
Solvothermal Synthesis of Rare Earth Bisphthalocyanines

 ,
,  , and
, and
Abstract

1. Introduction
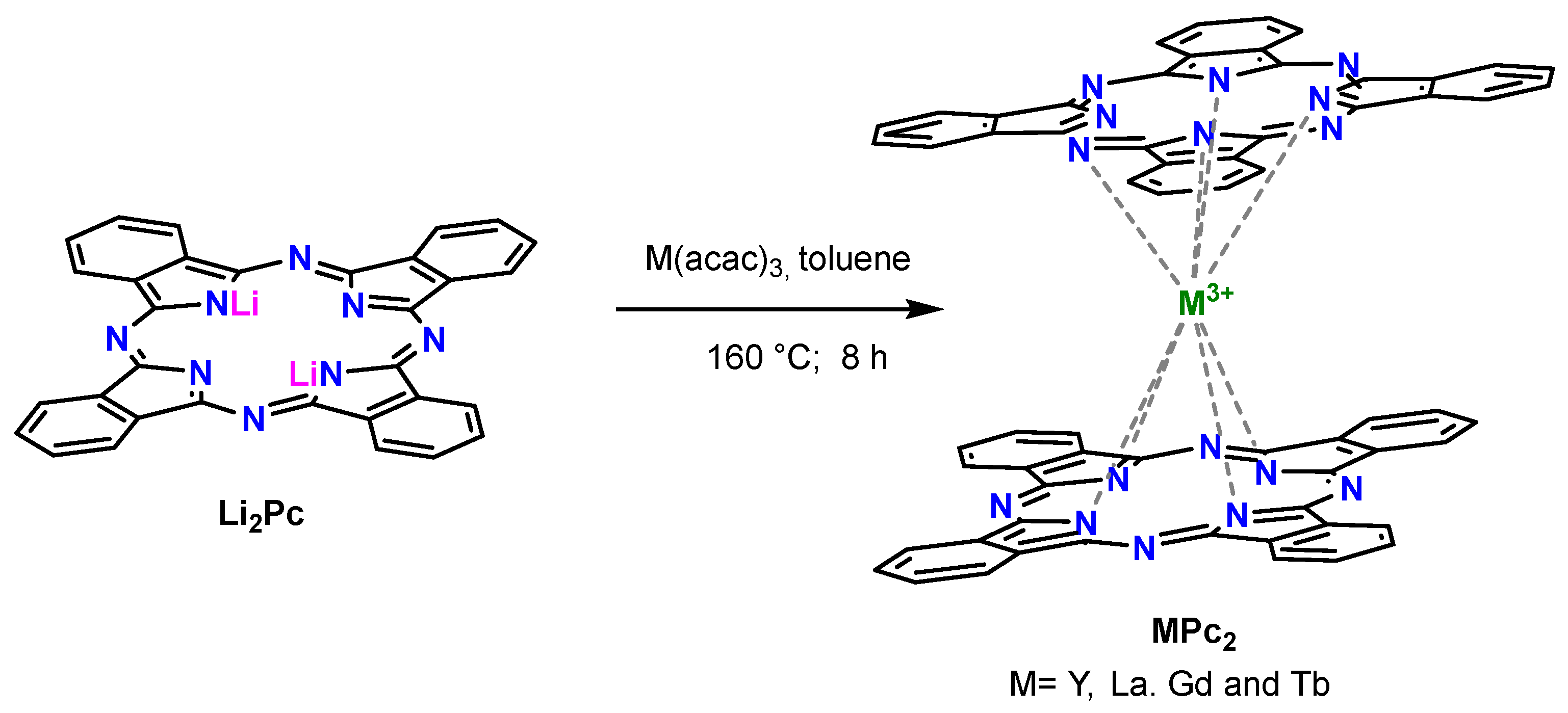
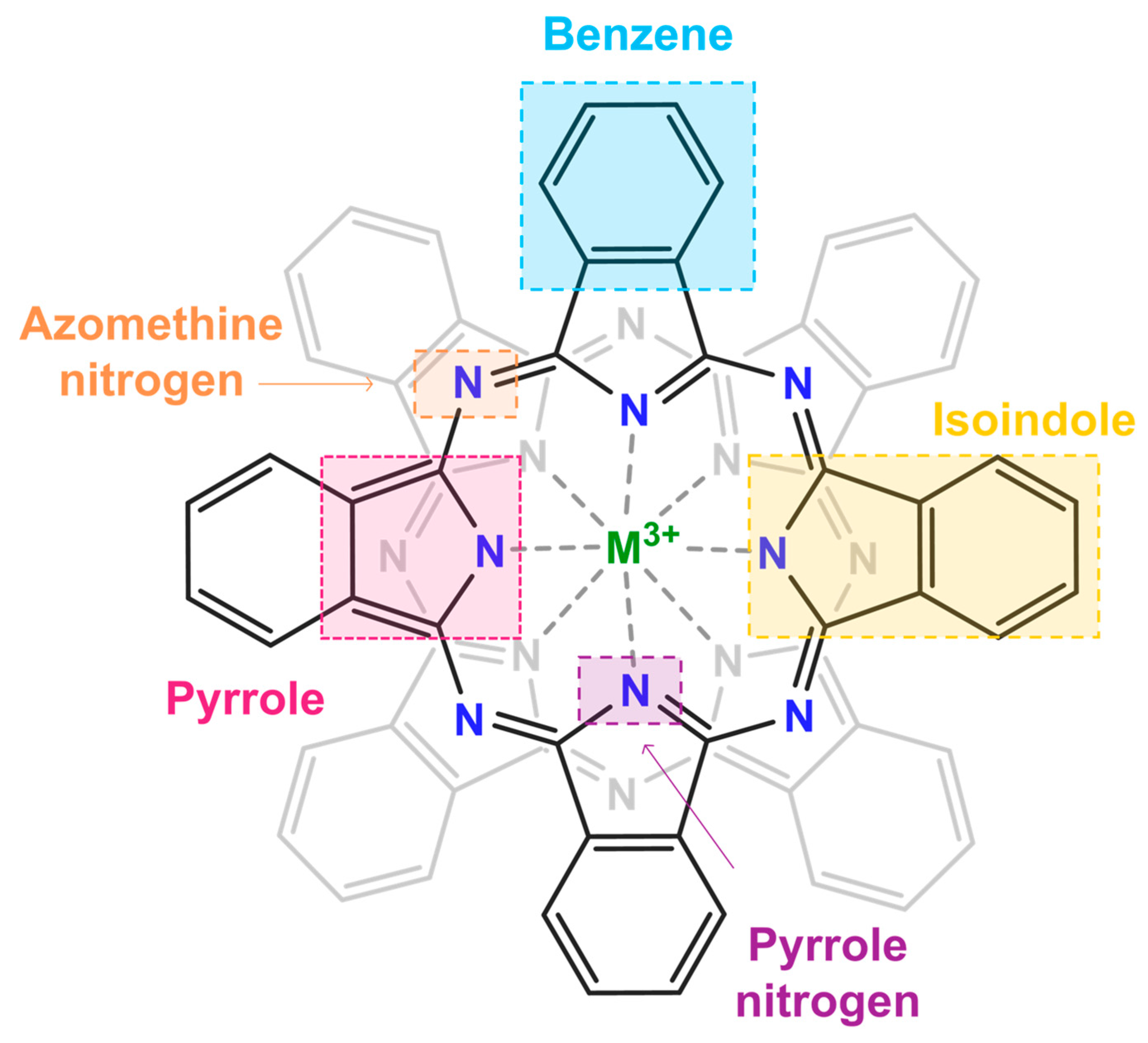
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Synthesis
3.3. Characterization
3.3.1. Infrared Spectroscopy
3.3.2. Raman Spectroscopy
3.3.3. Elemental Analysis
3.3.4. X ray Photoelectron Spectroscopy
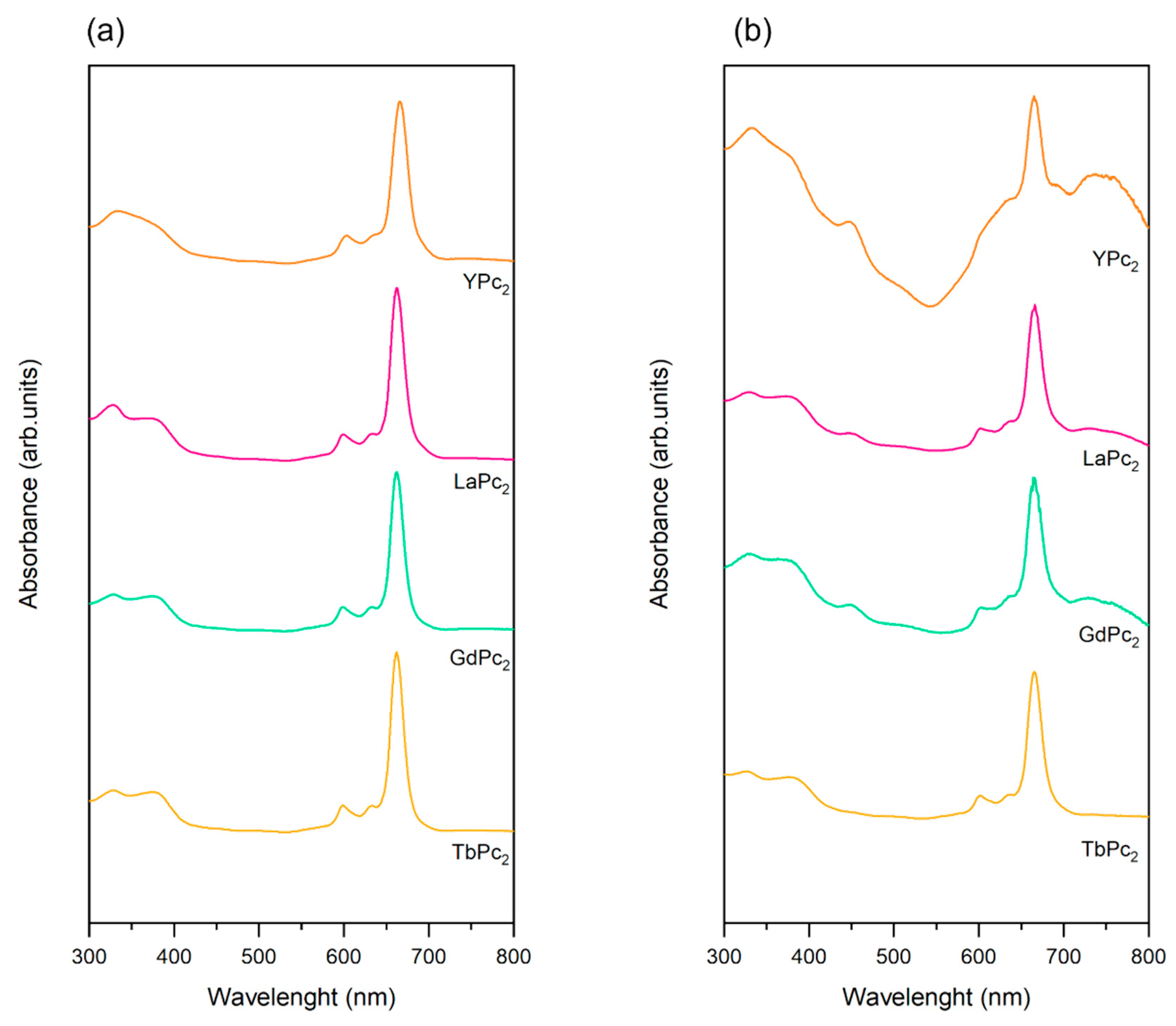
3.3.5. Ultraviolet–Visible Spectroscopy
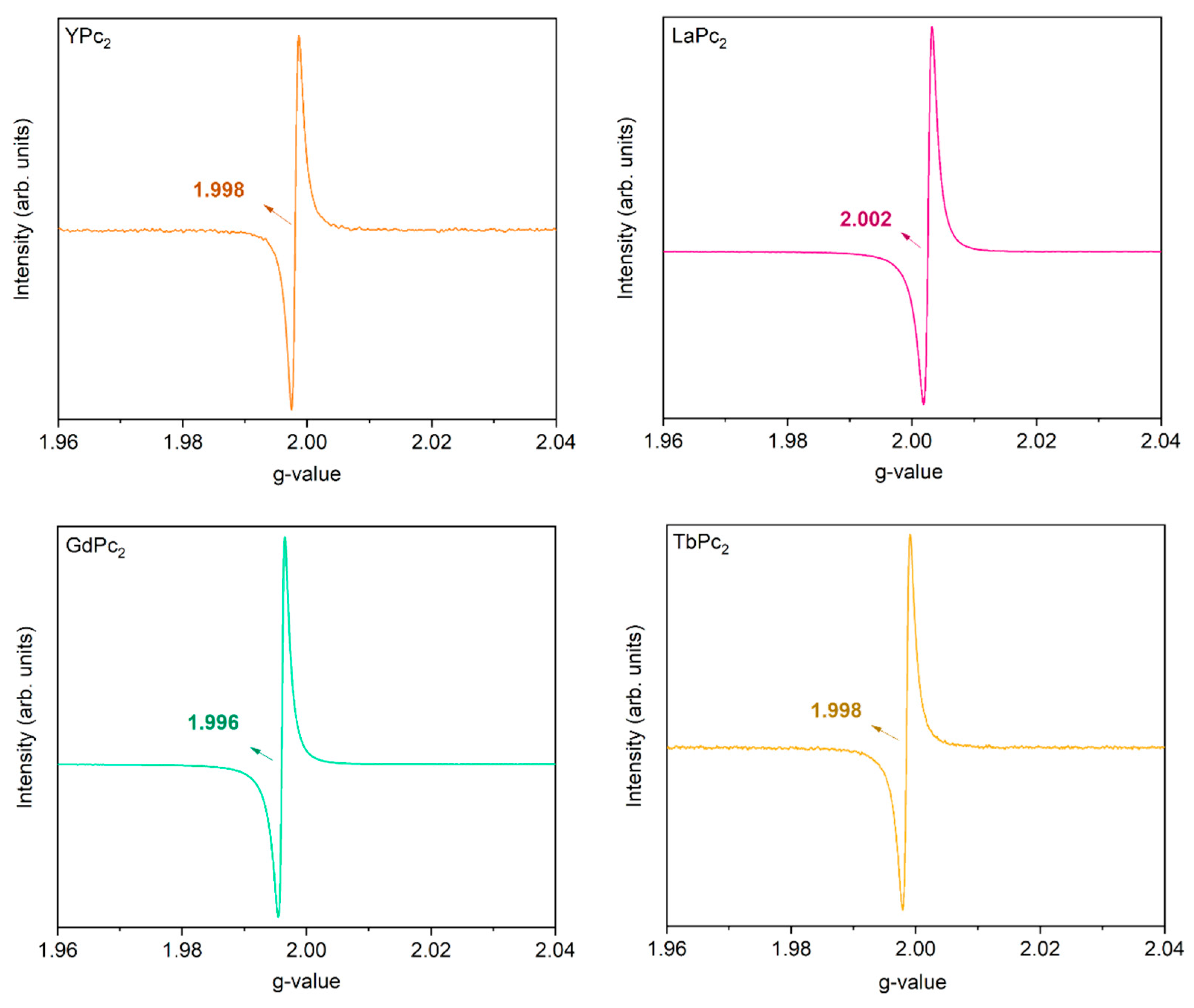
3.3.6. Electron Paramagnetic Resonance Spectroscopy
3.3.7. Thermal Analysis
3.3.8. Scanning Electron Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fukuda, T.; Biyajima, T.; Kobayashi, N. A Discrete Quadruple-Decker Phthalocyanine. J. Am. Chem. Soc. Commun. 2010, 132, 6278–6279. [Google Scholar] [CrossRef]
- Martynov, A.G.; Horii, Y.; Katoh, K.; Bian, Y.; Jiang, J.; Yamashita, M.; Gorbunova, Y.G. Rare-Earth Based Tetrapyrrolic Sandwiches: Chemistry, Materials and Applications. Chem. Soc. Rev. 2022, 51, 9262–9339. [Google Scholar] [CrossRef]
- De La Torre, G.; Claessens, C.G.; Torres, T. Phthalocyanines: Old Dyes, New Materials. Putting Color in Nanotechnology. Chem. Commun. 2007, 20, 2000–2015. [Google Scholar] [CrossRef]
- Robles, R.; Nicolás, L.; Lorente, N.; Isshiki, H.; Liu, J.; Katoh, K.; Breedlove, B.K.; Yamashita, M.; Komeda, T. Spin Doping of Individual Molecules by Using Single-Atom Manipulation. Nano Lett. 2012, 12, 3609–3612. [Google Scholar] [CrossRef]
- Woodruff, D.N.; Winpenny, R.E.P.; Layfield, R.A. Lanthanide Single-Molecule Magnets. Chem. Rev. 2013, 113, 5110–5148. [Google Scholar] [CrossRef]
- Zhang, J.L. Marriage of Phthalocyanine Chemistry with Lanthanides: A Single-Ion Magnet with a Blocking Temperature up to 25 K. Inorg. Chem. Front. 2017, 4, 1950–1952. [Google Scholar] [CrossRef]
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.Y.; Kaizu, Y. Lanthanide Double-Decker Complexes Functioning as Magnets at the Single-Molecular Level. J. Am. Chem. Soc. 2003, 125, 8694–8695. [Google Scholar] [CrossRef]
- Pushkarev, V.E.; Tomilova, L.G.; Nemykin, V.N. Historic Overview and New Developments in Synthetic Methods for Preparation of the Rare-Earth Tetrapyrrolic Complexes. Coord. Chem. Rev. 2016, 319, 110–179. [Google Scholar] [CrossRef]
- Pushkarev, V.E.; Tomilova, L.G.; Tomilov, Y.V. Synthetic Approaches to Lanthanide Complexes with Tetrapyrrole Type Ligands. Russ. Chem. Rev. 2008, 77, 875–907. [Google Scholar] [CrossRef]
- Yamamoto, S.; Dudkin, S.V.; Kimura, M.; Kobayashi, N. Phthalocyanine Synthesis. In Handbook of Porphyrin Science: With Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine—Volume 45: Phthalocyanine Synthesis and Computational Design of Functional Tetrapyrroles; World Scientific Publishing Co.: Singapore, 2019; pp. 1–168. ISBN 9789811201813. [Google Scholar]
- Bekaroglu, Ö. History, Development, and a New Concept of Phthalocyanines in Turkey. Turk. J. Chem. 2014, 38, 903–922. [Google Scholar] [CrossRef]
- Li, D.; Ge, S.; Sun, G.; He, Q.; Huang, B.; Tian, G.; Lu, W.; Li, G.; Chen, Y.; An, S.; et al. A Novel and Green Route for Solvothermal Synthesis of Manganese Phthalocyanine Crystals. Dye. Pigment. 2015, 113, 200–204. [Google Scholar] [CrossRef]
- Li, D.; Zhang, P.; Ge, S.; Sun, G.; He, Q.; Fa, W.; Li, Y.; Ma, J. A Green Route to Prepare Metal-Free Phthalocyanine Crystals with Controllable Structures by a Simple Solvothermal Method. RSC Adv. 2021, 11, 31226–31234. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Yu, S.; Shen, R.; Ma, C.; Cheng, C.; Ji, D.; Fan, Z.; Wang, X.; Du, G. A Novel Method for the Direct Synthesis of Crystals of Copper Phthalocyanine. Dye. Pigment. 2008, 78, 84–88. [Google Scholar] [CrossRef]
- Ge, S.; Zhang, Y.; Huang, B.; Huang, S.; Tie, W.; Lei, Y.; He, Q.; Tu, G.; Qin, Q.; Niu, S.; et al. Synthesis of Highly Crystalline Copper Phthalocyanine Needles by Solvothermal Method. Mater. Lett. 2016, 163, 61–64. [Google Scholar] [CrossRef]
- Xia, D.; Li, W.; Wang, X.; Yu, S.; Fan, C.-x.; Ma, C.; Cheng, C.; Fan, Z.; Du, G.; Cong, F.; et al. Preparation of Copper Phthalocyanine Crystals Using Solvothermal Synthesis. Chem. Res. Chin. Univ. 2008, 24, 407–410. [Google Scholar] [CrossRef]
- Clarisse, G.; Riou, M.T. Synthesis and Characterization of Some Lanthanide Phthalocyanines. Inorganica Chim. Acta 1987, 130, 139–144. [Google Scholar] [CrossRef]
- Jiang, J.; Arnold, D.P.; Yu, H. Infra-Red Spectra of Phthalocyanine and Naphthalocyanine in Sandwich-Type (Na)Phthalocyaninato and Porphyrinato Rare Earth Complexes. Polyhedron 1999, 18, 2129–2139. [Google Scholar] [CrossRef]
- M’Sadak, M.; Roncali, J.; Garnier, F. Rare-Earth Substitution Effect on the Electrochemical Properties of Lanthanide Diphthalocyanines. J. Electroanal. Chem. Interfacial Electrochem. 1985, 189, 99–111. [Google Scholar] [CrossRef]
- Lu, F.; Bao, M.; Ma, C.; Zhang, X.; Arnold, D.P.; Jiang, J. Infrared Spectra of Phthalocyanine and Naphthalocyanine in Sandwich-Type (Na)Phthalocyaninato and Porphyrinato Rare Earth Complexes. Part 3. The Effects of Substituents and Molecular Symmetry on the Infrared Characteristics of Phthalocyanine in Bis(Phthalocyaninato) Rare Earth Complexes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2003, 59, 3273–3286. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, X.; Zhou, Y.; Zhang, X.; Xu, H.; Liu, Z.; Li, X.; Jiang, J. Structures and Spectroscopic Properties of Bis(Phthalocyaninato) Yttrium and Lanthanum Complexes: Theoretical Study Based on Density Functional Theory Calculations. J. Phys. Chem. A 2007, 111, 392–400. [Google Scholar] [CrossRef]
- Yara, M.N.; Kandaz, M.; Koca, A.; Salih, B. Functional Alcohol-Soluble Double-Decker Phthalocyanines: Synthesis, Characterization, Electrochemistry and Peripheral Metal Ion Binding. J. Porphryns Phthalocyanines 2006, 10, 1022–1033. [Google Scholar] [CrossRef]
- Souto, J.; Tomilova, L.; Aroca, R.; Desaja, J.A. Spectroscopic Studies of Langmuir-Blodgett Monolayers of Praseodymium Bisphthalocyanines. Langmuir 1992, 8, 942–946. [Google Scholar] [CrossRef]
- Kratochvílová, I.; Šebera, J.; Paruzel, B.; Pfleger, J.; Toman, P.; Marešová, E.; Havlová, Š.; Hubík, P.; Buryi, M.; Vrňata, M.; et al. Electronic Functionality of Gd-Bisphthalocyanine: Charge Carrier Concentration, Charge Mobility, and Influence of Local Magnetic Field. Synth. Met. 2018, 236, 68–78. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Batov, M.S.; Martynov, A.G.; Nefedova, I.V.; Gorbunova, Y.G.; Otsuka, A.; Yamochi, H.; Kitagawa, H.; Lyubovskaya, R.N. Effect of One- and Two-Electron Reduction of Terbium(III) Double-Decker Phthalocyanine on Single-Ion Magnet Behavior and NIR Absorption. Inorg. Chem. 2019, 58, 5058–5068. [Google Scholar] [CrossRef] [PubMed]
- Guilard, R.; Kadish, K.M.; Smith, K.M. The Porphyrin Handbook; Academic Press: Cambridge, MA, USA, 2003; Volume 11, ISBN 0123932203. [Google Scholar]
- Chen, X.; Chen, Y.; Bai, M.; Wang, C.; Qi, D.; Liu, Q.; Xu, M.; Jiang, J. Distribution of the Unpaired Electron in Neutral Bis(Phthalocyaninato) Yttrium Double-Deckers: An Experimental and Theoretical Combinative Investigation. J. Porphyr. Phthalocyanines 2018, 22, 165–172. [Google Scholar] [CrossRef]
- Jiang, J.; Bao, M.; Rintoul, L.; Arnold, D.P. Vibrational Spectroscopy of Phthalocyanine and Naphthalocyanine in Sandwich-Type (Na)Phthalocyaninato and Porphyrinato Rare Earth Complexes. Coord. Chem. Rev. 2006, 250, 424–448. [Google Scholar] [CrossRef]
- Suzuki, A.; Oku, T. Effects of Central Metal on Electronic Structure, Magnetic Properties, Infrared and Raman Spectra of Double-Decker Phthalocyanine. Appl. Surf. Sci. 2016, 380, 127–134. [Google Scholar] [CrossRef]
- Zheng, W.; Wang, B.B.; Lai, J.C.; Wan, C.Z.; Lu, X.R.; Li, C.H.; You, X.Z. Electrochromic Properties of Novel Octa-Pinene Substituted Double-Decker Ln(III) (Ln = Eu, Er, Lu) Phthalocyanines with Distinctive near-IR Absorption. J. Mater. Chem. C 2015, 3, 3072–3080. [Google Scholar] [CrossRef]
- Åhlund, J.; Nilson, K.; Schiessling, J.; Kjeldgaard, L.; Berner, S.; Mårtensson, N.; Puglia, C.; Brena, B.; Nyberg, M.; Luo, Y. The Electronic Structure of Iron Phthalocyanine Probed by Photoelectron and X-Ray Absorption Spectroscopies and Density Functional Theory Calculations. J. Chem. Phys. 2006, 125, 034709. [Google Scholar] [CrossRef]
- Brena, B.; Luo, Y.; Nyberg, M.; Carniato, S.; Nilson, K.; Alfredsson, Y.; Åhlund, J.; Mårtensson, N.; Siegbahn, H.; Puglia, C. Equivalent Core-Hole Time-Dependent Density Functional Theory Calculations of Carbon 1s Shake-up States of Phthalocyanine. Phys. Rev. B 2004, 70, 195214. [Google Scholar] [CrossRef]
- Mannini, M.; Bertani, F.; Tudisco, C.; Malavolti, L.; Poggini, L.; Misztal, K.; Menozzi, D.; Motta, A.; Otero, E.; Ohresser, P.; et al. Magnetic Behaviour of TbPc2 Single-Molecule Magnets Chemically Grafted on Silicon Surface. Nat. Commun. 2014, 5, 6–13. [Google Scholar] [CrossRef]
- Alcón, I.; Gonidec, M.; Ajayakumar, M.R.; Mas-Torrent, M.; Veciana, J. A Surface Confined Yttrium(III) Bis-Phthalocyaninato Complex: A Colourful Switch Controlled by Electrons. Chem. Sci. 2016, 7, 4940–4944. [Google Scholar] [CrossRef]
- Xu, R.; Zhu, M.; Li, W.; Ding, J.; Zhang, Y. Excellent Nonlinear Optical Limiting Behavior of Tetra (C60) Lanthanum Phthalocyanine in Solid Poly(Methyl Methacrylate) Films. J. Phys. Chem. C 2023, 127, 4258–4265. [Google Scholar] [CrossRef]
- Serrano, G.; Sorrentino, A.L.; Poggini, L.; Cortigiani, B.; Goletti, C.; Sessoli, R.; Mannini, M. Substrate Mediated Interaction of Terbium(III) Double-Deckers with the TiO2(110) Surface. Phys. Chem. Chem. Phys. 2021, 23, 12060–12067. [Google Scholar] [CrossRef]
- Mele, G.; Garcìa-Lòpez, E.; Palmisano, L.; Dyrda, G.; Słota, R. Photocatalytic Degradation of 4-Nitrophenol in Aqueous Suspension by Using Polycrystalline TiO2 Impregnated with Lanthanide Double-Decker Phthalocyanine Complexes. J. Phys. Chem. C 2007, 111, 6581–6588. [Google Scholar] [CrossRef]
- Erzunov, D.A.; Botnar, A.A.; Domareva, N.P.; Tikhomirova, T.V.; Vashurin, A.S. Synthesis, Spectroscopic Properties and Redox Behavior Kinetics of Rare-Earth Bistetrakis-4-[3-(3,4-Dicyanophenoxy)Phenoxy]Phthalocyaninato Metal Complexes with Er, Lu and Yb. Molecules 2021, 26, 2181. [Google Scholar] [CrossRef]
- Dailey, M.; Besson, C. Selective Crystallization of Four Bis(Phthalocyaninato)Lanthanoid(III) Polymorphs. CrystEngComm 2021, 23, 7151–7161. [Google Scholar] [CrossRef]
- Ou, C.; Lv, W.; Chen, J.; Yu, T.; Song, Y.; Wang, Y.; Wang, S.; Yang, G. Structural, Photophysical and Nonlinear Optical Limiting Properties of Sandwich Phthalocyanines with Different Rare Earth Metals. Dye. Pigment. 2021, 184, 108862. [Google Scholar] [CrossRef]
- Dyrda, G.; Zakrzyk, M.; Broda, M.A.; Pedziński, T.; Mele, G.; Słota, R. Hydrogen Bond-Mediated Conjugates Involving Lanthanide Diphthalocyanines and Trifluoroacetic Acid (LnPc2@TFA): Structure, Photoactivity, and Stability. Molecules 2020, 25, 3638. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, R.C.W.; Mak, T.C.W.; Chan, T.W.D.; Ng, D.K.P. Synthesis, Spectroscopic and Electrochemical Properties of Substituted Bis(Phthalocyaninato)Lanthanide(III) Complexes. Polyhedron 1997, 16, 515–520. [Google Scholar] [CrossRef]
- Słota, R.; Dyrda, G.; Hofer, M.; Mele, G.; Bloise, E.; Del Sole, R. Novel Lipophilic Lanthanide Bis-Phthalocyanines Functionalized by Pentadecylphenoxy Groups: Synthesis, Characterization and UV-Photostability. Molecules 2012, 17, 10738–10753. [Google Scholar] [CrossRef]
- Komijani, D.; Ghirri, A.; Bonizzoni, C.; Klyatskaya, S.; Moreno-Pineda, E.; Ruben, M.; Soncini, A.; Affronte, M.; Hill, S. Radical-Lanthanide Ferromagnetic Interaction in a TbIII Bis-phthalocyaninato Complex. Phys. Rev. Mater. 2018, 2, 024405. [Google Scholar] [CrossRef]
- Seoudi, R.; El-Bahy, G.S.; El Sayed, Z.A. FTIR, TGA and DC Electrical Conductivity Studies of Phthalocyanine and Its Complexes. J. Mol. Struct. 2005, 753, 119–126. [Google Scholar] [CrossRef]
- Ivashenko, O.; Logtenberg, H.; Areephong, J.; Coleman, A.C.; Wesenhagen, P.V.; Geertsema, E.M.; Heureux, N.; Feringa, B.L.; Rudolf, P.; Browne, W.R. Remarkable Stability of High Energy Conformers in Self-Assembled Monolayers of a Bistable Electro-and Photoswitchable Overcrowded Alkene. J. Phys. Chem. C 2011, 115, 22965–22975. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MPc2 Complexes | Yield (%) a | H2Pc (%) b | Elemental Analysis (%) | |||
|---|---|---|---|---|---|---|
| C | H | N | M | |||
| YPc2 | 68.1 | 5.0 | 69.37 (69.00) c | 2.92 (2.89) | 20.17 (20.12) | 7.54 (7.98) |
| LaPc2 | 43.0 | 33.6 | 66.09 (66.04) | 2.69 (2.65) | 19.28 (19.25) | 12.04 (11.93) |
| GdPc2 | 63.1 | 16.7 | 65.08 (65.04) | 2.80 (2.73) | 19.53 (18.96) | 12.64 (13.3) |
| TbPc2 | 61.8 | 18.3 | 65.44 (64.92) | 2.98 (2.72) | 18.96 (18.93) | 12.63 (13.4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolivar-Pineda, L.M.; Mendoza-Domínguez, C.U.; Rudolf, P.; Basiuk, E.V.; Basiuk, V.A. Solvothermal Synthesis of Rare Earth Bisphthalocyanines. Molecules 2024, 29, 2690. https://doi.org/10.3390/molecules29112690
Bolivar-Pineda LM, Mendoza-Domínguez CU, Rudolf P, Basiuk EV, Basiuk VA. Solvothermal Synthesis of Rare Earth Bisphthalocyanines. Molecules. 2024; 29(11):2690. https://doi.org/10.3390/molecules29112690
Chicago/Turabian StyleBolivar-Pineda, Lina M., Carlos U. Mendoza-Domínguez, Petra Rudolf, Elena V. Basiuk, and Vladimir A. Basiuk. 2024. "Solvothermal Synthesis of Rare Earth Bisphthalocyanines" Molecules 29, no. 11: 2690. https://doi.org/10.3390/molecules29112690
APA StyleBolivar-Pineda, L. M., Mendoza-Domínguez, C. U., Rudolf, P., Basiuk, E. V., & Basiuk, V. A. (2024). Solvothermal Synthesis of Rare Earth Bisphthalocyanines. Molecules, 29(11), 2690. https://doi.org/10.3390/molecules29112690