the Association Free Energy. Biophys. J. 2004, 86, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; David, L.; Gilson, M.K. Accelerated Poisson–Boltzmann calculations for static and dynamic systems. J. Comput. Chem. 2002, 23, 1244–1253. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A. Normal mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 1994, 4, 285–290. [Google Scholar] [CrossRef]
- Cui, Q.; Sulea, T.; Schrag, J.D.; Munger, C.; Hung, M.N.; Naïm, M.; Cygler, M.E.; Purisima, O. Molecular dynamics-Solvated interaction energy studies of protein–protein interactions: The MP1–p14 scaffolding complex. J. Mol. Biol. 2008, 379, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Hünenberger, P.; Mark, A.; van Gunsteren, W. Fluctuation and Cross-correlation Analysis of Protein Motions Observed in Nanosecond Molecular Dynamics Simulations. J. Mol. Biol. 1995, 252, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Balsera, M.A.; Wriggers, W.; Oono, Y.; Schulten, K. Principal Component Analysis and Long Time Protein Dynamics. J. Phys. Chem. 1996, 100, 2567–2572. [Google Scholar] [CrossRef]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Principal Component Analysis for Protein Folding Dynamics. J. Mol. Biol. 2009, 385, 312–329. [Google Scholar] [CrossRef]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Protein Dyn. Methods Protoc. 2014, 1084, 193–226. [Google Scholar]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | hERRγ-4sec | hERRγ-2nap | hERRγ-BPMe |
|---|---|---|---|
| ΔEvdW | −23.95 ± 2.28 | −23.54 ± 2.20 | −46.47 ± 2.64 |
| ΔEele | −13.02 ± 3.29 | −12.54 ± 6.15 | −5.59 ± 4.66 |
| ΔE(MM) | −36.97 ± 2.83 | −36.08 ± 4.62 | −52.06 ± 3.79 |
| ΔGPB | 17.49 ± 2.98 | 16.30 ± 3.50 | 32.29 ± 4.45 |
| ΔGSA | −1.67 ± 0.08 | −1.56 ± 0.09 | −3.73 ± 0.12 |
| ΔGsol | 15.82 ± 2.11 | 14.74 ± 2.48 | 28.56 ± 3.15 |
| ΔGpolar | 4.47 ± 3.14 | 3.76 ± 5.00 | 26.70 ± 4.56 |
| ΔGnonpol | −25.62 ± 1.61 | −25.10 ± 1.55 | −50.20 ± 1.87 |
| ΔH | −21.15 ± 3.29 | −21.33 ± 3.82 | −23.49 ± 3.79 |
| −TΔS | 15.21 ± 3.04 | 15.66 ± 3.10 | 20.52 ± 3.91 |
| ΔGbind | −5.94 ± 3.17 | −5.67 ± 3.31 | −2.97 ± 3.85 |
| ΔGexp | −9.49 | −8.98 |
| System | hERRγ-4sec | hERRγ-2nap | hERRγ-BPMe |
|---|---|---|---|
| ΔEvdW | −27.63 ± 2.17 | −25.93 ± 2.26 | −28.37 ± 2.83 |
| ΔECoul | −7.21 ± 1.42 | −8.16 ± 2.74 | −2.56 ± 2.37 |
| ΔGcav | −5.49 ± 0.24 | −6.11 ± 0.27 | −5.68 ± 0.61 |
| ΔGR | 4.17 ± 0.74 | 5.34 ± 0.99 | 9.96 ± 1.69 |
| ΔGnonpol | −33.12 ± 1.13 | −32.04 ± 1.61 | −34.05 ± 2.06 |
| ΔGpol | −3.04 ± 1.54 | −2.82 ± 1.94 | 7.4 ± 2.05 |
| ΔGbind | −6.68 ± 0.21 | −6.54 ± 0.24 | −5.68 ± 0.37 |
| ΔGexp | −9.49 | −8.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bu, F.; Chen, L.; Sun, Y.; Zhao, B.; Wang, R. Insight into the Binding Interaction between PEDCs and hERRγ Utilizing Molecular Docking and Molecular Dynamics Simulations. Molecules 2024, 29, 3256. https://doi.org/10.3390/molecules29143256
Bu F, Chen L, Sun Y, Zhao B, Wang R. Insight into the Binding Interaction between PEDCs and hERRγ Utilizing Molecular Docking and Molecular Dynamics Simulations. Molecules. 2024; 29(14):3256. https://doi.org/10.3390/molecules29143256
Chicago/Turabian StyleBu, Fanqiang, Lin Chen, Ying Sun, Bing Zhao, and Ruige Wang. 2024. "Insight into the Binding Interaction between PEDCs and hERRγ Utilizing Molecular Docking and Molecular Dynamics Simulations" Molecules 29, no. 14: 3256. https://doi.org/10.3390/molecules29143256
APA StyleBu, F., Chen, L., Sun, Y., Zhao, B., & Wang, R. (2024). Insight into the Binding Interaction between PEDCs and hERRγ Utilizing Molecular Docking and Molecular Dynamics Simulations. Molecules, 29(14), 3256. https://doi.org/10.3390/molecules29143256