
Infrared Photodissociation Spectroscopy of Dinuclear Vanadium-Group Metal Carbonyl Complexes: Diatomic Synergistic Activation of Carbon Monoxide


Abstract
1. Introduction
2. Results
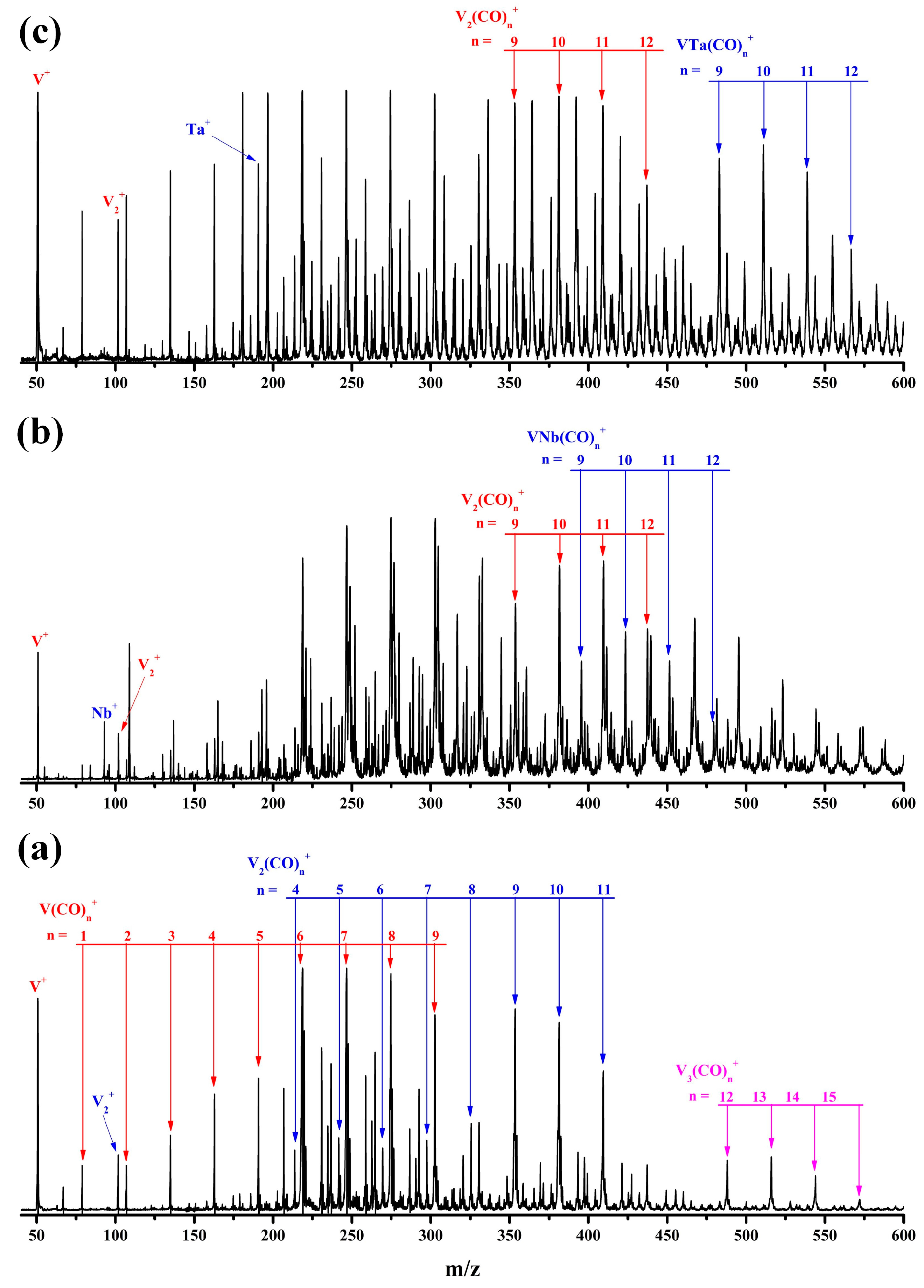
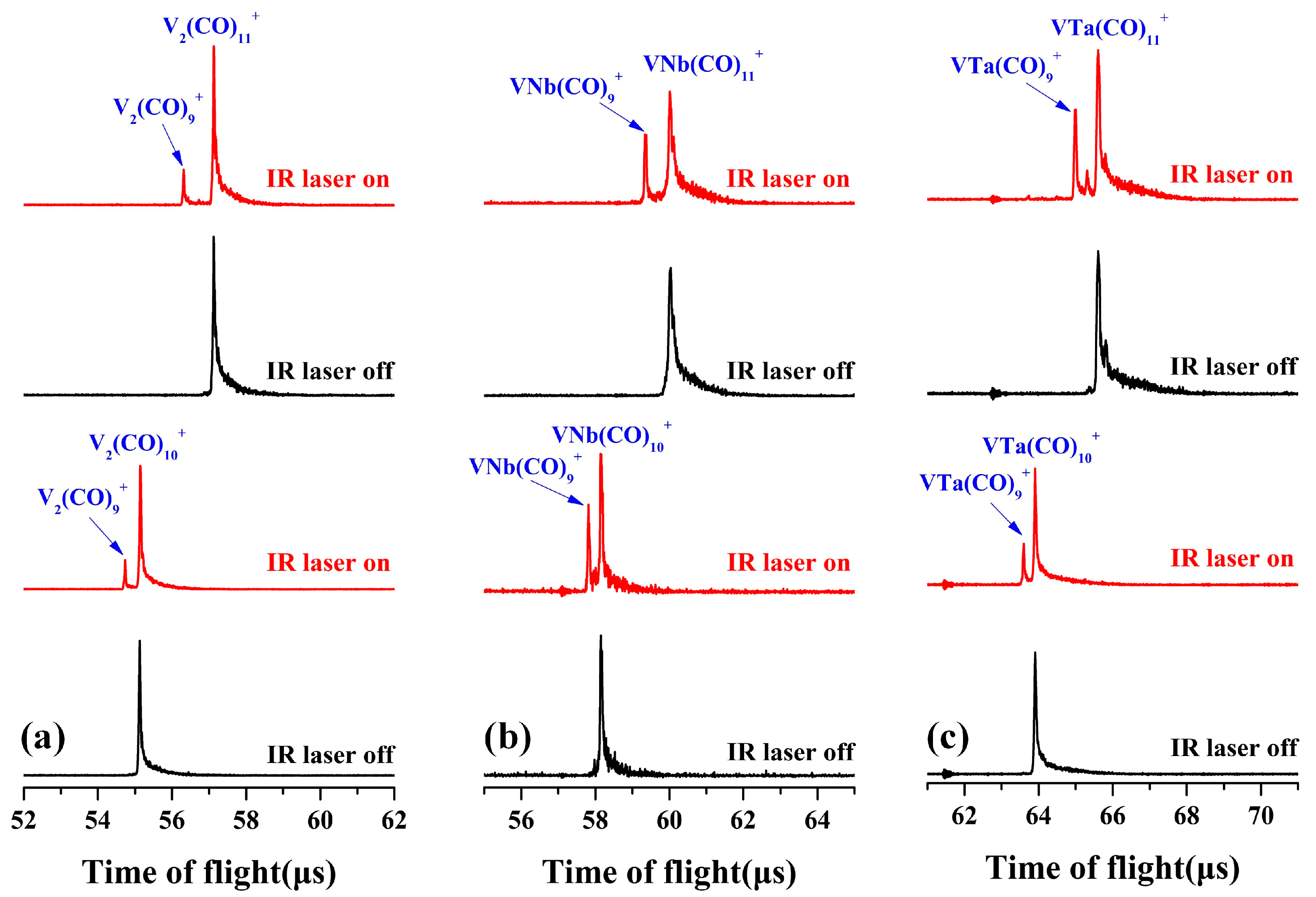
2.1. Mass Spectra
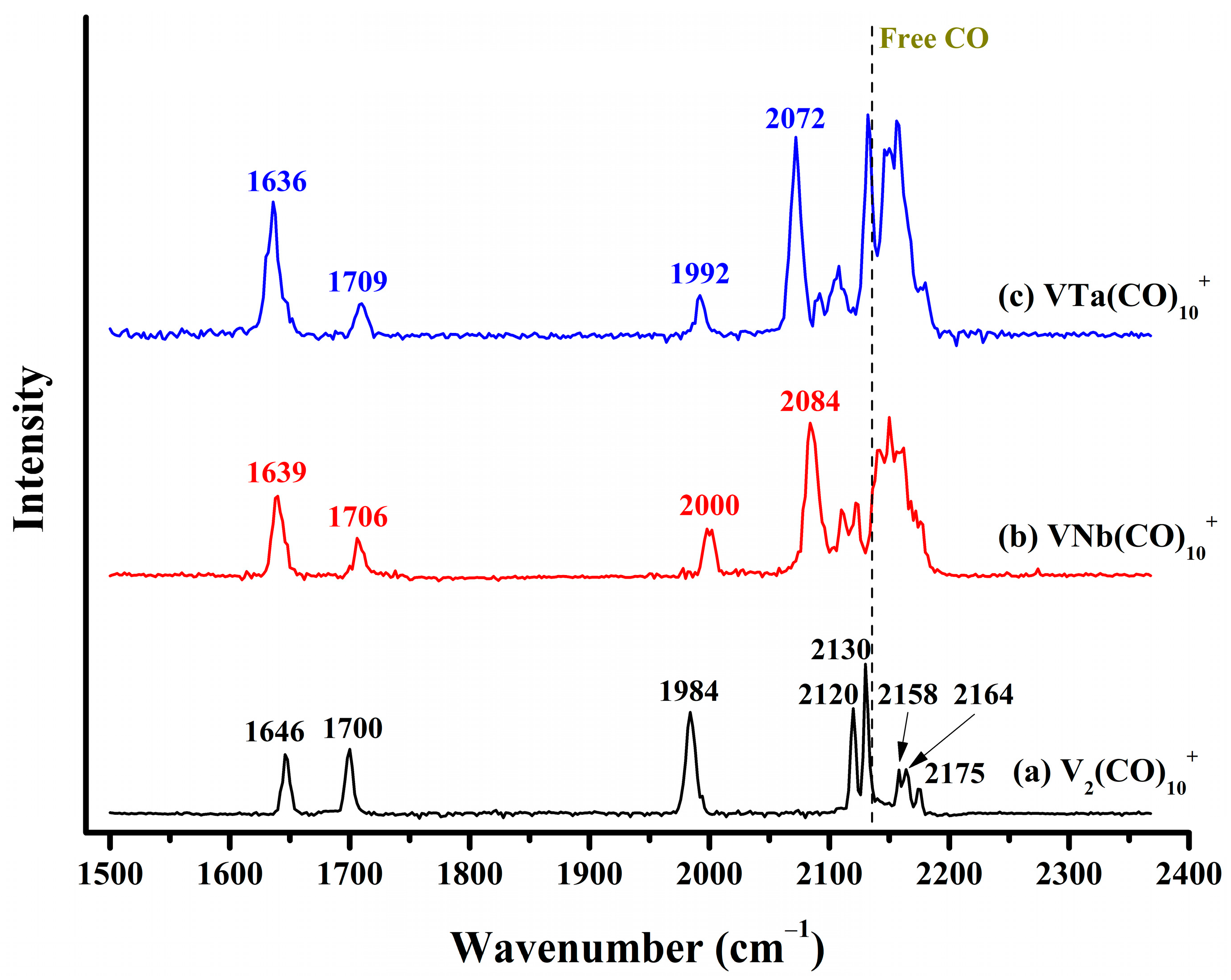
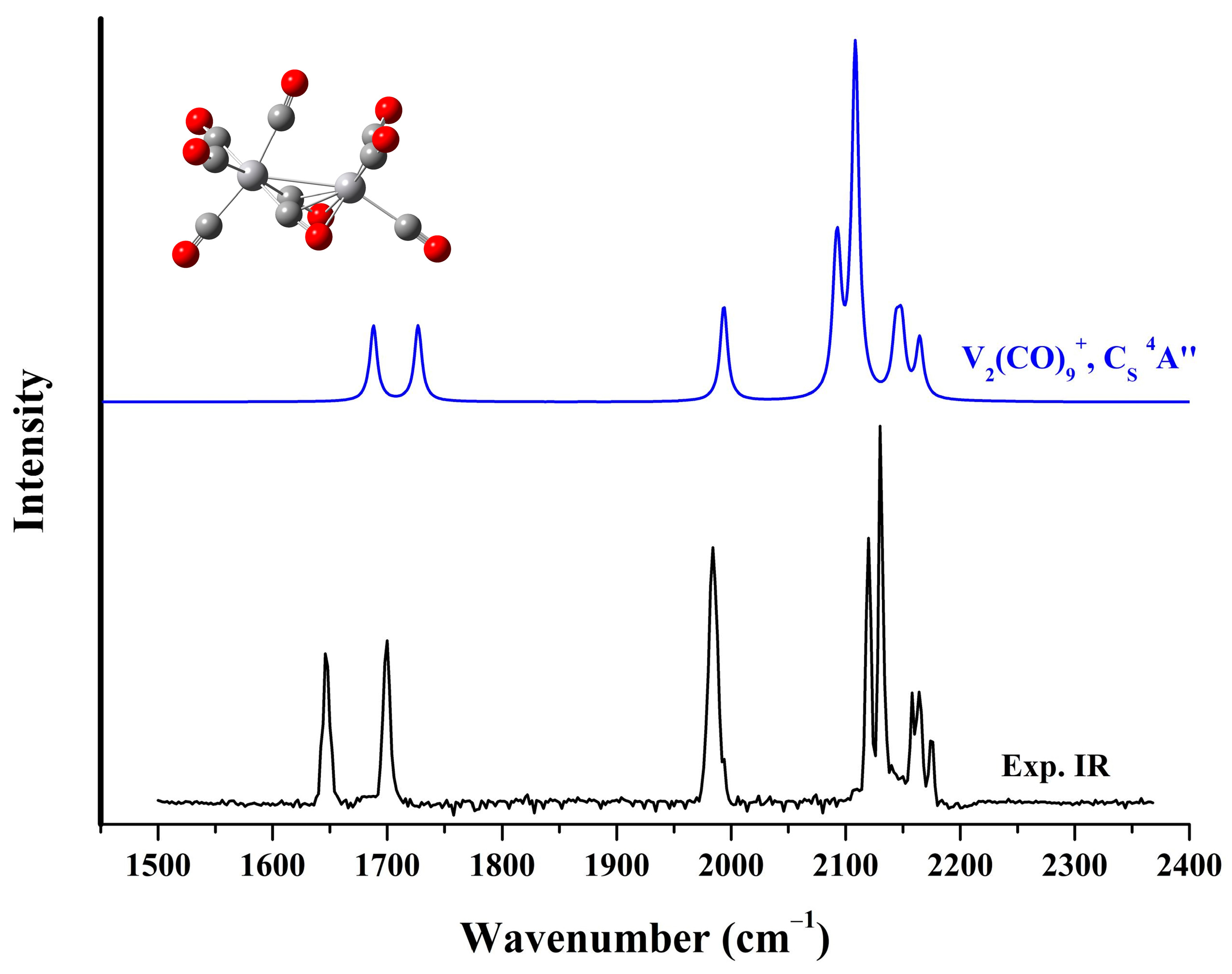
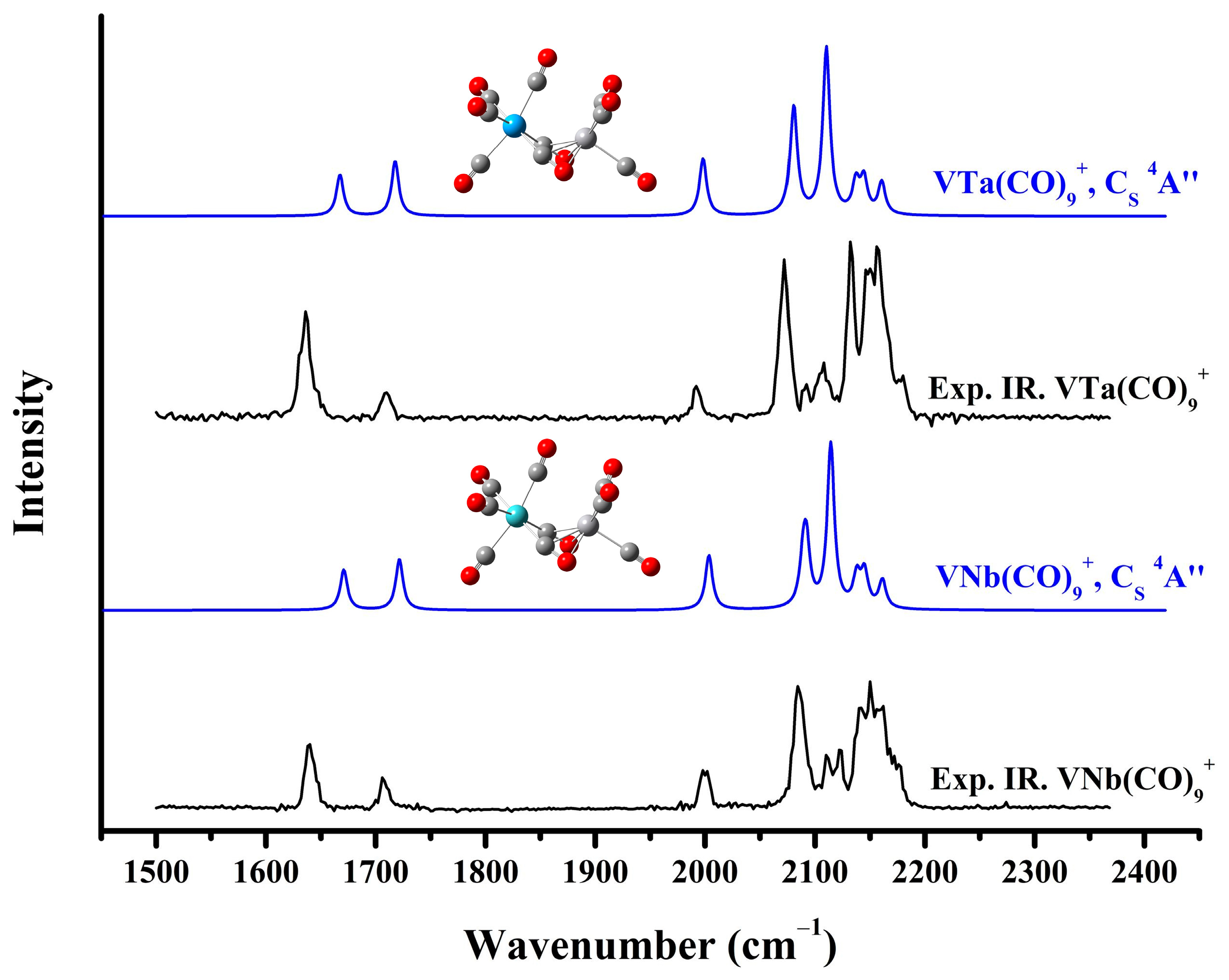
2.2. Infrared Photodissociation Spectra
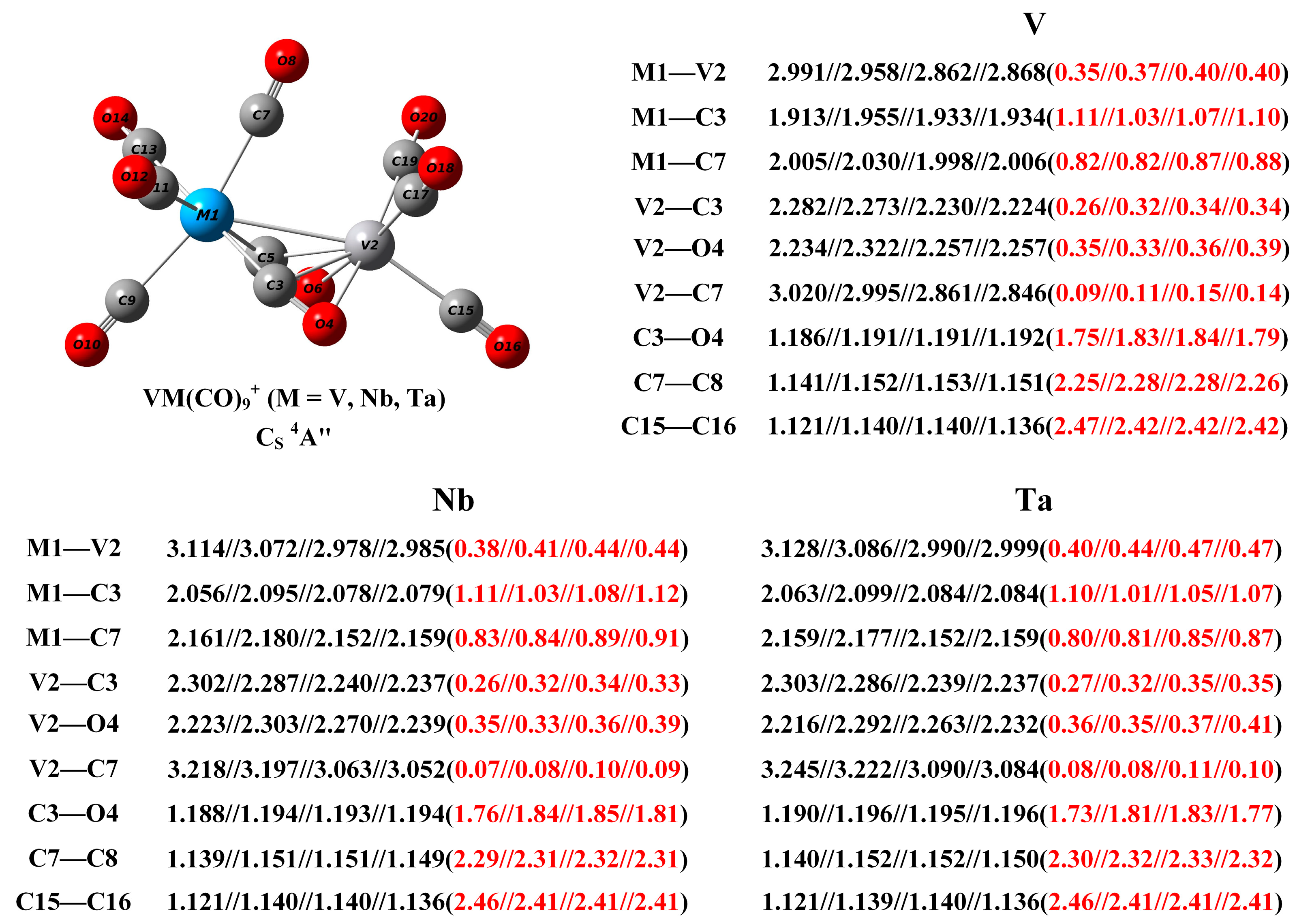
2.3. Structure of the VM(CO)9+ Cations
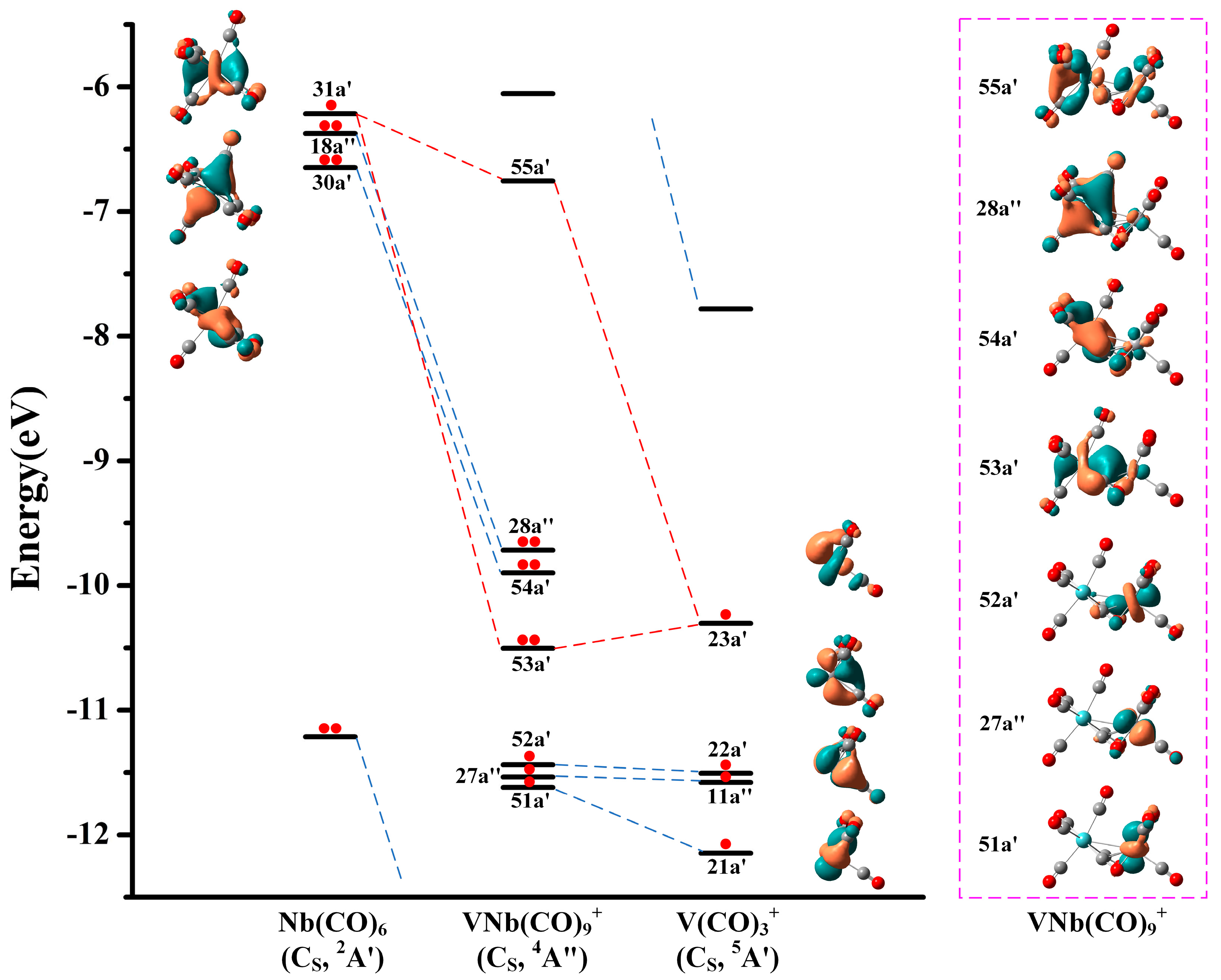
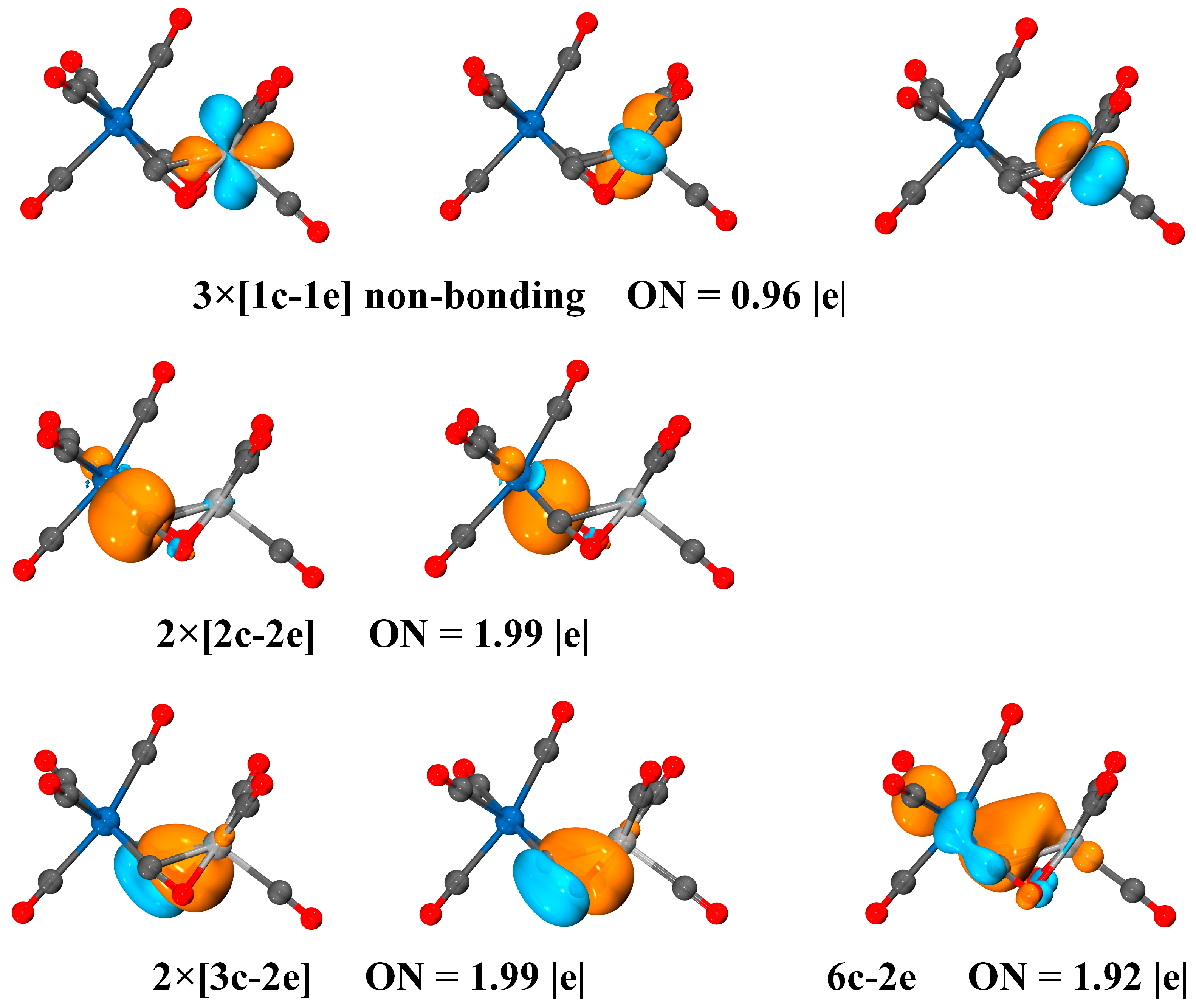
3. Discussion
4. Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fei, H.; Dong, J.; Chen, D.; Hu, T.; Duan, X.; Shakir, I.; Huang, Y.; Duan, X. Single atom electrocatalysts supported on graphene or graphene-like carbons. Chem. Soc. Rev. 2019, 48, 5207–5241. [Google Scholar] [CrossRef]
- Zhang, T.; Walsh, A.G.; Yu, J.; Zhang, P. Single-atom alloy catalysts: Structural analysis, electronic properties and catalytic activities. Chem. Soc. Rev. 2021, 50, 569–588. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Pan, H.; Mei, H.; Liu, X.; Lu, G.; Lou, C.; Li, Z.; Zhang, J. Emerging single atom catalysts in gas sensors. Chem. Soc. Rev. 2022, 51, 7260–7280. [Google Scholar] [CrossRef]
- An, Q.; Yang, C.; Xu, Y.; Yu, F.; Jiang, J.; Gong, C.; Li, B.; Zhang, J.; Liu, Q. The superiority and perspectives in single-atom site and multi-atom site catalysts for energy conversion. APL Mater. 2022, 10, 120901. [Google Scholar] [CrossRef]
- Sun, L.B.; Reddu, V.; Wang, X. Multi-atom cluster catalysts for efficient electrocatalysis. Chem. Soc. Rev. 2022, 51, 8923–8956. [Google Scholar] [CrossRef]
- Wang, Z.; Zou, G.; Park, J.H.; Zhang, K. Progress in design and preparation of multi-atom catalysts for photocatalytic CO2 reduction. Sci. China-Mater. 2024, 67, 397–423. [Google Scholar] [CrossRef]
- Ying, Y.; Luo, X.; Qiao, J.; Huang, H. “More is different”: Synergistic effect and structural engineering in double-atom catalysts. Adv. Funct. Mater. 2021, 31, 2007423. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, Q.-a.; Wang, J.; Wang, J.; Zhang, J.; Zhao, Y. Supported dual-atom catalysts: Preparation, characterization, and potential applications. Chin. J. Catal. 2020, 41, 783–798. [Google Scholar] [CrossRef]
- Pedersen, A.; Barrio, J.; Li, A.; Jervis, R.; Brett, D.J.L.; Titirici, M.M.; Stephens, I.E.L. Dual-metal atom electrocatalysts: Theory, synthesis, characterization, and applications. Adv. Energy Mater. 2022, 12, 2102715. [Google Scholar] [CrossRef]
- Ricks, A.M.; Reed, Z.E.; Duncan, M.A. Infrared spectroscopy of mass-selected metal carbonyl cations. J. Mol. Spectrosc. 2011, 266, 63–74. [Google Scholar] [CrossRef]
- Frenking, G.; Fernandez, I.; Holzmann, N.; Pan, S.; Krossing, I.; Zhou, M. Metal-CO bonding in mononuclear transition metal carbonyl complexes. JACS Au 2021, 1, 623–645. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-J.; Zhou, M.-F. Infrared spectra, structures and bonding of binuclear transition metal carbonyl cluster ions. Chin. J. Chem. Phys. 2018, 31, 1–11. [Google Scholar] [CrossRef]
- Wang, G.J.; Zhou, M.F.; Goettel, J.T.; Schrobilgen, G.J.; Su, J.; Li, J.; Schloder, T.; Riedel, S. Identification of an iridium-containing compound with a formal oxidation state of IX. Nature 2014, 514, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Wang, J.-Q.; Qu, H.; Li, W.-L.; Meng, L.; Luo, M.; Li, J.; Zhou, M. Preparation and characterization of uranium-iron triple-bonded UFe(CO)3− and OUFe(CO)3− complexes. Angew. Chem. Int. Ed. 2017, 56, 6932–6936. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhao, L.; Jin, J.; Pan, S.; Li, W.; Jin, X.; Wang, G.; Zhou, M.; Frenking, G. Observation of alkaline earth complexes M(CO)8 (M = Ca, Sr, or Ba) that mimic transition metals. Science 2018, 361, 912–916. [Google Scholar] [CrossRef]
- Chi, C.; Wang, J.-Q.; Hu, H.-S.; Zhang, Y.-Y.; Li, W.-L.; Meng, L.; Luo, M.; Zhou, M.; Li, J. Quadruple bonding between iron and boron in the BFe(CO)3− complex. Nat. Commun. 2019, 10, 4713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Q.; Chi, C.; Hu, H.-S.; Li, X.; Luo, M.; Li, J.; Zhou, M. Multiple bonding between group-3 metals and Fe(CO)3−. Angew. Chem. Int. Ed. 2020, 59, 2344–2348. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Bai, Y.; Zhou, Y.; Wang, G.; Zhao, L.; Zhou, M.; Frenking, G. Highly coordinated heteronuclear calcium-iron carbonyl cation complexes CaFe(CO)n+ (n = 5–12) with d-d bonding. Angew. Chem. Int. Ed. 2021, 60, 13865–13870. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, J.; Hu, H.-S.; Li, J.; Zhou, M. Formation and characterization of BeFe(CO)3− anion with beryllium-iron bonding. Angew. Chem. Int. Ed. 2021, 60, 9334–9338. [Google Scholar] [CrossRef]
- Wrighton, M. Photochemistry of metal carbonyls. Chem. Rev. 1974, 74, 401–430. [Google Scholar] [CrossRef]
- Aubke, F.; Wang, C. Carbon monoxide as a σ-donor ligand in coordination chemistry. Coord. Chem. Rev. 1994, 137, 483–524. [Google Scholar] [CrossRef]
- Wang, G.; Cui, J.; Chi, C.; Zhou, X.; Li, Z.H.; Xing, X.; Zhou, M. Bonding in homoleptic iron carbonyl cluster cations: A combined infrared photodissociation spectroscopic and theoretical study. Chem. Sci. 2012, 3, 3272–3279. [Google Scholar] [CrossRef]
- Chi, C.; Cui, J.; Li, Z.H.; Xing, X.; Wang, G.; Zhou, M. Infrared photodissociation spectra of mass selected homoleptic dinuclear iron carbonyl cluster anions in the gas phase. Chem. Sci. 2012, 3, 1698–1706. [Google Scholar] [CrossRef]
- Cui, J.; Wang, G.; Zhou, X.; Chi, C.; Hua Li, Z.; Liu, Z.; Zhou, M. Infrared photodissociation spectra of mass selected homoleptic nickel carbonyl cluster cations in the gas phase. Phys. Chem. Chem. Phys. 2013, 15, 10224–10232. [Google Scholar] [CrossRef]
- Cui, J.; Zhou, X.; Wang, G.; Chi, C.; Liu, Z.; Zhou, M. Infrared photodissociation spectroscopy of mass selected homoleptic copper carbonyl cluster cations in the gas phase. J. Phys. Chem. A 2013, 117, 7810–7817. [Google Scholar] [CrossRef]
- Zhang, N.; Luo, M.; Chi, C.; Wang, G.; Cui, J.; Zhou, M. Infrared photodissociation spectroscopy of mass-selected heteronuclear iron-copper carbonyl cluster anions in the gas phase. J. Phys. Chem. A 2015, 119, 4142–4150. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Kong, F.; Wang, G.; Zhou, M. Infrared photodissociation spectroscopy of heterodinuclear iron-zinc and cobalt-zinc carbonyl cation complexes. J. Phys. Chem. A 2017, 121, 1628–1633. [Google Scholar] [CrossRef]
- Qu, H.; Kong, F.; Wang, G.; Zhou, M. Infrared photodissociation spectroscopic and theoretical study of heteronuclear transition metal carbonyl cluster cations in the gas phase. J. Phys. Chem. A 2016, 120, 7287–7293. [Google Scholar] [CrossRef]
- Zhou, X.; Cui, J.; Li, Z.H.; Wang, G.; Zhou, M. Infrared photodissociation spectroscopic and theoretical study of homoleptic dinuclear chromium carbonyl cluster cations with a linear bridging carbonyl group. J. Phys. Chem. A 2012, 116, 12349–12356. [Google Scholar] [CrossRef]
- Zhou, X.; Cui, J.; Li, Z.H.; Wang, G.; Liu, Z.; Zhou, M. Carbonyl bonding on oxophilic metal centers: Infrared photodissociation spectroscopy of mononuclear and dinuclear titanium carbonyl cation complexes. J. Phys. Chem. A 2013, 117, 1514–1521. [Google Scholar] [CrossRef]
- Langeslay, R.R.; Kaphan, D.M.; Marshall, C.L.; Stair, P.C.; Sattelberger, A.P.; Delferro, M. Catalytic applications of vanadium: A mechanistic perspective. Chem. Rev. 2019, 119, 2128–2191. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhao, Y.-X.; Jiang, L.-X.; Li, H.-F.; Chen, J.-J.; Zhang, T.; Liu, Q.-Y.; He, S.-G. Thermal activation of methane by vanadium boride cluster cations VBn+ (n = 3–6). Phys. Chem. Chem. Phys. 2018, 20, 4641–4645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Y.; Liu, A.-A.; Jiao, L.; Zhang, S.; Jiang, L.-H.; Kong, X.; Pang, D.-W. Capture of small clusters by ligand-solvent interaction. J. Chem. Phys. 2023, 159, 064301. [Google Scholar] [CrossRef]
- Gan, W.; Geng, L.; Huang, B.; Hansen, K.; Luo, Z. Dehydrogenation of diborane on small Nbn+ clusters. Phys. Chem. Chem. Phys. 2024, 26, 9586–9592. [Google Scholar] [CrossRef]
- Brathwaite, A.D.; Ricks, A.M.; Duncan, M.A. Infrared photodissociation spectroscopy of vanadium oxide-carbonyl cations. J. Phys. Chem. A 2013, 117, 13435–13442. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhou, Z.; Zhuang, J.; Li, Z.H.; Fan, K.; Zhao, Y.; Zheng, X. Carbon dioxide coordination and activation by niobium oxide molecules. J. Phys. Chem. A 2011, 115, 14361–14369. [Google Scholar] [CrossRef]
- Iskra, A.; Gentleman, A.S.; Cunningham, E.M.; Mackenzie, S.R. Carbon dioxide binding to metal oxides: Infrared spectroscopy of NbO2+(CO2)n and TaO2+(CO2)n complexes. Int. J. Mass Spectrom. 2019, 435, 93–100. [Google Scholar] [CrossRef]
- Jian, X.; Xin, K.; Hu, J.; Zhang, L.; Wang, X.; Wang, G. Infrared spectroscopy of solvation in the TaO2+ hydrolysis reaction. J. Phys. Chem. A 2021, 125, 5054–5060. [Google Scholar] [CrossRef]
- Xin, K.; Chen, Y.; Zhang, L.; Xu, B.; Wang, X.; Wang, G. Infrared photodissociation spectroscopic investigation on VO+ and NbO+ hydrolysis catalyzed by water molecules. Phys. Chem. Chem. Phys. 2021, 23, 528–535. [Google Scholar] [CrossRef]
- Ricks, A.M.; Reed, Z.D.; Duncan, M.A. Seven-coordinate homoleptic metal carbonyls in the gas phase. J. Am. Chem. Soc. 2009, 131, 9176–9177. [Google Scholar] [CrossRef]
- Ricks, A.M.; Brathwaite, A.D.; Duncan, M.A. Coordination and spin states in vanadium carbonyl complexes V(CO)n+, n = 1–7 revealed with ir spectroscopy. J. Phys. Chem. A 2013, 117, 1001–1010. [Google Scholar] [CrossRef]
- Russon, L.M.; Heidecke, S.A.; Birke, M.K.; Conceicao, J.; Morse, M.D.; Armentrout, P.B. Photodissociation measurements of bond dissociation energies: Ti2+, V2+, Co2+, and Co3+. J. Chem. Phys. 1994, 100, 4747–4755. [Google Scholar] [CrossRef]
- Pyykko, P. Relativistic effects in structural chemistry. Chem. Rev. 1988, 88, 563–594. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision a.2; Gaussian Inc.: Pittsburgh, PA, USA, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens Matter. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. 3. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. General performance of density functionals. J. Phys. Chem. A 2007, 111, 10439–10452. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.J.; Truhlar, D.G. Density functional theory for transition metals and transition metal chemistry. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816. [Google Scholar] [PubMed]
- Katari, M.; Nicol, E.; Steinmetz, V.; van der Rest, G.; Carmichael, D.; Frison, G. Improved infrared spectra prediction by DFT from a new experimental database. Chem. Eur. J. 2017, 23, 8414–8423. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the 2nd and 3rd row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Schultz, N.E.; Zhao, Y.; Truhlar, D.G. Databases for transition element bonding: Metal–metal bond energies and bond lengths and their use to test hybrid, hybrid meta, and meta density functionals and generalized gradient approximations. J. Phys. Chem. A 2005, 109, 4388–4403. [Google Scholar] [CrossRef] [PubMed]
- Buehl, M.; Kabrede, H. Geometries of transition-metal complexes from density-functional theory. J. Chem. Theory Comput. 2006, 2, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Aoto, Y.A.; de Lima Batista, A.P.; Köhn, A.; de Oliveira-Filho, A.G.S. How to arrive at accurate benchmark values for transition metal compounds: Computation or experiment? J. Chem. Theory Comput. 2017, 13, 5291–5316. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the perdew–burke–ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta--generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef]
- Mayer, I. Charge, bond order and valence in the ab initio SCF theory. Chem. Phys. Lett. 1983, 97, 270–274. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Dapprich, S.; Frenking, G. Investigation of donor-acceptor interactions—A charge decomposition analysis using fragment molecular-orbitals. J. Phys. Chem. 1995, 99, 9352–9362. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Calculation of molecular orbital composition. Acta Chim. Sin. 2011, 69, 2393–2406. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Exptl. | Calc. | Mode a |
|---|---|---|---|
| V2(CO)10+ | 1646 | 1688 (501) | C–O (side-on bridge) asym-str. |
| 1700 | 1727 (503) | C–O (side-on bridge) sym-str. | |
| 1984 | 1994 (627) | C–O (end-on semi-bridge) str. | |
| 2120 | 2091 (478), 2094 (628) | C–O (end-on at left-hand V) asym-str. | |
| 2130 | 2108 (2332) | C–O (end-on at left-hand V) sym-str. | |
| 2158 | 2143 (397) | C–O (end-on at right-hand V) asym-str. | |
| 2164 | 2149 (428) | C–O (end-on at right-hand V) asym-str. | |
| 2175 | 2164 (387) | C–O (end-on at right-hand V) sym-str. | |
| VNb(CO)10+ | 1639 | 1671 (531) | C–O (side-on bridge) asym-str. |
| 1706 | 1721 (673) | C–O (side-on bridge) sym-str. | |
| 2000 | 2004 (618) | C–O (end-on semi-bridge) str. | |
| 2084, 2111, 2123, 2142, 2151, 2161, 2178 | 2089 (524), 2092 (773), 2114 (2161), 2137 (409), 2145 (458), 2162 (367) | C–O (terminal bound to V and Nb) asym-str./sym-str. | |
| VTa(CO)10+ | 1636 | 1668 (589) | C–O (side-on bridge) asym-str. |
| 1709 | 1718 (662) | C–O (side-on bridge) sym-str. | |
| 1992 | 1994 (605) | C–O (end-on semi-bridge) str. | |
| 2072, 2092, 2107, 2133, 2149, 2158, 2180 | 2084 (553), 2090 (759), 2111 (2184), 2136 (421), 2142 (441), 2159 (338) | C–O (terminal bound to V and Nb) asym-str./sym-str. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.; Wang, X. Infrared Photodissociation Spectroscopy of Dinuclear Vanadium-Group Metal Carbonyl Complexes: Diatomic Synergistic Activation of Carbon Monoxide. Molecules 2024, 29, 2831. https://doi.org/10.3390/molecules29122831
Hu J, Wang X. Infrared Photodissociation Spectroscopy of Dinuclear Vanadium-Group Metal Carbonyl Complexes: Diatomic Synergistic Activation of Carbon Monoxide. Molecules. 2024; 29(12):2831. https://doi.org/10.3390/molecules29122831
Chicago/Turabian StyleHu, Jin, and Xuefeng Wang. 2024. "Infrared Photodissociation Spectroscopy of Dinuclear Vanadium-Group Metal Carbonyl Complexes: Diatomic Synergistic Activation of Carbon Monoxide" Molecules 29, no. 12: 2831. https://doi.org/10.3390/molecules29122831
APA StyleHu, J., & Wang, X. (2024). Infrared Photodissociation Spectroscopy of Dinuclear Vanadium-Group Metal Carbonyl Complexes: Diatomic Synergistic Activation of Carbon Monoxide. Molecules, 29(12), 2831. https://doi.org/10.3390/molecules29122831