
Synthesis and Biological Evaluation of Novel Biased Mu-Opioid Receptor Agonists

Abstract
1. Introduction
2. Results and Discussion
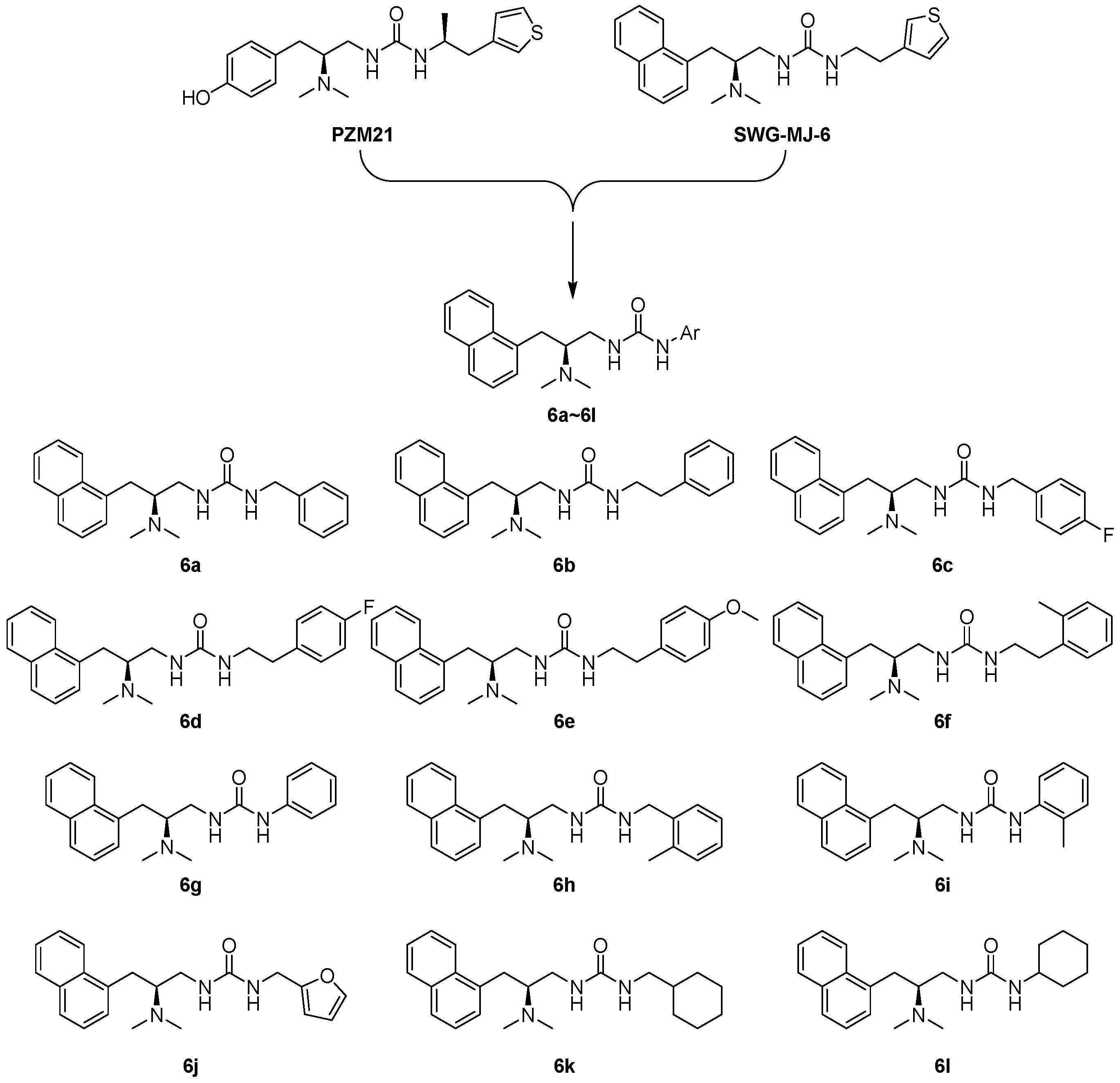
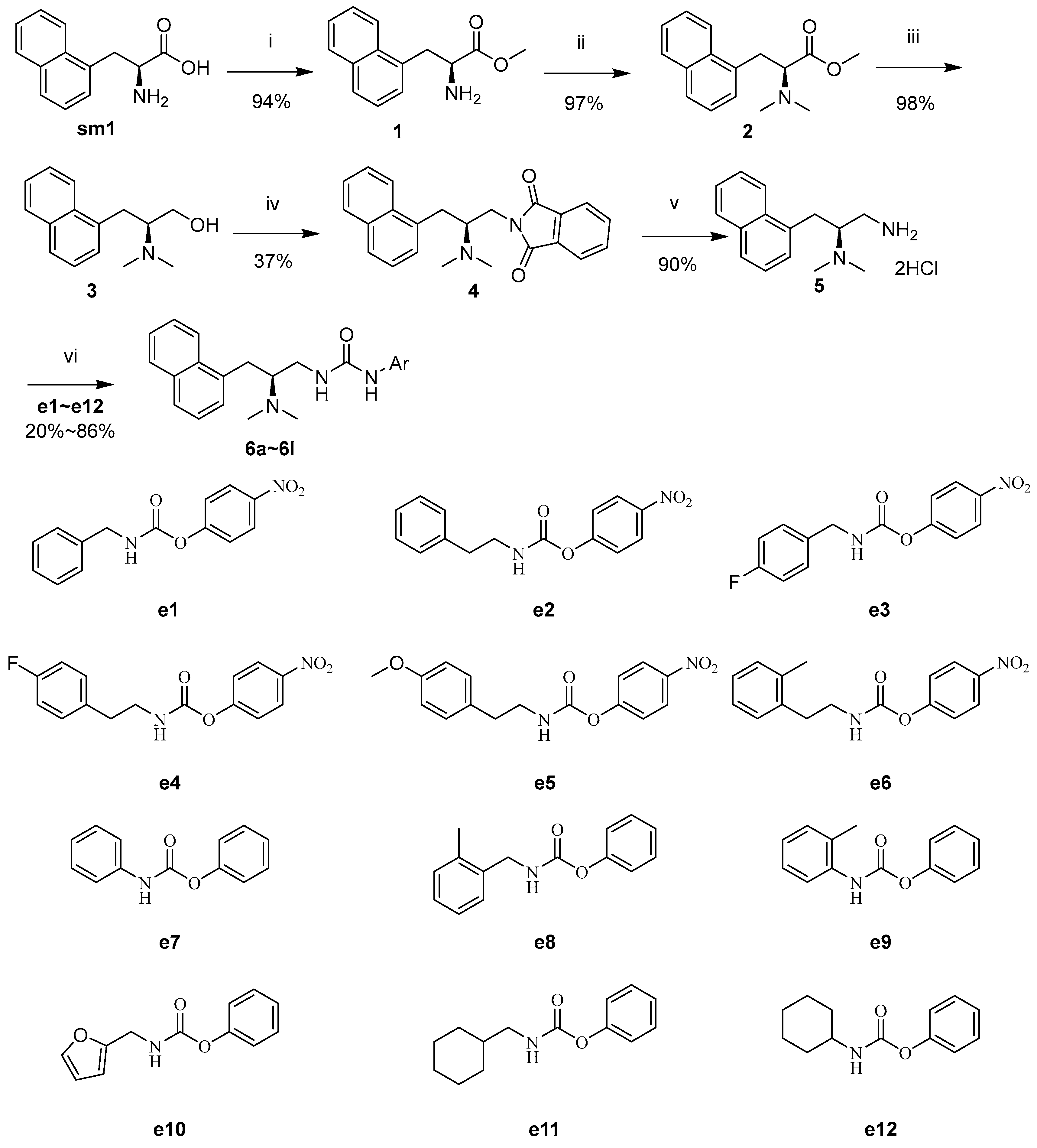
2.1. Synthesis
2.2. The Selective Gi/O-Biased μOR Agonist Activities and the β-Arrestin Recruitment Assay In Vitro
2.2.1. μOR Gi/o-Mediated cAMP Inhibition
2.2.2. β-arrestin Recruitment Assay
2.3. In Vivo Antinociceptive Effects
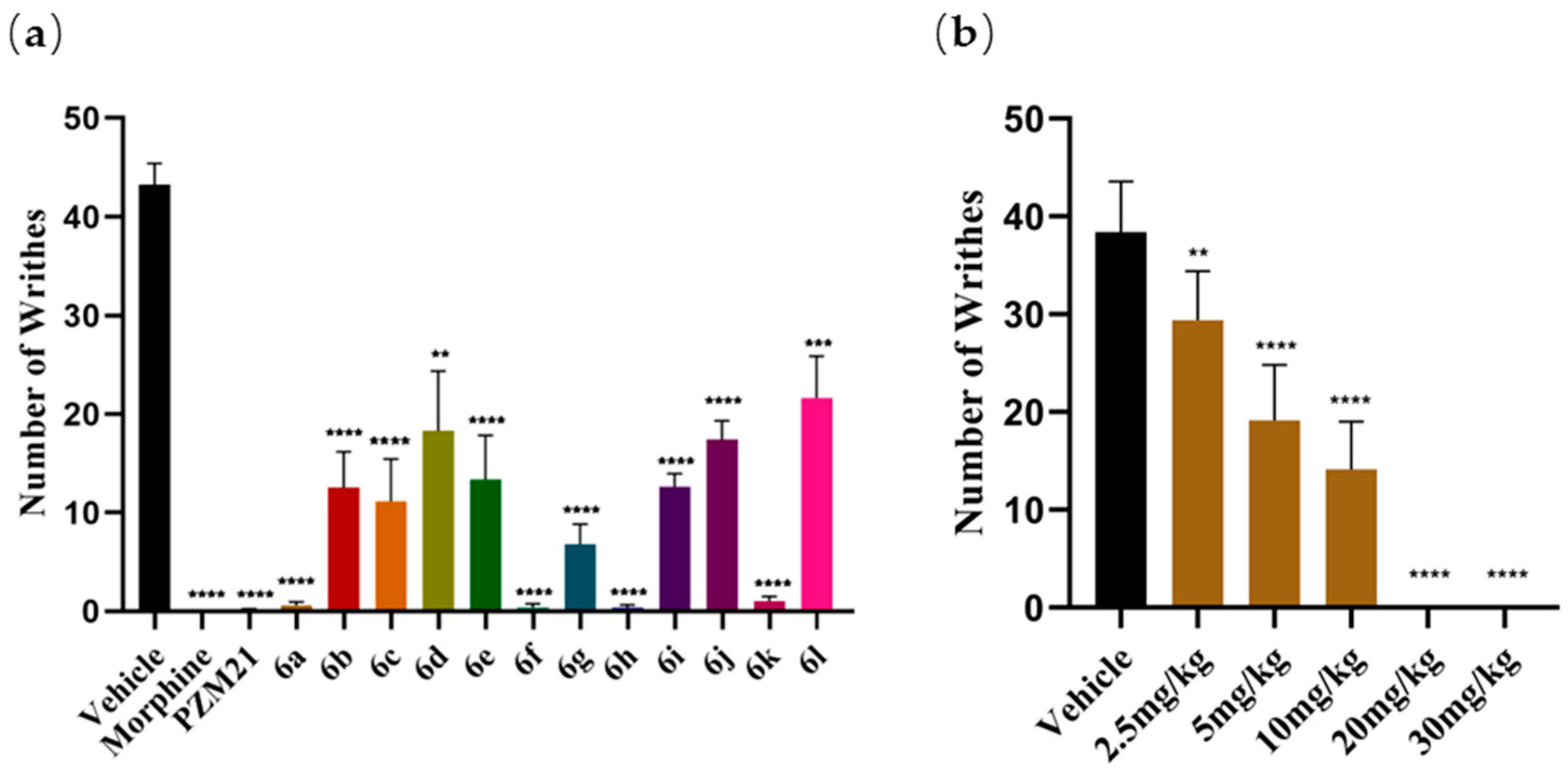
2.3.1. Evaluation of Antinociceptive Activities of the Novel Compounds in the Acetic Acid-Induced Writhing Assay
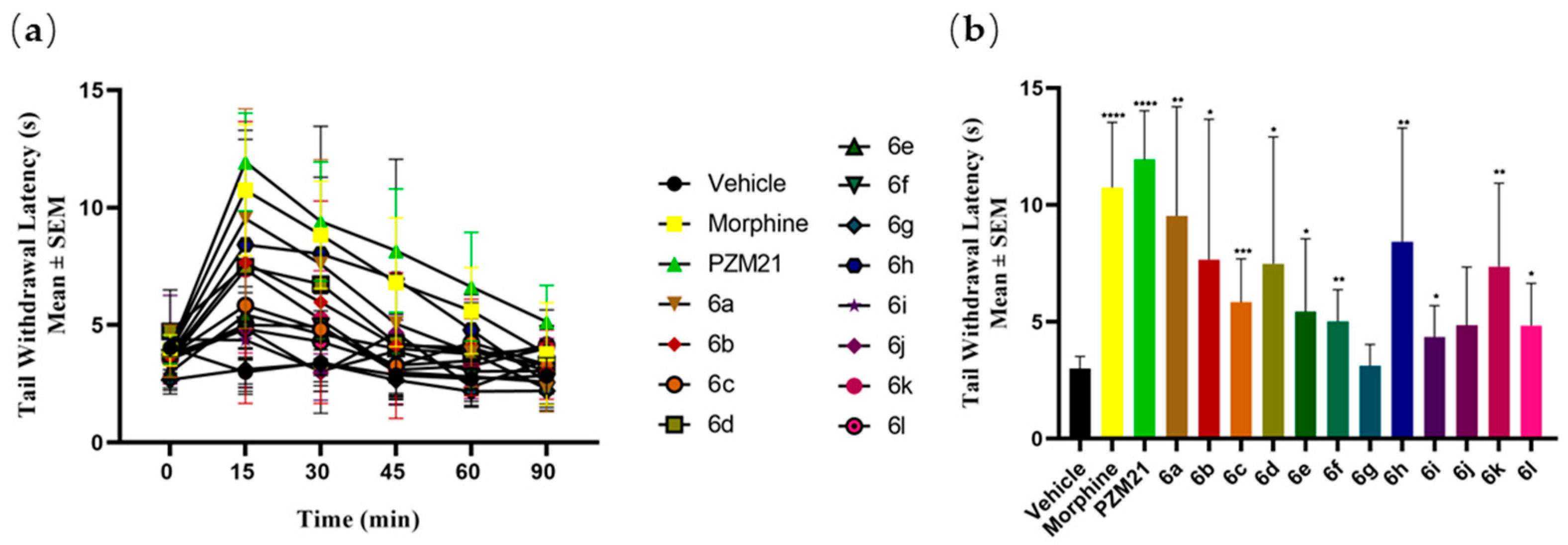
2.3.2. Antinociceptive Activities of the Novel Compounds in the Hot-Water Tail Withdrawal Test
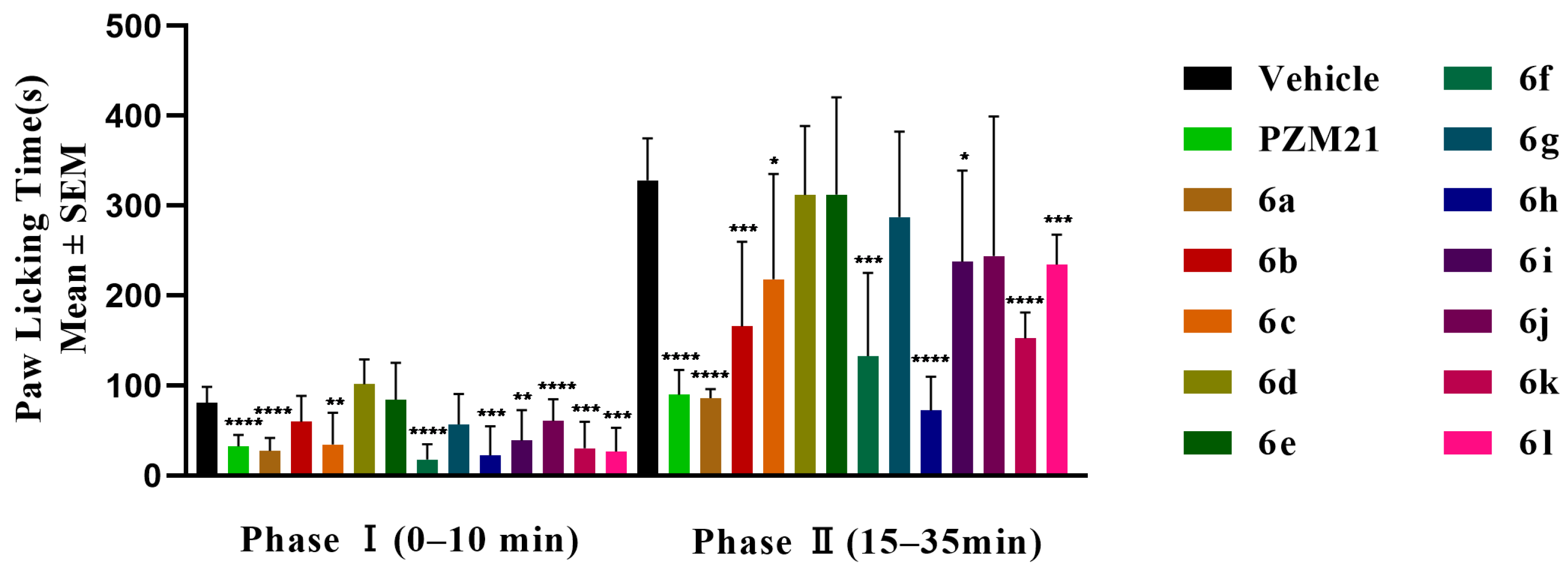
2.3.3. Antinociceptive Activities of the Novel Compounds in a Formalin-Induced Nociception Assay
2.4. In Vivo Adverse Effects Studies
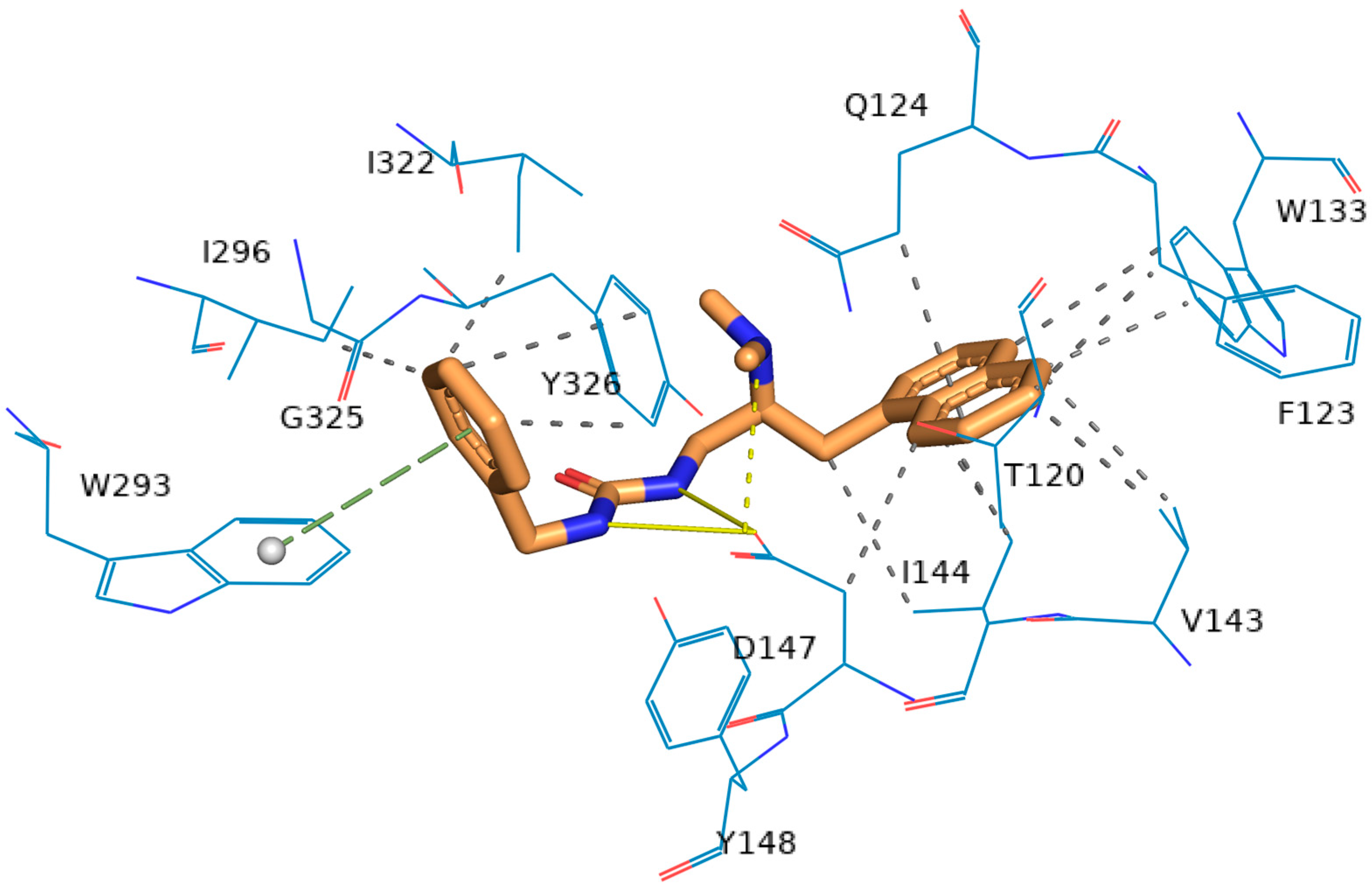
2.5. Molecular Modeling
3. Materials and Methods
3.1. General Information
3.2. Chemical and Reagents
3.3. Animals
3.4. Chemistry
3.4.1. Synthesis of e1~e6
3.4.2. Synthesis of e7~e12
3.4.3. Synthesis of 5
3.4.4. Synthesis of 6a~6l
- (S)-1-benzyl-3-(2-(dimethylamino)-3-(naphthalen-1-yl)propyl) urea (6a): 1H NMR (600 MHz, DMSO-d6) δ 8.07 (d, J = 8.3 Hz, 1H), 7.93 (d, J = 8.1 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.56–7.50 (m, 2H), 7.43 (t, J = 7.4 Hz, 1H), 7.38 (br d, J = 6.9 Hz, 1H), 7.27–7.25 (m, 2H), 7.19–7.17 (m, 3H), 6.54 (t, J = 6.0 Hz, 1H), 5.81–5.80 (m, 1H), 4.11 (d, J = 6.0 Hz, 2H), 3.37–3.34 (m, 1H), 3.07–2.97 (m, 2H), 2.87–2.81 (m, 2H), 2.39 (s, 6H). 13C NMR (150 MHz, DMSO-d6) δ 157.8, 140.9, 136.0, 133.5, 131.6, 128.7, 128.1, 127.5, 126.9, 126.6, 126.5, 126.0, 125.5, 123.5, 64.0, 42.8, 40.0, 39.5, 28.5. HR-ESI-MS m/z 362.2227 [M + H]+ (calcd. for C23H28N3O, 362.2232).
- (S)-1-(2-(dimethylamino)-3-(naphthalen-1-yl)propyl)-3-phenethyl urea (6b): 1H NMR (600 MHz, DMSO-d6) δ 8.07 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.58–7.49 (m, 2H), 7.46–7.41 (m, 1H), 7.38 (br d, J = 6.8 Hz, 1H), 7.26–7.23 (m, 2H), 7.18–7.12 (m, 3H), 6.08 (t, J = 5.4 Hz, 1H), 5.74 (br s, 1H), 3.37–3.34 (m, 1H), 3.13 (dd, J = 13.7, 6.6 Hz, 2H), 3.05–2.93 (m, 2H), 2.88–2.79 (m, 2H), 2.60 (t, J = 7.3 Hz, 2H), 2.38 (s, 6H). 13C NMR (150 MHz, DMSO-d6) δ 157.7, 139.7, 136.0, 133.6, 131.6, 128.7, 128.6, 128.2, 127.6, 126.6, 126.0, 125.9, 125.5, 125.5, 123.5, 64.1, 40.8, 40.0, 39.3, 36.2, 28.5. HR-ESI-MS m/z 376.2383 [M + H]+ (calcd. for C24H30N3O, 376.2389).
- (S)-1-(2-(dimethylamino)-3-(naphthalen-1-yl)propyl)-3-(4-fluoro benzyl) urea (6c): 1H NMR (600 MHz, DMSO-d6) δ 8.07 (d, J = 8.3 Hz, 1H), 7.93 (d, J = 7.9 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.57–7.49 (m, 2H), 7.45–7.36 (m, 2H), 7.22–7.17 (m, 2H), 7.10–7.05 (m, 2H), 6.58–6.53 (m, 1H), 5.79 (br s, 1H), 4.08 (d, J = 5.9 Hz, 2H), 3.37–3.29 (m, 1H), 3.06–2.92 (m, 2H), 2.90–2.78 (m, 2H), 2.39 (s, 6H). 13C NMR (150 MHz, DMSO-d6) δ 161.0 (d, J = 241.8 Hz), 157.8, 137.1, 133.5, 131.6, 128.8 (d, J = 8.0 Hz), 128.7, 127.6, 126.6, 126.6, 126.0, 125.5 (d, J = 2.7 Hz), 123.5, 114.8 (d, J = 21.1 Hz), 64.0, 42.1, 40.1, 40.0, 28.4. HR-ESI-MS m/z 380.2133 [M + H]+ (calcd. for C23H27FN3O, 380.2138).
- (S)-1-(2-(dimethylamino)-3-(naphthalen-1-yl)propyl)-3-(4-fluoro phenethyl) urea (6d): 1H NMR (600 MHz, DMSO-d6) δ 8.07 (d, J = 8.3 Hz, 1H), 7.93 (d, J = 7.7 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.58–7.49 (m, 2H), 7.46–7.41 (m, 1H), 7.38 (br d, J = 6.8 Hz, 1H), 7.20–7.15 (m, 2H), 7.09–7.03 (m, 2H), 6.09–6.04 (m, 1H), 5.71 (br s, 1H), 3.38–3.29 (m, 1H), 3.11 (dd, J = 13.3, 6.7 Hz, 2H), 3.06–2.98 (m, 1H), 2.98–2.78 (m, 1H), 2.88–2.78 (m, 2H), 2.58 (t, J = 7.2 Hz, 2H), 2.38 (s, 6H). 13C NMR (150 MHz, DMSO-d6) δ 160.7 (d, J = 241.4 Hz), 157.7, 135.9, 135.8, 133.5, 131.6, 130.3 (d, J = 7.6 Hz), 128.7, 127.6, 126.6, 126.0, 125.5 (d, J = 3.0 Hz), 123.5, 114.9 (d, J = 21.0 Hz), 64.1, 40.8, 40.1, 35.2, 28.4. HR-ESI-MS m/z 394.2289 [M + H]+ (calcd. for C24H29FN3O, 394.2295).
- (S)-1-(2-(dimethylamino)-3-(naphthalen-1-yl)propyl)-3-(4-methoxy phenethyl) urea (6e): 1H NMR (600 MHz, DMSO-d6) δ 8.07 (d, J = 8.3 Hz, 1H), 7.93 (d, J = 7.8 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.58–7.49 (m, 2H), 7.47–7.41 (m, 1H), 7.38 (br d, J = 6.7 Hz, 1H), 7.07–7.04 (m, 2H), 6.84–6.78 (m, 2H), 6.04 (t, J = 4.8 Hz, 1H), 5.72 (br s, 1H), 3.69 (s, 3H), 3.39–3.27 (m, 1H), 3.08 (dd, J = 13.5, 6.7 Hz, 2H), 3.05–2.99 (m, 1H), 2.99–2.89 (m, 1H), 2.90–2.77 (m, 2H), 2.52 (t, J = 7.3 Hz, 2H), 2.38 (s, 6H). 13C NMR (150 MHz, DMSO-d6) δ 157.5, 133.5, 131.6, 129.5, 128.7, 127.6, 126.6, 126.0, 125.5, 125.5, 123.5, 113.7, 64.1, 54.9, 41.1, 40.1, 35.2, 28.5. HR-ESI-MS m/z 406.2489 [M + H]+ (calcd. for C25H32N3O2, 406.2495).
- (S)-1-(2-(dimethylamino)-3-(naphthalen-1-yl) propyl)-3-(2-methyl phenethyl) urea (6f): 1H NMR (600 MHz, DMSO-d6) δ 8.08 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 7.8 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.58–7.49 (m, 2H), 7.47–7.42 (m, 1H), 7.39 (br d, J = 6.9 Hz, 1H), 7.14–7.08 (m, 1H), 7.07–7.03 (m, 3H), 6.15 (t, J = 5.4 Hz, 1H), 5.73 (br s, 1H), 3.39–3.33 (m, 1H), 3.07 (dd, J = 14.6, 6.2 Hz, 2H), 3.05–3.00 (m, 1H), 2.99–2.93 (m, 1H), 2.90–2.78 (m, 2H), 2.62–2.56 (m, 2H), 2.40 (s, 6H), 2.22 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 157.8, 137.8, 135.9, 133.6, 131.6, 129.9, 129.0, 128.7, 127.6, 126.6, 126.0, 126.0, 125. 8, 125.5, 123.5, 64.1, 40.1, 39.3, 33.8, 28.5, 18.9. HR-ESI-MS m/z 390.2540 [M + H]+ (calcd. for C25H32N3O, 390.2545).
- (S)-1-(2-(dimethylamino)-3-(naphthalen-1-yl) propyl)-3-phenyl urea (6g): 1H NMR (600 MHz, DMSO-d6) δ 8.67 (s, 1H), 8.09 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 8.1 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.58–7.54 (m, 1H), 7.53–7.49 (m, 1H), 7.45 (t, J =7.5 Hz, 1H), 7.41 (br d, J = 6.9 Hz, 1H), 7.29 (d, J = 8.0 Hz, 2H), 7.15 (t, J = 8.0 Hz, 2H), 6.83 (t, J = 7.3 Hz, 1H), 6.05 (br d, J = 5.8 Hz, 1H), 3.42 (dd, J = 13.3, 3.2 Hz, 1H), 3.10 (ddd, J = 12.0, 7.1, 4.5 Hz, 1H), 3.04–2.96 (m, 1H), 2.95–2.88 (m, 1H), 2.87–2.80 (m, 1H), 2.43 (s, 6H). 13C NMR (150 MHz, DMSO-d6) δ 154.9, 140.6, 135.9, 133.6, 131.6, 128.8, 128. 6, 127.6, 126.7, 126.0, 125.6, 125.5, 123.5, 120.8, 117.4, 63.9, 40.1, 39.9, 28.0. HR-ESI-MS m/z 348.2070 [M + H]+ (calcd. for C22H26N3O, 348.2076).
- (S)-1-(2-(dimethylamino)-3-(naphthalen-1-yl) propyl)-3-(2-methyl benzyl) urea (6h): 1H NMR (600 MHz, DMSO-d6) δ 8.08 (d, J = 8.3 Hz, 1H), 7.93 (d, J = 7.6 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.59–7.48 (m, 2H), 7.47–7.41 (m, 1H), 7.39 (br d, J = 6.8 Hz, 1H), 7.17–7.06 (m, 4H), 6.44 (t, J = 5.5 Hz, 1H), 5.83 (br s, 1H), 4.14–4.05 (m, 2H), 3.42–3.33 (m, 1H), 3.09–2.95 (m, 2H), 2.94–2.79 (m, 2H), 2.40 (s, 6H), 2.21 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 157.7, 138.4, 136.0, 135.3, 133.5, 131.6, 129.8, 128.7, 127.5, 127.2, 126.6, 126.5, 126.0, 125.6, 125.5, 125.5, 123.5, 64.1, 40.9, 40.0, 28.5, 18.5. HR-ESI-MS m/z 376.2383 [M + H]+ (calcd. for C24H30N3O, 376.2389).
- (S)-1-(2-(dimethylamino)-3-(naphthalen-1-yl) propyl)-3-(o-tolyl) urea (6i): 1H NMR (600 MHz, DMSO-d6) δ 8.08 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.83 (s, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.70 (d, J = 8.1 Hz, 1H), 7.58–7.54 (m, 1H), 7.53–7.49 (m, 1H), 7.47–7.43 (m, 1H), 7.41 (br d, J = 6.8 Hz, 1H), 7.06 (d, J = 7.4 Hz, 1H), 7.03–6.99 (m, 1H), 6.82 (t, J = 7.4 Hz, 1H), 6.53 (br d, J = 4.8 Hz, 1H), 3.42 (dd, J = 13.4, 3.5 Hz, 1H), 3.18–3.10 (m, 1H), 3.09–3.01 (m, 1H), 2.98–2.89 (m, 1H), 2.88–2.82 (m, 1H), 2.44 (s, 6H), 2.14 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 155.2, 138.3, 135.9, 133.6, 131.6, 130.0, 128.8, 127.6, 126.7, 126.6, 126.0, 125.9, 125.5, 125.5, 123.5, 121.7, 120.6, 64.0, 40.0, 39.3, 28.3, 18.0. HR-ESI-MS m/z 362.2227 [M + H]+ (calcd. for C23H28N3O, 362.2232).
- (S)-1-(2-(dimethylamino)-3-(naphthalen-1-yl) propyl)-3-(furan-2-yl methyl) urea (6j): 1H NMR (600 MHz, DMSO-d6) δ 8.07 (d, J = 8.3 Hz, 1H), 7.93 (d, J = 7.7 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.58–7.48 (m, 3H), 7.46–7.41 (m, 1H), 7.38 (br d, J = 6.8 Hz, 1H), 6.47 (t, J = 5.6 Hz, 1H), 6.33 (dd, J = 3.3, 1.8 Hz, 1H), 6.12 (m, 1H), 5.82 (br d, J = 4.3 Hz, 1H), 4.09 (d, J = 5.7 Hz, 2H), 3.35 (dd, J = 12.8, 3.1 Hz, 1H), 3.04 (ddd, J = 11.9, 7.1, 4.6 Hz, 1H), 3.01–2.93 (m, 1H), 2.91–2.79 (m, 2H), 2.38 (s, 6H). 13C NMR (150 MHz, DMSO-d6) δ 157.4, 153.7, 141.8, 136.0, 133.5, 131.6, 128.7, 127.5, 126.6, 126.0, 125.5, 125.5, 123.5, 110.3, 106.0, 64.0, 40.1, 40.0, 36.2, 28.4. HR-ESI-MS m/z 352.2020 [M + H]+ (calcd. for C21H26N3O2, 352.2025).
- (S)-1-(cyclohexylmethyl)-3-(2-(dimethylamino)-3-(naphthalen-1-yl) propyl) urea (6k): 1H NMR (600 MHz, CDCl3) δ 7.98 (d, J = 8.4 Hz, 1H), 7.83–7.78 (m, 1H), 7.68 (d, J = 8.2 Hz, 1H), 7.50–7.42 (m, 2H), 7.31–7.35 (m, 1H), 7.28–7.26 (m, 1H), 5.44 (br s, 1H), 5.19 (br s, 1H), 3.46 (dd, J = 13.6, 2.6 Hz, 1H), 3.20–3.09 (m, 1H), 3.07–2.99 (m, 2H), 2.85 (t, J = 6.3 Hz, 2H), 2.78–2.73 (m, 1H), 2.42 (s, 6H), 2.11–1.98 (m, 2H), 1.67–1.58 (m, 3H), 1.32–1.25 (m, 1H), 1.18–1.05 (m, 3H), 0.85–0.75 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 158.9, 134.7, 134.1, 131.8, 129.0, 127.8, 127.3, 126.0, 125.6, 125.6, 123.3, 64.3, 46.7, 40.5, 40.1, 38.4, 32.2, 30.8, 26.5, 25.9. HR-ESI-MS m/z 368.2696 [M + H]+ (calcd. for C23H34N3O, 368.2702).
- (S)-1-cyclohexyl-3-(2-(dimethylamino)-3-(naphthalen-1-yl) propyl) urea (6l): 1H NMR (600 MHz, CDCl3) δ 7.99 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.71 (d, J = 8.1 Hz, 1H), 7.52–7.44 (m, 2H), 7.38–7.34 (m, 1H), 7.30 (br d, J = 6.9 Hz, 1H), 5.21 (br s, 1H), 4.77 (br s, 1H), 3.51 (dd, J = 13.6, 3.1 Hz, 1H), 3.34–3.41 (m, 1H), 3.20–3.13 (m, 1H), 3.11–3.01 (m, 2H), 2.79 (dd, J = 13.6, 9.9 Hz, 1H), 2.47 (s, 6H), 1.85–1.77 (m, 2H), 1.66–1.59 (m, 2H), 1.57–1.51 (m, 1H), 1.32–1.22 (m, 2H), 1.13–1.05 (m, 1H), 1.04–0.96 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 158.0, 134.7, 134.2, 131.9, 129.2, 127.9, 127.5, 126.2, 125.7, 125.7, 123.3, 64.4, 49.0, 40.7, 40.2, 33.9, 29.1, 25.7, 25.0. HR-ESI-MS m/z 354.2542 [M + H]+ (calcd. for C22H32N3O, 354.2545).
3.5. In Vitro Pharmacology
3.5.1. μOR Gi/o-Mediated Cyclic Adenosine Monophosphate (cAMP) Inhibition Assay
Cell Culture
3.5.2. β-Arrestin Recruitment Assay
Cell Culture
3.6. In Vivo Pharmacological Experiments
3.6.1. Acetic Acid-Induced Writhing Test
3.6.2. Hot-Water Tail Withdrawal Test
3.6.3. Formalin Paw-Licking Test
3.7. Physical Dependence and Respiratory Depression Assessments
3.7.1. Physical Dependence
3.7.2. Respiratory Depression
3.8. Docking Simulations
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.J.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A. The revised International Association for the Study of Pain definition of pain: Concepts, challenges, and compromises. Pain 2020, 161, 1976–1982. [Google Scholar] [CrossRef]
- Ayad, S.; Demitrack, M.A.; Burt, D.A.; Michalsky, C.; Wase, L.; Fossler, M.J.; Khanna, A.K. Evaluating the incidence of opioid-induced respiratory depression associated with oliceridine and morphine as measured by the frequency and average cumulative duration of dosing interruption in patients treated for acute postoperative pain. Clin. Drug Investig. 2020, 40, 755–764. [Google Scholar] [CrossRef]
- Machelska, H.; Celik, M.Ö. Advances in achieving opioid analgesia without side effects. Front. Pharmacol. 2018, 9, 1388. [Google Scholar] [CrossRef]
- Montgomery, D.; Anand, J.P.; Baber, M.A.; Twarozynski, J.J.; Hartman, J.G.; Delong, L.J.; Traynor, J.R.; Mosberg, H.I. Structure–Activity Relationships of 7-Substituted Dimethyltyrosine-Tetrahydroisoquinoline Opioid Peptidomimetics. Molecules 2019, 24, 4302. [Google Scholar] [CrossRef]
- Wheeler, M.; Oderda, G.M.; Ashburn, M.A.; Lipman, A.G. Adverse events associated with postoperative opioid analgesia: A systematic review. J. Pain 2002, 3, 159–180. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, X.; Liu, C.; Kang, J.; Yang, J.; Pei, G.; Wu, C. Improvement of morphine-mediated analgesia by inhibition of β-arrestin 2 expression in mice periaqueductal gray matter. Int. J. Mol. Sci. 2009, 10, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Walker, J.K.; Bohn, L.M. Morphine side effects in β-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.-T. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves first BCMA-targeted therapeutic. Nat. Rev. Drug Discov. 2020, 19, 659–660. [Google Scholar] [CrossRef]
- Viscusi, E.R.; Skobieranda, F.; Soergel, D.G.; Cook, E.; Burt, D.A.; Singla, N. APOLLO-1: A randomized placebo and active-controlled phase III study investigating oliceridine (TRV130), a G protein-biased ligand at the µ-opioid receptor, for management of moderate-to-severe acute pain following bunionectomy. J. Pain Res. 2019, 12, 927–943. [Google Scholar] [CrossRef]
- Urits, I.; Viswanath, O.; Orhurhu, V.; Gress, K.; Charipova, K.; Kaye, A.D.; Ngo, A. The utilization of mu-opioid receptor biased agonists: Oliceridine, an opioid analgesic with reduced adverse effects. Curr. Pain Headache Rep. 2019, 23, 31. [Google Scholar] [CrossRef] [PubMed]
- Fossler, M.J.; Sadler, B.M.; Farrell, C.; Burt, D.A.; Pitsiu, M.; Skobieranda, F.; Soergel, D.G. Oliceridine (TRV130), a novel G protein–biased ligand at the μ-opioid receptor, demonstrates a predictable relationship between plasma concentrations and pain relief. I: Development of a pharmacokinetic/pharmacodynamic model. J. Clin. Pharmacol. 2018, 58, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Singla, N.; Minkowitz, H.S.; Soergel, D.G.; Burt, D.A.; Subach, R.A.; Salamea, M.Y.; Fossler, M.J.; Skobieranda, F. A randomized, Phase IIb study investigating oliceridine (TRV130), a novel µ-receptor G-protein pathway selective (μ-GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty. J. Pain Res. 2017, 10, 2413–2424. [Google Scholar] [CrossRef] [PubMed]
- Altarifi, A.A.; David, B.; Muchhala, K.H.; Blough, B.E.; Akbarali, H.; Negus, S.S. Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J. Psychopharmacol. 2017, 31, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Kingwell, K. Pioneering biased ligand offers efficacy with reduced on-target toxicity. Nat. Rev. Drug Discov. 2015, 14, 809. [Google Scholar] [CrossRef]
- DeWire, S.M.; Yamashita, D.S.; Rominger, D.H.; Liu, G.; Cowan, C.L.; Graczyk, T.M.; Chen, X.-T.; Pitis, P.M.; Gotchev, D.; Yuan, C. AG protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 2013, 344, 708–717. [Google Scholar] [CrossRef]
- Shi, R.; Chai, Y.; Feng, H.; Xie, L.; Zhang, L.; Zhong, T.; Chen, J.; Yan, P.; Zhu, B.; Zhao, J.; et al. Study of the mass balance, biotransformation and safety of [14C]SHR8554, a novel μ-opioid receptor injection, in healthy Chinese subjects. Front. Pharmacol. 2023, 14, 1231102. [Google Scholar] [CrossRef]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y.; Zuo, A.; Li, C.; Wang, W.; Jiang, W.; Zhang, X.; Che, X.; Zhang, Y.; Wu, W.; et al. Synthesis, biological, and structural explorations of a series of mu-opioid receptor (MOR) agonists with high G protein signaling bias. Eur. J. Med. Chem. 2022, 228, 113986. [Google Scholar] [CrossRef]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hubner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef]
- Kingwell, K. Screening for cleaner pain relief. Nat. Rev. Drug Discov. 2016, 15, 677. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, B.L. Designing the ideal opioid. Nature 2016, 537, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Sun, J.; Li, M.; Yu, Z.; Cheng, J.; Zhong, B.; Shi, W. Synthesis and Evaluation of Novel Biased μ-Opioid-Receptor (μOR) Agonists. Molecules 2019, 24, 259. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Ren, F.; Jia, H.; Yu, Z.; Cheng, J.; Zhang, T.; Shi, W.; Feng, X. Synthesis and biological evaluation of a peptide-remifentanil conjugate as a novel bifunctional mu/delta-opioid receptor agonist for the treatment of pain. Arab. J. Chem. 2023, 16, 105018. [Google Scholar] [CrossRef]
- Li, J.; Zhang, T.; Sun, J.; Ren, F.; Jia, H.; Yu, Z.; Cheng, J.; Shi, W. Synthesis and evaluation of peptide–fentanyl analogue conjugates as dual μ/δ-opioid receptor agonists for the treatment of pain. Chin. Chem. Lett. 2021, 33, 4107–4110. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EC50 (nM) (95% CI) | Max Effect ± SEM (%) |
|---|---|---|
| DAMGO | 2.196 (1.955–3.361) | 100.85 ± 0.56 (2 μM) |
| Morphine | 21.83 (15.07–31.61) | 100.99 ± 2.03 (10 μM) |
| PZM21 | 1.615 (1.302–2.002) | 89.75 ± 2.93 (10 μM) |
| 6a | 10.82 (4.381–26.77) | 51.86 ± 1.06 (500 nM) |
| 6b | 109.6 (68.77–174.7) | 73.91 ± 2.38 (50 μM) |
| 6c | 72.83 (30.81–130.0) | 42.67 ± 0.27 (500 nM) |
| 6d | 87.98 (65.80–117.6) | 87.85 ± 1.18 (10 μM) |
| 6e | - | −1.41 ± 5.21 (500 nM) |
| 6f | 106.6 (83.14–136.6) | 84.82 ± 1.86 (10 μM) |
| 6g | 59.49 (25.48–138.9) | 24.64 ± 0.40 (1 μM) |
| 6h | 13.12 (4.196–41.01) | 41.70 ± 0.07 (10 μM) |
| 6i | - | 7.10 ± 0.77 (500 nM) |
| 6j | 97.83 (39.61–241.61) | 27.20 ± 1.71 (10 μM) |
| 6k | - | 20.35 ± 10.61 (100 pM) |
| 6l | - | 18.09 ± 1.81 (1 nM) |
| Compound | EC50 (nM) (95% CI) | Max Effect ± SEM (%) |
|---|---|---|
| DAMGO | 100.6 (86.13–153.8) | 91.19 ± 5.50 (3 μM) |
| Morphine | 621.5 (438.7–880.4) | 21.41 ± 4.00 (3 μM) |
| PZM21 | 36.96 (14.28–95.63) | 3.09 ± 0.42 (1 μM) |
| 6a | - | 0.38 ± 0.21 (100 μM) |
| 6b | - | 1.99 ± 1.77(100 μM) |
| 6c | - | 1.06 ± 0.93 (50 μM) |
| 6d | - | 7.72 ± 2.88 (100 μM) |
| 6e | - | 2.95 ± 1.37 (100 μM) |
| 6f | - | 5.45 ± 1.75 (100 μM) |
| 6g | - | 1.12 ± 0.52 (50 μM) |
| 6h | - | 2.19 ± 1.91 (100 μM) |
| 6i | - | 1.96 ± 0.72 (100 μM) |
| 6j | - | 1.63 ± 0.32 (50 μM) |
| 6k | - | 4.15 ± 0.02 (50 μM) |
| 6l | - | 1.96 ± 0.45 (50 μM) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Yu, R.; Zhang, T.; Ren, F.; Yu, Z.; Cheng, J.; Jia, H.; Shi, W.; Zhang, Y. Synthesis and Biological Evaluation of Novel Biased Mu-Opioid Receptor Agonists. Molecules 2024, 29, 2961. https://doi.org/10.3390/molecules29132961
Guo Y, Yu R, Zhang T, Ren F, Yu Z, Cheng J, Jia H, Shi W, Zhang Y. Synthesis and Biological Evaluation of Novel Biased Mu-Opioid Receptor Agonists. Molecules. 2024; 29(13):2961. https://doi.org/10.3390/molecules29132961
Chicago/Turabian StyleGuo, Yanhao, Ruimin Yu, Tao Zhang, Fengxia Ren, Zixing Yu, Jingchao Cheng, Hongxin Jia, Weiguo Shi, and Yatong Zhang. 2024. "Synthesis and Biological Evaluation of Novel Biased Mu-Opioid Receptor Agonists" Molecules 29, no. 13: 2961. https://doi.org/10.3390/molecules29132961
APA StyleGuo, Y., Yu, R., Zhang, T., Ren, F., Yu, Z., Cheng, J., Jia, H., Shi, W., & Zhang, Y. (2024). Synthesis and Biological Evaluation of Novel Biased Mu-Opioid Receptor Agonists. Molecules, 29(13), 2961. https://doi.org/10.3390/molecules29132961