
Abietane Diterpenes from Medusantha martiusii and Their Anti-Neuroinflammatory Activity

 , , , ,
, , , ,  , ,
, ,  , , , and
, , , and
Abstract

1. Introduction
2. Results and Discussion
2.1. Structure Elucidation of the Compounds
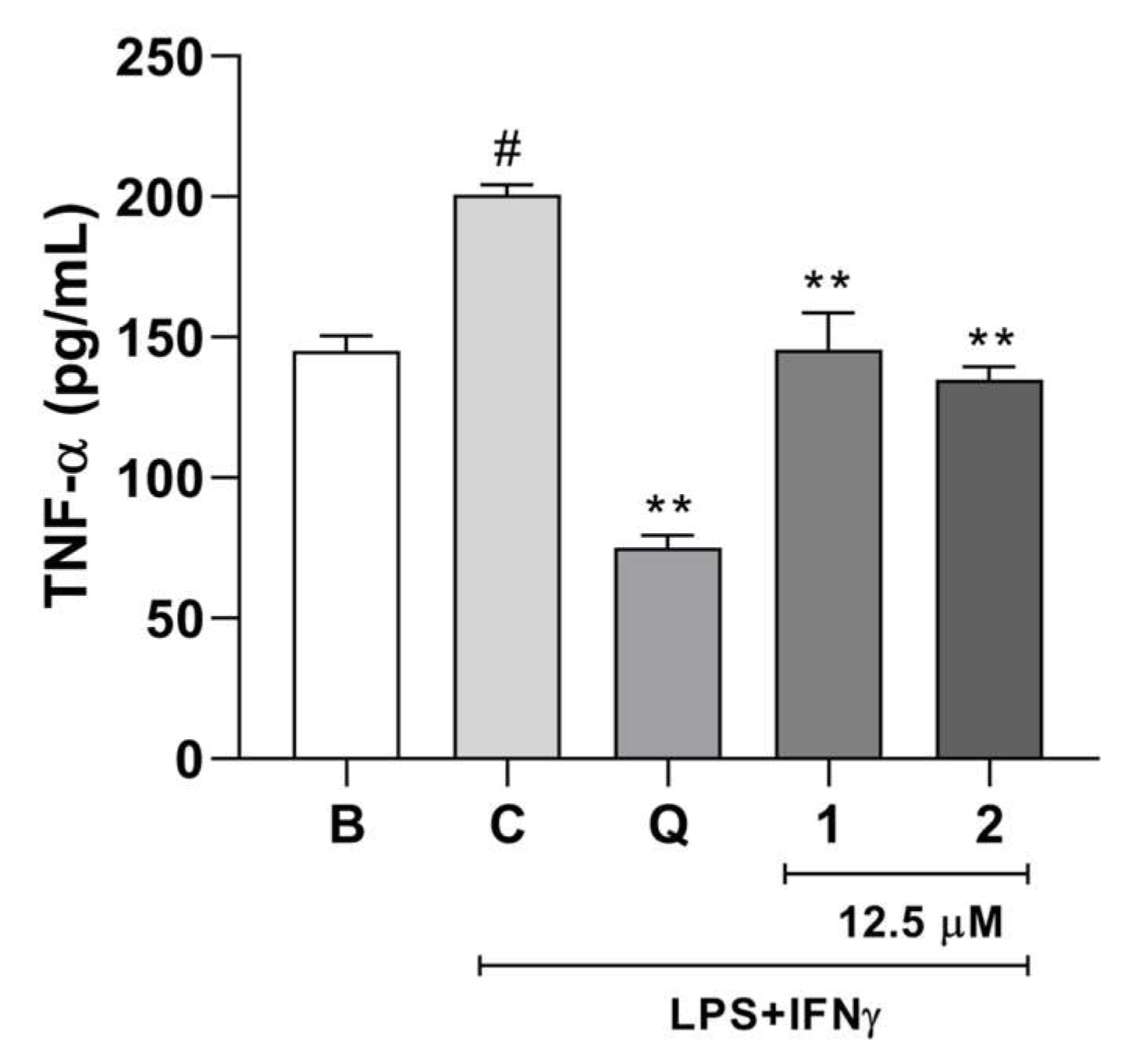
2.2. Biological Activity
Anti-Neuroinflammatory Activity
3. Experimental Section
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction, Isolation, and Purification Process
3.4. Characterization Data
3.5. NMR and ECD Calculations
3.6. Anti-Neuroinflammatory Assay
3.6.1. Cell Viability (MTT Assay)
3.6.2. Nitric Oxide (NO) and TNF-α Measurement
3.6.3. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harley, R.M.; Pastore, J.F.B. A Generic Revision and New Combinations in the Hyptidinae (Lamiaceae), Based on Molecular and Morphological Evidence. Phytotaxa 2012, 58, 1. [Google Scholar] [CrossRef]
- Monteiro, F.K.D.S.; Melo, J.I.M.D. Flora da Paraíba, Brasil: Subfamília Nepetoideae (Lamiaceae). Rodriguésia 2020, 71, e01762018. [Google Scholar] [CrossRef]
- Bornowski, N.; Hamilton, J.P.; Liao, P.; Wood, J.C.; Dudareva, N.; Buell, C.R. Genome Sequencing of Four Culinary Herbs Reveals Terpenoid Genes Underlying Chemodiversity in the Nepetoideae. DNA Res. 2020, 27, dsaa016. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Mendoza, N.; Martínez-Gordillo, M.J.; Martínez-Ambriz, E.; Basurto-Peña, F.A.; González-Trujano, M.E.; Aguirre-Hernández, E. Ethnobotanical, Phytochemical, and Pharmacological Properties of the Subfamily Nepetoideae (Lamiaceae) in Inflammatory Diseases. Plants 2023, 12, 3752. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yang, H.-Y.; Huang, P.-Z.; Zhang, L.-J.; Feng, W.-J.; Li, Y.; Gao, K. Abietane Diterpenoids with Anti-Inflammatory Activities from Callicarpa Bodinieri. Phytochemistry 2023, 214, 113825. [Google Scholar] [CrossRef] [PubMed]
- Kolsi, L.E.; Leal, A.S.; Yli-Kauhaluoma, J.; Liby, K.T.; Moreira, V.M. Dehydroabietic Oximes Halt Pancreatic Cancer Cell Growth in the G1 Phase through Induction of P27 and Downregulation of Cyclin D1. Sci. Rep. 2018, 8, 15923. [Google Scholar] [CrossRef] [PubMed]
- Abdissa, N.; Frese, M.; Sewald, N. Antimicrobial Abietane-Type Diterpenoids from Plectranthus punctatus. Molecules 2017, 22, 1919. [Google Scholar] [CrossRef] [PubMed]
- Tabefam, M.; Farimani, M.M.; Danton, O.; Ramseyer, J.; Kaiser, M.; Ebrahimi, S.N.; Salehi, P.; Batooli, H.; Potterat, O.; Hamburger, M. Antiprotozoal Diterpenes from Perovskia abrotanoides. Planta Med. 2018, 84, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Agra, M.D.F.; Silva, K.N.; Basílio, I.J.L.D.; Freitas, P.F.D.; Barbosa-Filho, J.M. Survey of Medicinal Plants Used in the Region Northeast of Brazil. Rev. Bras. Farmacogn. 2008, 18, 472–508. [Google Scholar] [CrossRef]
- Ranzato Filardi, F.L.; Barros, F.D.; Baumgratz, J.F.A.; Bicudo, C.E.M.; Cavalcanti, T.B.; Nadruz Coelho, M.A.; Costa, A.; Costa, D.; Goldenburg, R.; Labiak, P.H.; et al. BFG Brazilian Flora 2020: Innovation and Collaboration to Meet Target 1 of the Global Strategy for Plant Conservation (GSPC). Rodriguésia 2018, 69, 1513–1527. [Google Scholar] [CrossRef]
- Araújo, E.C.C.; Lima, M.A.S.; Silveira, E.R. Spectral Assignments of New Diterpenes from Hyptis martiusii Benth. Magn. Reson. Chem. 2004, 42, 1049–1052. [Google Scholar] [CrossRef]
- Araújo, E.C.C.; Lima, M.A.S.; Montenegro, R.C.; Nogueira, M.A.S.; Costa-Lotufo, L.V.; Pessoa, C.; Moraes, M.O.; Silveira, E.R. Cytotoxic Abietane Diterpenes from Hyptis martiusii Benth. Z. Für Naturforschung C 2006, 61, 177–183. [Google Scholar] [CrossRef]
- Cavalcanti, B.C.; Moura, D.J.; Rosa, R.M.; Moraes, M.O.; Araújo, E.C.C.; Lima, M.A.S.; Silveira, E.R.; Saffi, J.; Henriques, J.A.P.; Pessoa, C.; et al. Genotoxic Effects of Tanshinones from Hyptis martiusii in V79 Cell Line. Food Chem. Toxicol. 2008, 46, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.G.R.; Tintino, C.D.M.O.; Pessoa, R.T.; de Lacerda Neto, L.J.; Martins, A.O.B.P.B.; de Oliveira, M.R.C.; Coutinho, H.D.M.; Cruz-Martins, N.; Quintans, L.J., Jr.; Wilairatana, P.; et al. Anti-Inflammatory and Antinociceptive Effect of Hyptis martiusii BENTH Leaves Essential Oil. Biotechnol. Rep. 2022, 35, e00756. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.C.; Lichter, R.L.; Nelson, G.L. Carbon-13 Nuclear Magnetic Resonance Spectroscopy, 2nd ed.; Wiley & Sons: New York, NY, USA, 1980. [Google Scholar]
- Han, D.; Li, W.; Hou, Z.; Lin, C.; Xie, Y.; Zhou, X.; Gao, Y.; Huang, J.; Lai, J.; Wang, L.; et al. The Chromosome-Scale Assembly of the Salvia rosmarinus Genome Provides Insight into Carnosic Acid Biosynthesis. Plant J. 2023, 113, 819–832. [Google Scholar] [CrossRef]
- Lima, K.S.B.D.; Pimenta, A.T.A.; Guedes, M.L.S.; Lima, M.A.S.; Silveira, E.R. Abietane Diterpenes from Hyptis carvalhoi Harley. Biochem. Syst. Ecol. 2012, 44, 240–242. [Google Scholar] [CrossRef]
- Costa-Lotufo, L.V.; Araújo, E.C.C.; Lima, M.A.S.; Moraes, M.E.A.; Pessoa, C.; Silviera, E.R.; Morais, M.O. Antiproliferative Effects of Abietane Diterpenoids Isolated from Hyptis martiusii Benth (Labiatae). Pharmazie 2004, 59, 78–79. [Google Scholar] [PubMed]
- González, A.G.; Herrera, J.R.; Luis, J.G.; Ravelo, A.G.; Ferro, E.A. Terpenes and Flavones of Salvia cardiophylla. Phytochemistry 1988, 27, 1540–1541. [Google Scholar] [CrossRef]
- Zhao, H.; Li, H.; Huang, G.; Chen, Y. A New Abietane Mono-Norditerpenoid from Podocarpus nagi. Nat. Prod. Res. 2017, 31, 844–848. [Google Scholar] [CrossRef]
- Hirata, A.; Kim, S.-Y.; Kobayakawa, N.; Tanaka, N.; Kashiwada, Y. Miltiorins A–D, Diterpenes from Radix Salviae miltiorrhizae. Fitoterapia 2015, 102, 49–55. [Google Scholar] [CrossRef]
- Lin, S.; Zhang, Y.; Liu, M.; Yang, S.; Gan, M.; Zi, J.; Song, W.; Fan, X.; Wang, S.; Liu, Y.; et al. Abietane and C20-Norabietane Diterpenes from the Stem Bark of Fraxinus sieboldiana and Their Biological Activities. J. Nat. Prod. 2010, 73, 1914–1921. [Google Scholar] [CrossRef]
- Zheng, T.-L.; Liu, S.-Z.; Huo, C.-Y.; Li, J.; Wang, B.-W.; Jin, D.-P.; Cheng, F.; Chen, X.-M.; Zhang, X.-M.; Xu, X.-T.; et al. Au-Catalyzed 1,3-Acyloxy Migration/Cyclization Cascade: A Direct Strategy toward the Synthesis of Functionalized Abietane-Type Diterpenes. CCS Chem. 2021, 3, 2795–2802. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of Neuroinflammation in Neurodegeneration Development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Oliveira, M.R. The Dietary Components Carnosic Acid and Carnosol as Neuroprotective Agents: A Mechanistic View. Mol. Neurobiol. 2016, 53, 6155–6168. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.R.; Souza, I.C.C.; Fürstenau, C.R. Carnosic Acid Induces Anti-Inflammatory Effects in Paraquat-Treated SH-SY5Y Cells Through a Mechanism Involving a Crosstalk Between the Nrf2/HO-1 Axis and NF-κB. Mol. Neurobiol. 2018, 55, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Justo, A.F.O.; Suemoto, C.K. The Modulation of Neuroinflammation by Inducible Nitric Oxide Synthase. J. Cell Commun. Signal. 2022, 16, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Konsman, J.P. Cytokines in the Brain and Neuroinflammation: We Didn’t Starve the Fire! Pharmaceuticals 2022, 15, 140. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H.; Wang, J.; Wang, Q.; Ma, Q.; Chen, Y. Protocatechuic Acid Inhibits Inflammatory Responses in LPS-Stimulated BV2 Microglia via NF-κB and MAPKs Signaling Pathways. Neurochem. Res. 2015, 40, 1655–1660. [Google Scholar] [CrossRef]
- ChemSketch, Version 2022.1.2; Advanced Chemistry Development, Inc. (ACD/Labs): Toronto, ON, Canada, 2022. Available online: www.acdlabs.com (accessed on 19 October 2023).
- Spartan’ 10, Version 1.1.0; Wavefunction Inc.: Irvine, CA, USA, 2011.
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An Improved Probability for the Stereochemical Assignment of Isomeric Compounds Using Quantum Chemical Calculations of NMR Shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef]
- Origin(Pro), Version 2023; OriginLab Corporation: Northampton, MA, USA, 2023.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Griess, P. Bemerkungen Zu Der Abhandlung Der HH. Weselsky Und Benedikt “Ueber Einige Azoverbindungen”. Berichte Dtsch. Chem. Ges. 1879, 12, 426–428. [Google Scholar] [CrossRef]
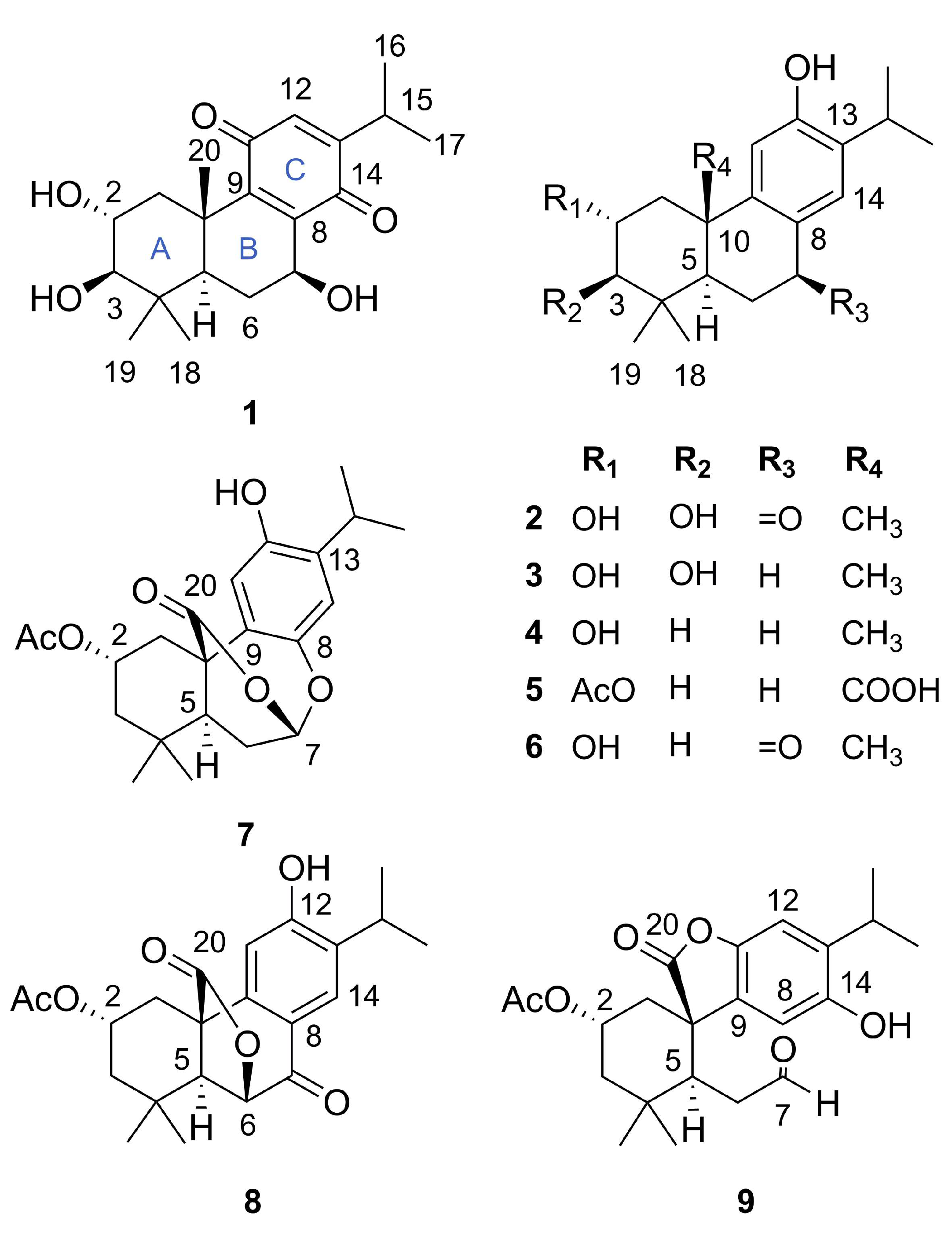
 ) and HMBC (
) and HMBC ( ) of compounds 1–3, 5, and 7–9.
) of compounds 1–3, 5, and 7–9.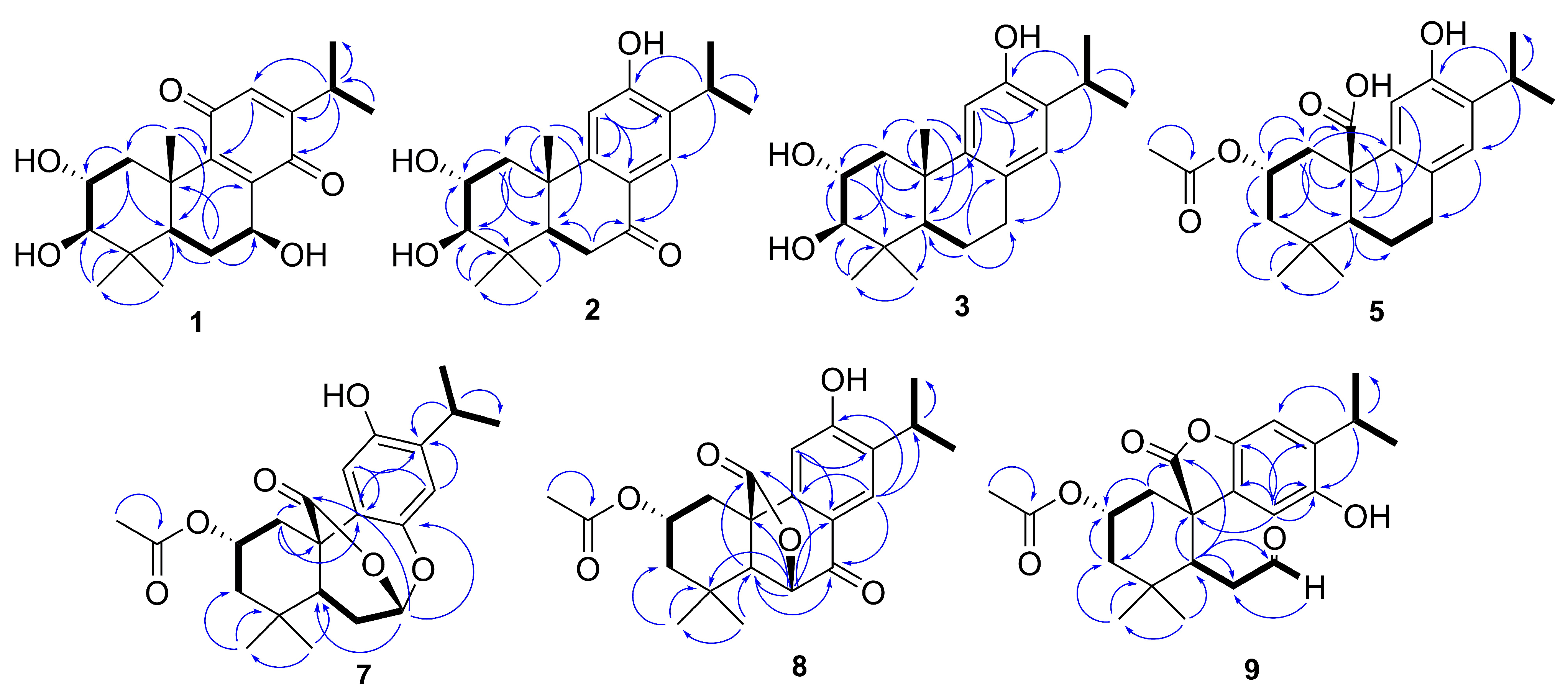
 ) of compounds 1, 2, 5, and 7.
) of compounds 1, 2, 5, and 7.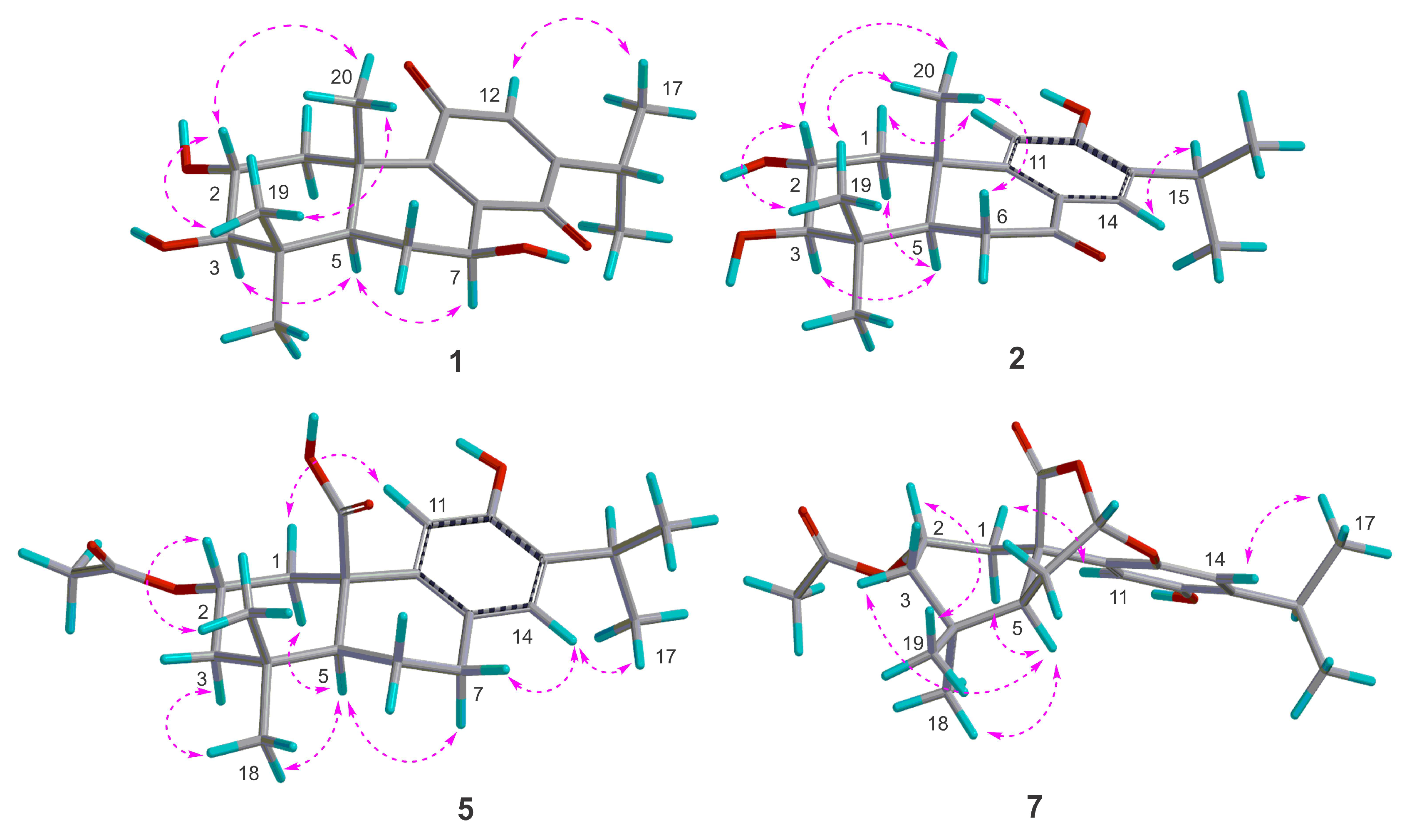



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 b | 3 b | 5 b | 7 a | 8 a | 9 a |
|---|---|---|---|---|---|---|---|
| 1 | 42.3 | 45.6 | 46.6 | 43.0 | 36.8 | 32.0 | 40.0 |
| 2 | 68.8 | 69.4 | 70.0 | 70.8 | 67.9 | 67.3 | 66.4 |
| 3 | 82.8 | 83.5 | 84.3 | 47.7 | 45.3 | 43.6 | 46.1 |
| 4 | 38.9 | 40.5 | 40.5 | 35.8 | 36.3 | 34.0 | 36.3 |
| 5 | 48.3 | 50.3 | 51.5 | 53.2 | 50.6 | 59.5 | 45.7 |
| 6 | 26.1 | 36.6 | 20.4 | 19.6 | 26.7 | 81.4 | 41.8 |
| 7 | 67.9 | 200.6 | 31.1 | 30.5 | 95.4 | 189.1 | 199.5 |
| 8 | 141.9 | 123.7 | 126.5 | 128.7 | 145.8 | 121.8 | 110.2 |
| 9 | 150.6 | 157.2 | 148.0 | 139.1 | 121.7 | 143.8 | 128.3 |
| 10 | 40.0 | 39.9 | 39.5 | 49.5 | 47.3 | 49.2 | 53.1 |
| 11 | 188.0 | 110.6 | 111.6 | 112.4 | 112.3 | 111.0 | 146.6 |
| 12 | 132.4 | 162.6 | 153.4 | 153.5 | 148.1 | 159.1 | 108.6 |
| 13 | 153.6 | 135.2 | 133.8 | 135.3 | 136.7 | 136.1 | 137.1 |
| 14 | 190.1 | 127.3 | 127.4 | 128.2 | 117.3 | 128.2 | 150.2 |
| 15 | 26.4 | 27.9 | 27.7 | 27.9 | 26.8 | 27.0 | 27.4 |
| 16 | 21.5 | 22.8 | 23.3 | 23.1 | 22.3 | 22.3 | 22.5 |
| 17 | 21.4 | 22.9 | 23.2 | 23.2 | 22.2 | 22.4 | 22.8 |
| 18 | 28.8 | 28.6 | 29.4 | 32.6 | 30.8 | 31.4 | 33.3 |
| 19 | 17.0 | 16.9 | 17.4 | 21.5 | 20.8 | 22.7 | 22.0 |
| 20 | 21.1 | 24.6 | 26.3 | 179.0 | 170.9 | 176.0 | 177.9 |
| 2-OCOCH3 | - | - | - | 21.4 | 21.3 | 21.4 | 21.4 |
| 2-OCOCH3 | - | - | - | 172.6 | 169.9 | 170.0 | 170.3 |
| No. | 1 a | 2 b | 3 b | 5 b | 7 c | 8 c | 9 c |
|---|---|---|---|---|---|---|---|
| 1 | 1.15, m 3.06, dd (12.7, 4.4) | 1.56, t (11.9) 2.55, dd (12.5, 4.3) | 1.40, t (12.0) 2.49, dd (12.4, 4.4) | 1.27, overlap 3.09, ddd (12.1, 4.5, 2.8) | 1.85, t (11.7) 2.77 ddd (11.8, 3.7, 2.4) | 1.76, t (12.0) 3.01, dd (12.6, 4.3) | 1.71, dd (13.2, 11.7) 2.23, ddd (13.3, 4.0, 2.5) |
| 2 | 3.82, ddd (11.5, 9.6, 4.4) | 3.83, ddd (11.7, 9.6, 4.3) | 3.78, ddd (11.6, 9.6, 4.4) | 5.42, tt (11.7, 4.5) | 5.22, tt (11.5, 3.9) | 5.04, tt (11.6, 4.1) | 5.50, tt (11.7, 3.9) |
| 3 | 3.01, d (9.6) | 3.02, d (9.6) | 2.99, d (9.6) | 1.86, overlap 1.27, overlap | 1.14–1.23, m 1.97, ddd (12.4, 4.1, 2.4) | 1.88, m 1.24, overlap | 2.03, ddd (12.7, 4.0, 2.5) 1.49, t (12.3) |
| 5 | 1.15, m | 1.89, dd (13.4, 4.2) | 1.32, dd (12.4, 2.4) | 1.50, dd (12.8, 2.4) | 2.15, dd (10.5, 2.0) | 2.41, s | 2.34, dd (6.1, 4.6) |
| 6 | 2.20, m 1.64, m | 2.64, m 2.64, m | 1.84, m 1.71, m | 2.54, m 1.86, overlap | 2.24 ddd (15.9, 6.5, 2.0) 2.35 dd (15.9, 10.5) | 4.77, s | 2.46, ddd (17.8, 4.6, 1.5) 1.94, ddd (17.8, 6.1, 1.5) |
| 7 | 4.79, dd (10.2, 7.5) | - | 2.71, m 2.82, m | 2.87, m 2.76, m | 5.94 d (6.5) | - | 9.19, t (1.5) |
| 8 | - | - | - | - | - | - | 6.57, s |
| 11 | - | 6.76, s | 6.65, s | 6.68, s | 6.69, s | 6.71, s | - |
| 12 | 6.36, d (1.2) | - | - | - | - | - | 6.86, s |
| 14 | - | 7.80, s | 6.75, s | 6.85, s | 6.70, s | 7.91, s | - |
| 15 | 2.97, m | 3.22, sept (6.8) | 3.16, sept (6.8) | 3.18, sept (6.8) | 3.08, sept (6.8) | 3.16, sept (6.8) | 3.16, sept (6.9) |
| 16 | 1.10, s | 1.19, d (6.8) | 1.16, d (6.8) | 1.17, d (6.8) | 1.18, d (6.8) | 1.23, s | 1.20, d (6.9) |
| 17 | 1.08, s | 1.21, d (6.8) | 1.15, d (6.8) | 1.17, d (6.8) | 1.19, d (6.8) | 1.24, s | 1.18, d (6.9) |
| 18 | 1.07, s | 1.06, s | 1.07, s | 1.02, s | 0.86, s | 1.07, s | 0.94, s |
| 19 | 0.91, s | 0.98, s | 0.89, s | 0.93, s | 0.96, s | 1.05, s | 1.23, s |
| 20 | 1.40, s | 1.27, s | 1.19, s | - | - | - | - |
| 2-OCOCH3 | - | - | - | 2.02, s | 2.05, s | 2.06, s | 1.99, s |
| Compound | Cell Viability (%) | ||
|---|---|---|---|
| 12.5 µM | 25 µM | 50 µM | |
| 1 | 87.95 ± 2.57 | 85.09 ± 1.54 | 76.28 ± 2.16 |
| 2 | 81.93 ± 0.65 | 85.84 ± 0.85 | 74.95 ± 3.11 |
| 3 | 87.39 ± 2.35 | 82.19 ± 0.96 | 76.37 ± 3.14 |
| 4 | 92.29 ± 0.60 | 85.41 ± 0.82 | 73.61 ± 2.73 |
| 5 | 86.18 ± 4.53 | 86.80 ± 2.52 | 82.90 ± 3.63 |
| 6 | 85.71 ± 1.11 | 81.71 ± 1.37 | 66.81 ± 3.61 |
| 7 | 82.74 ± 0.62 | 84.91 ± 1.40 | 81.83 ± 1.24 |
| Compound | IC50 (µM) 2 |
|---|---|
| 1 | 3.12 ± 0.75 |
| 2 | 15.53 ± 7.56 |
| Quercetin 1 | 11.8 ± 1.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Assis, E.B.d.; Andrade, R.S.d.; Silva, J.P.R.e.; Martorano, L.H.; Amorim, G.M.W.; Loureiro, P.B.A.; Abreu, L.S.; Sobral, M.V.; Scotti, M.T.; Santos Junior, F.M.d.; et al. Abietane Diterpenes from Medusantha martiusii and Their Anti-Neuroinflammatory Activity. Molecules 2024, 29, 2723. https://doi.org/10.3390/molecules29122723
Assis EBd, Andrade RSd, Silva JPRe, Martorano LH, Amorim GMW, Loureiro PBA, Abreu LS, Sobral MV, Scotti MT, Santos Junior FMd, et al. Abietane Diterpenes from Medusantha martiusii and Their Anti-Neuroinflammatory Activity. Molecules. 2024; 29(12):2723. https://doi.org/10.3390/molecules29122723
Chicago/Turabian StyleAssis, Edileuza B. de, Rodrigo S. de Andrade, Joanda P. R. e Silva, Lucas H. Martorano, Geraldo M. W. Amorim, Paulo B. A. Loureiro, Lucas S. Abreu, Marianna V. Sobral, Marcus T. Scotti, Fernando M. dos Santos Junior, and et al. 2024. "Abietane Diterpenes from Medusantha martiusii and Their Anti-Neuroinflammatory Activity" Molecules 29, no. 12: 2723. https://doi.org/10.3390/molecules29122723
APA StyleAssis, E. B. d., Andrade, R. S. d., Silva, J. P. R. e., Martorano, L. H., Amorim, G. M. W., Loureiro, P. B. A., Abreu, L. S., Sobral, M. V., Scotti, M. T., Santos Junior, F. M. d., Agra, M. d. F., Tavares, J. F., & Silva, M. S. d. (2024). Abietane Diterpenes from Medusantha martiusii and Their Anti-Neuroinflammatory Activity. Molecules, 29(12), 2723. https://doi.org/10.3390/molecules29122723