
Development of Multifunctional Catalysts for the Direct Hydrogenation of Carbon Dioxide to Higher Alcohols

Abstract
1. Introduction
2. Multifunctional Catalysts for CO2 Hydrogenation to Higher Alcohols
2.1. Noble Metal Catalysts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enter | Catalysts | T (°C) | P (MPa) | GHSV a /L g−1 h−1 | H2 /CO2 | X b CO2 (%) | S c CO (%) | S d HC (%) | S e MeOH (%) | S f HA (%) | STY g HA (mmol gcat−1 h−1) | Refs. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Rh-TiO2 | 300 | 10 | 6 | 3 | / | / | 69.8 | 21.1 | 9.3 | 0.7 | [52] |
| 2 | RhLi-SiO2 | 240 | 5 | 6 | 3 | 7.0 | 15.7 | 63.5 | 5.2 | 15.5 | 0.36 | [57] |
| 3 | Rh1/CeTiOx h | 250 | 3 | / | 2 | 6.3 | / | / | / | 99.1 | 5.73 j | [61] |
| 4 | 1Pd2Ce@Si16 | 250 | 3 | 3 | / | 5.9 | / | / | / | 98.7 | 11.6 j | [62] |
| 5 | Pt/Co3O4-p | 200 | 2 | 6 | 3 | 22.4 | / | 80.1 | 19.9 i | 0.56 | [63] | |
| 6 | PtRu/Fe2O3 h | 200 | 20 | / | / | / | 0.4 | 57.3 | 6.3 | 36.0 | 2.4 | [64] |
2.2. Co-Based Catalysts
| Enter | Catalysts | T (°C) | P (MPa) | GHSV a /L g−1 h−1 | H2 /CO2 | X b CO2 (%) | S c CO (%) | S d HC (%) | S e MeOH (%) | S f HA (%) | STY g HA (mmol gcat−1 h−1) | Refs. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Na-Co/SiO2 | 220 | 1.9 | 3 | 3 | 12.4 | / | 26.8 | / | 73.2 | / | [74] |
| 2 | Co3O4 | 200 | 2 | 6 | 3 | 28.9 | 0 | 53.6 | / | 19.2 | 1.6 | [75] |
| 3 | Co/La4Ga2O9 | 280 | 3 | 3 | 3 | 9.6 | 10.8 | 52.2 | 13.7 | 23.3 | / | [76] |
| 4 | Na-Co/SiO2 | 250 | 5 | 6 | 3 | 21.5 | 26.3 | 61.2 | 1.7 | 10.8 | 0.47 | [77] |
| 5 | Mo-Co-K | 320 | 5 | 3 | 3 | 23.5 | / | 21 | 70 | 8.9 | / | [78] |
| 6 | CoMoCx-800 h | 180 | 2 | / | 3 | / | / | / | / | / | 0.53 | [79] |
2.3. Cu-Based Catalysts
| Enter | Catalysts | T (°C) | P (MPa) | GHSV a /L g−1 h−1 | H2/ CO2 | X b CO2 (%) | S c CO (%) | S d HC (%) | S e MeOH (%) | S f HA (%) | STY g HA (mmol gcat−1h−1) | Refs. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Cu@ Na-Beta | 300 | 1.3 | 12 | 1 | 7.9 | 30.5 | / | / | 69.5 | 17.1 | [89] |
| 2 | Cu@m-Beta h | 200 | 4 | / | 3 | 17 | 10 | / | 20 | 68 | 50.13 i | [90] |
| 3 | Cu-CoGa0.4 | 220 | 3 | 6 | 3 | 17.8 | 2.3 | 43.5 | 27.5 | 23.8 | 1.35 | [91] |
| 4 | Na-CuCo-9 | 330 | 4 | 5 | 1 | 20.1 | 26.5 | 53.8 | 0.5 | 26.3 | 1.09 j | [92] |
| 5 | Cs-C0.8F1.0Z1.0 | 330 | 5 | 4.5 | 3 | 36.6 | / | / | / | 19.8 | 1.47 | [93] |
| 6 | Cr-CuFe | 320 | 4 | 6 | 3 | 38.4 | 14.8 | / | / | 29.2 | 104.1 k | [94] |
| 7 | sp-CuNaFe | 310 | 3 | 28.8 | / | 32.3 | / | 55 | / | 10 | 153 k | [95] |
2.4. Fe-Based Catalysts
| Enter | Catalysts | T (°C) | P (MPa) | GHSV a /L g−1 h−1 | H2 /CO2 | X b CO2 (%) | S c CO (%) | S d HC (%) | S e MeOH (%) | S f HA (%) | STY g HA (mg gcat−1 h−1) | Refs. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | FeNaS-0.6 | 320 | 3 | 8 | / | 32.0 | 20.7 | / | / | 12.8 | 78.5 | [107] |
| 2 | Na/Fe3O4 | 300 | 3.0 | 2.5 | 3 | 30.6 | 4.1 | 53.9 | 5.3 | 36.7 | / | [108] |
| 3 | 10Mn1K-FeC | 300 | 3.0 | 6 | 3 | 40.5 | 33.4 | 55.9 | 0.2 | 10.5 | / | [109] |
| 4 | KFeIn/ Ce-ZrO2 | 300 | 10.0 | 4.5 | 3 | 29.6 | 13.4 | 53.2 | 4.7 | 28.7 | / | [110] |
| 5 | In2Fe/ K-Al2O3 | 300 | 2 | 4 | 3 | 36.7 | 7.4 | 37.9 | 2.3 | 42 | / | [111] |
| 6 | PdFe-6.9 | 320 | 5.0 | 6 | 4 | 35.6 | 20.7 | / | / | 18.2 | 87.8 | [112] |
| 7 | Na-ZnFe@C | 320 | 5 | 0.9 | 3 | 38.4 | 7.6 | 43.2 | 1.8 | 20.3 | 158.1 | [113] |
2.5. Tandem Catalysts
| Enter | Catalysts | T (°C) | P (MPa) | GHSV a /L g−1 h−1 | H2 /CO2 | X b CO2 (%) | S c CO (%) | S d HC (%) | S e MeOH (%) | S f HA (%) | STY g HA (mmol gcat−1 h−1) | Refs. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PdGaCuZnAlK /CuFeAlK | 330 | 8 | 20 | 3 | 54.5 | 9.7 | 64.5 | 5.2 | 17.0 | / | [129] |
| 2 | CZK||CFCK | 350 | 6 | 5 | 3 | 32.4 | 45.3 | 42.9 | 5.3 | 6.5 | 1.37 | [130] |
| 3 | Na-Fe@C /KCuZnAl | 320 | 5 | / | 2.8 | 39.2 | 9.4 | 54.5 | 4.6 | 35 | / | [131] |
| 4 | CuZnAl/K-CuMgZnFe | 320 | 5 | 6 | 3 | 42.3 | 13.8 | 67.6 | 1.3 | 17.4 | 2.24 | [132] |
| 5 | MnCuK-FeC /CuZnAlZr | 300 | 3 | 6 | 3 | 42.1 | 22.7 | 60.6 | 1.2 | 15.5 | / | [133] |
| 6 | 4.7KCuFeZn /CuZnAlZr | 300 | 5 | 3 | 3 | 29.4 | 26.1 | 66.7 | 2.1 | 31.2 | 84.0 h | [134] |
| 7 | K-CuZnAl /Zr-CuFe | 320 | 4 | 24 | 3 | 28.8 | / | / | / | 19.7 | 195.1 h | [135] |
| 8 | Co2C||CuZnAl | 250 | 5 | 12 | 3 | 21.2 | / | / | / | 18.3 | 2.20 | [136] |
3. The Regulation Mechanism of CO2 Hydrogenation to Higher Alcohols
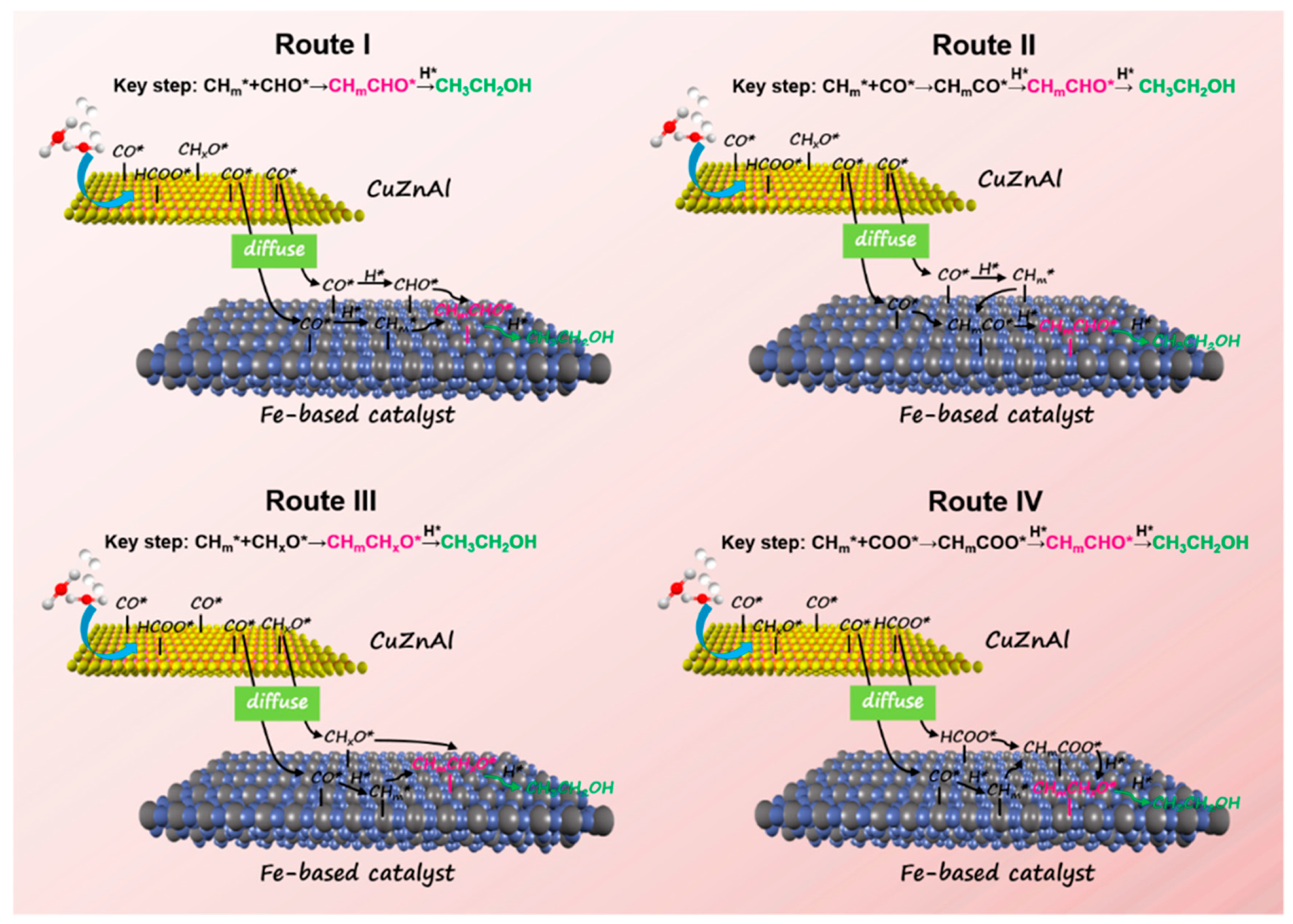
3.1. CO-Mediating Mechanism
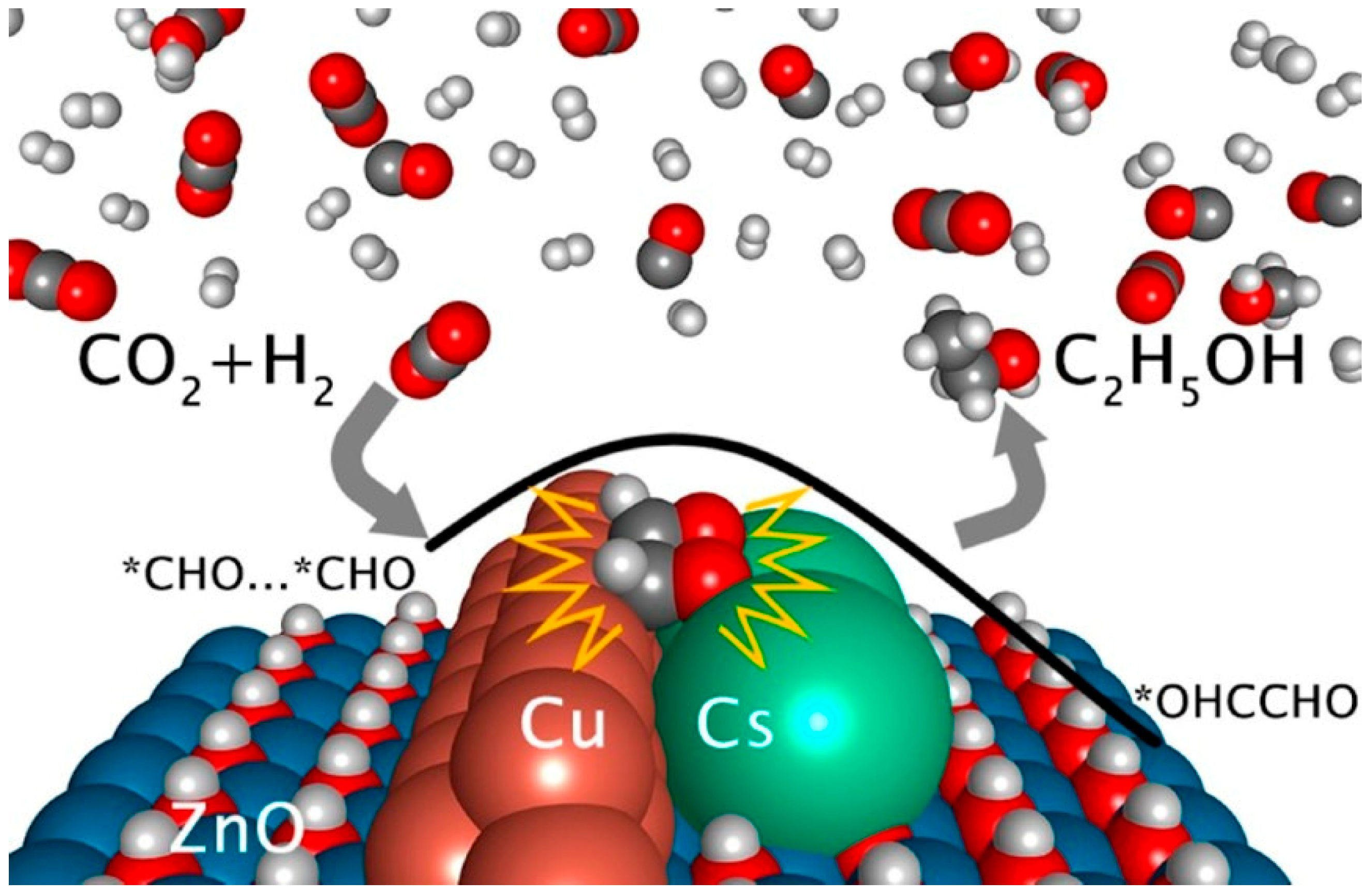
3.2. Formate/Methoxy-Mediated Mechanism
4. Effect of the Reaction Conditions on the Catalytic Performance of CO2 Hydrogenation
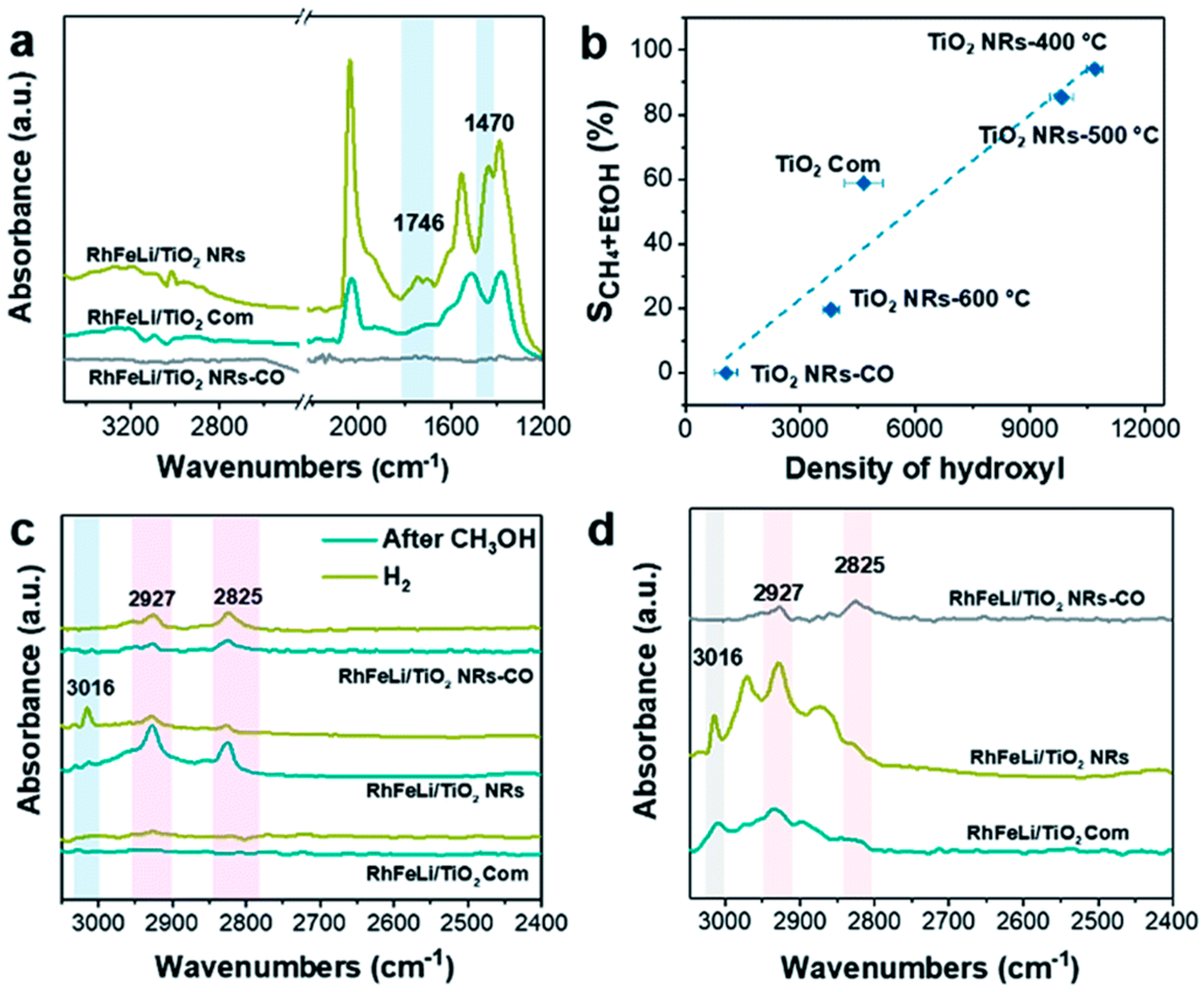
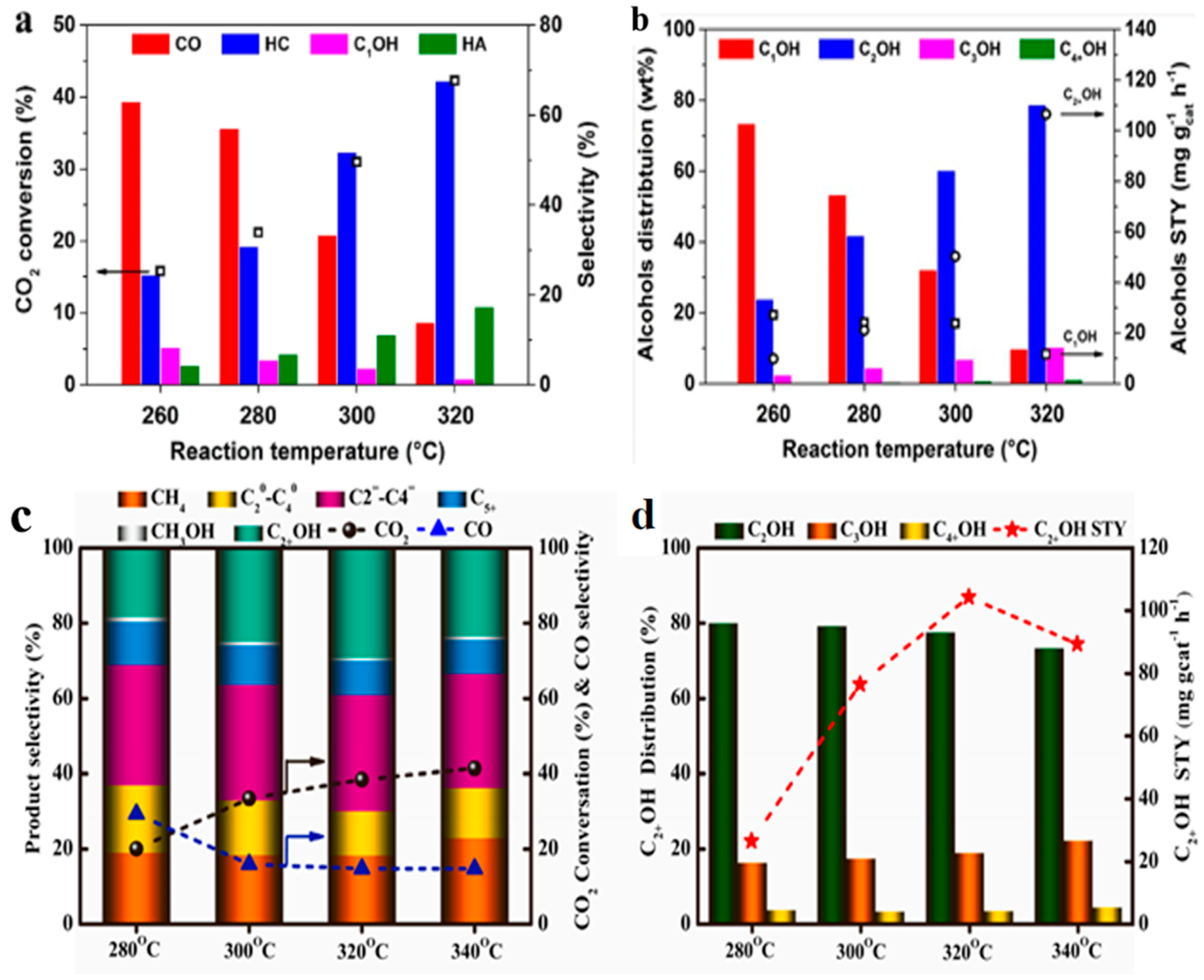
4.1. Reaction Temperature
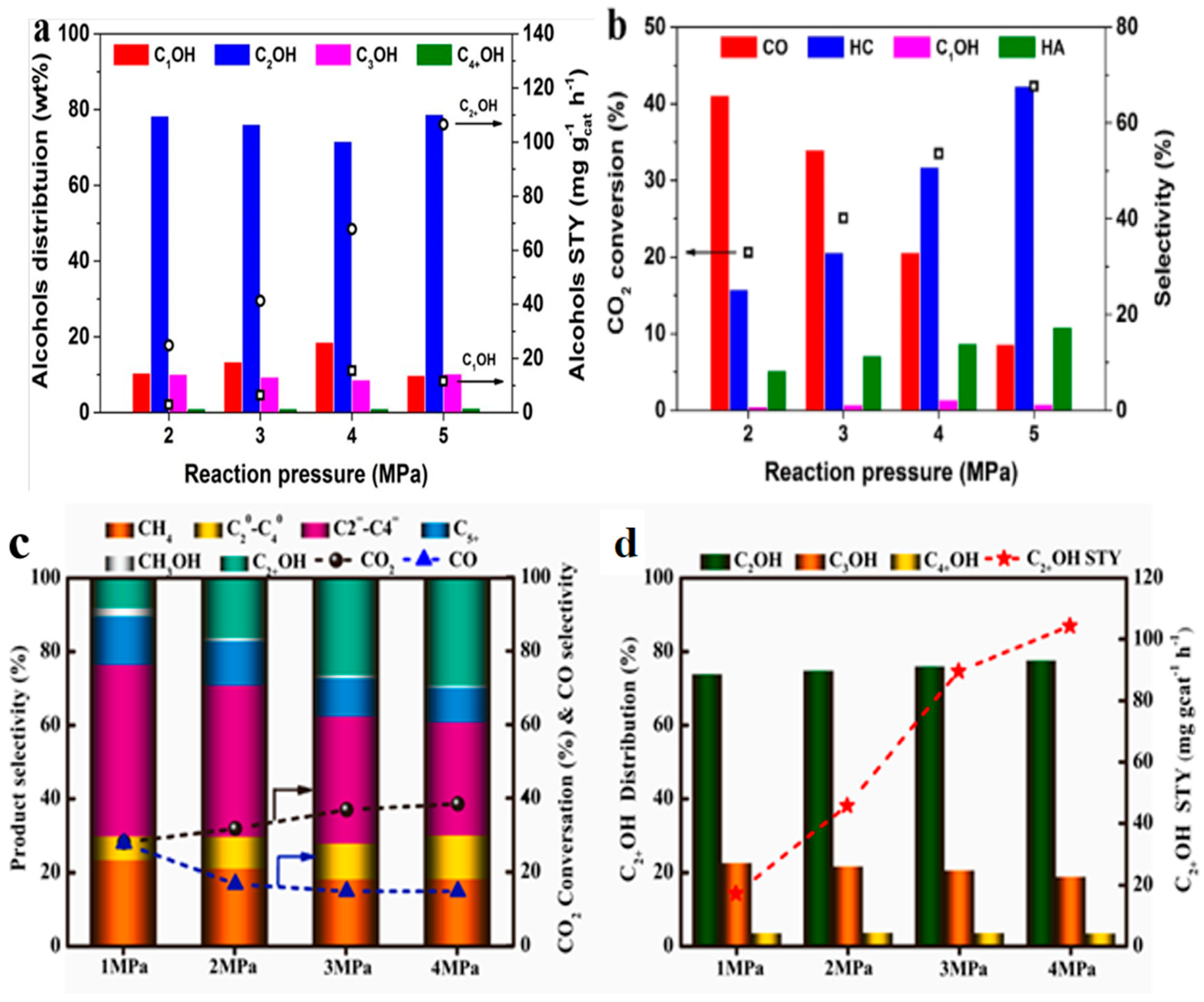
4.2. Reaction Pressure
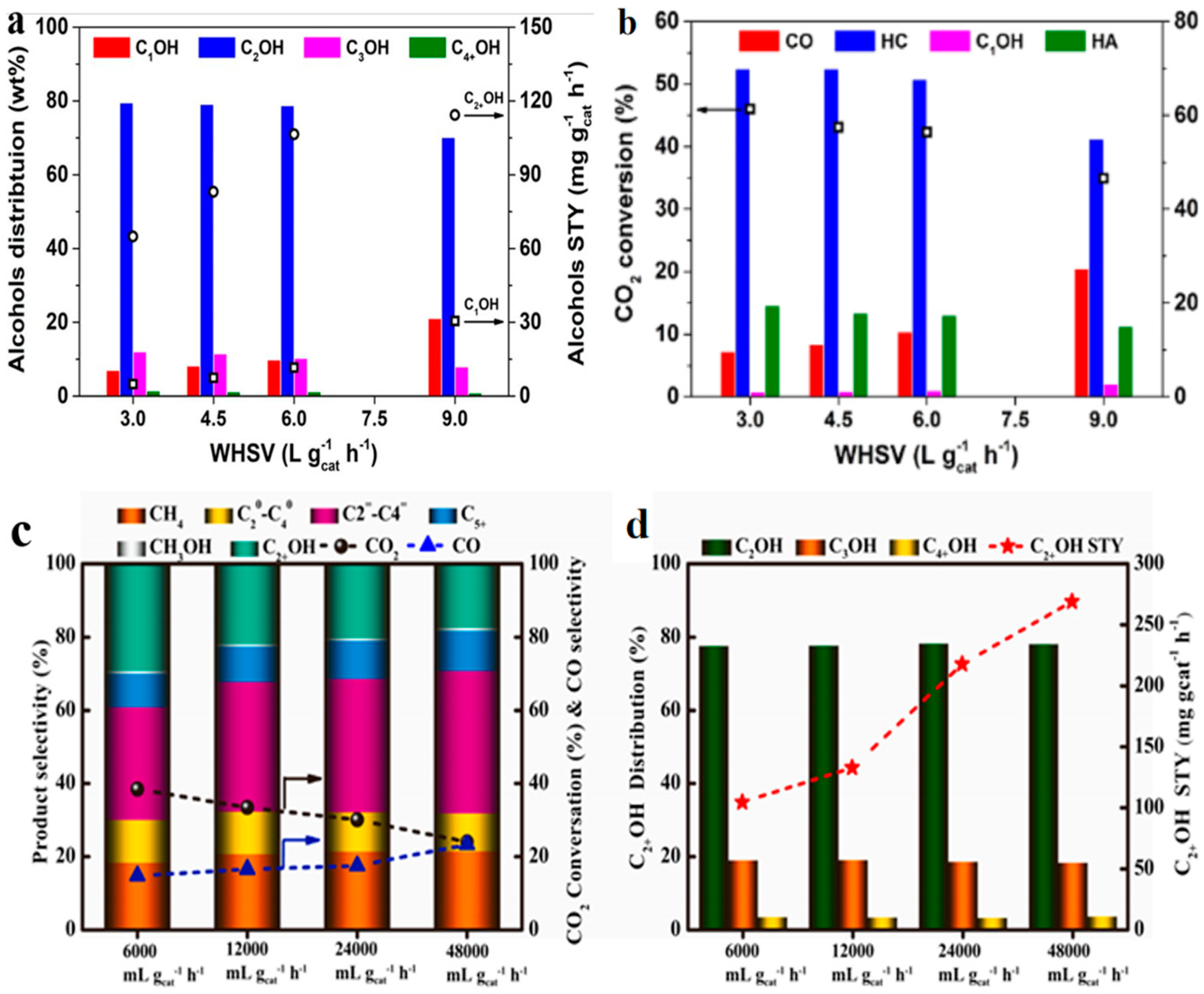
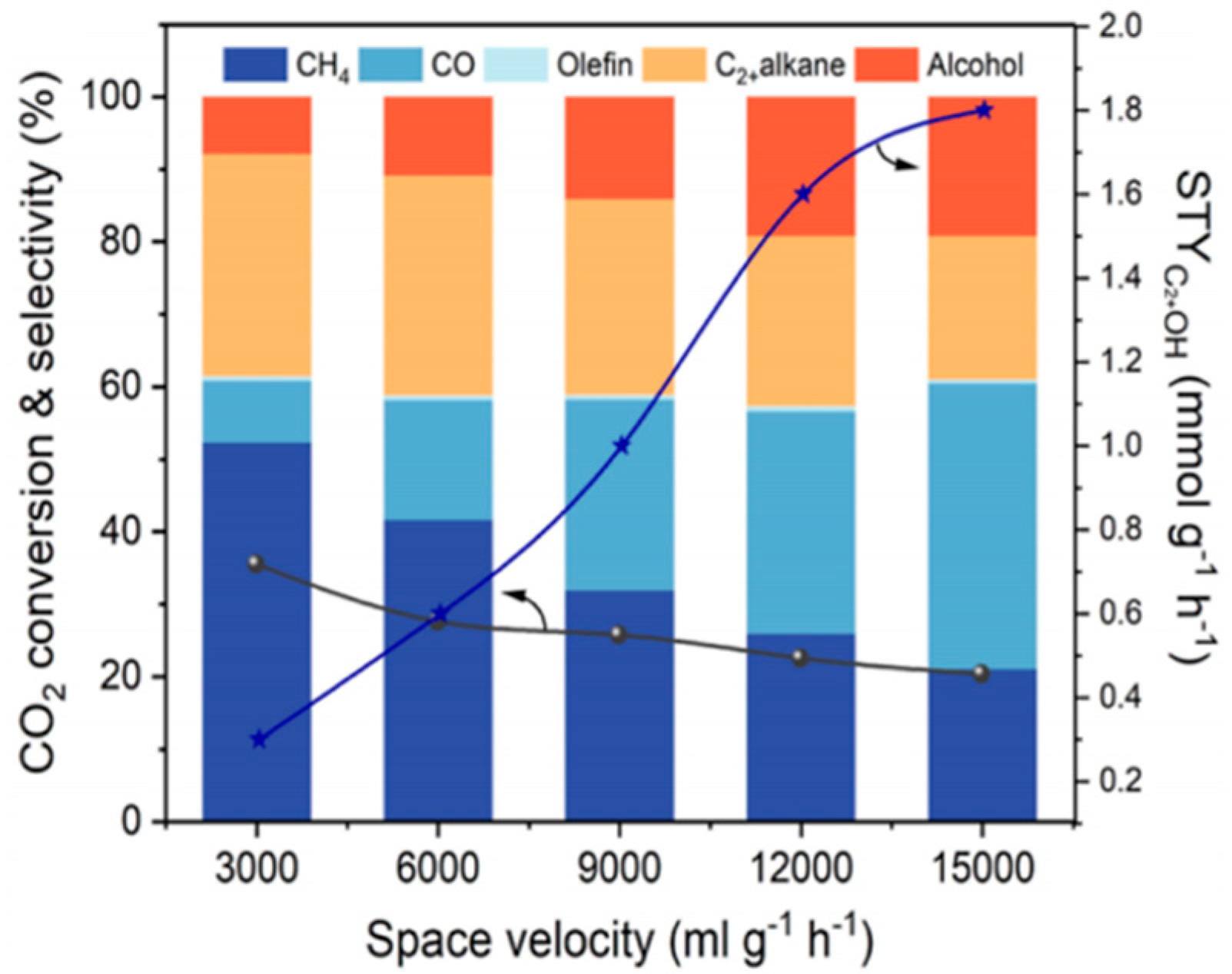
4.3. Space Velocity
4.4. H2/CO2 Ratio
4.5. Relative Humidity
4.6. Catalyst Stability
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Z.E.; Pan, S.Y.; Li, H.; Cai, J.C.; Olabi, A.G.; Anthony, E.J.; Manovic, V. Recent advances in carbon dioxide utilization. Renew. Sustain. Energy Rev. 2020, 125, 109799. [Google Scholar] [CrossRef]
- Lacis, A.A.; Schmidt, G.A.; Rind, D.; Ruedy, R.A. Atmospheric CO2: Principal Control Knob Governing Earth’s Temperature. Science 2010, 330, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.; Plattner, G.K.; Knutti, R.; Friedlingstein, P. Irreversible climate change due to carbon dioxide emissions. Proc. Natl. Acad. Sci. USA 2009, 106, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.E.; Wang, T.; Blunt, M.J.; Anthony, E.J.; Park, A.H.A.; Hughes, R.W.; Webley, P.A.; Yan, J.Y. Advances in carbon capture, utilization and storage. Appl. Energy 2020, 278, 115627. [Google Scholar] [CrossRef]
- Corma, A. Preface to Special Issue of ChemSusChem on Green Carbon Science: CO2 Capture and Conversion. ChemSusChem 2020, 13, 6054–6055. [Google Scholar] [CrossRef]
- Jiang, X.; Nie, X.W.; Guo, X.W.; Song, C.S.; Chen, J.G.G. Recent Advances in Carbon Dioxide Hydrogenation to Methanol via Heterogeneous Catalysis. Chem. Rev. 2020, 120, 7984–8034. [Google Scholar] [CrossRef]
- Hepburn, C.; Adlen, E.; Beddington, J.; Carter, E.A.; Fuss, S.; Mac Dowell, N.; Minx, J.C.; Smith, P.; Williams, C.K. The technological and economic prospects for CO2 utilization and removal. Nature 2019, 575, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Tapia, J.F.D.; Lee, J.Y.; Ooi, R.E.H.; Foo, D.C.Y.; Tan, R.R. A review of optimization and decision-making models for the planning of CO2 capture, utilization and storage (CCUS) systems. Sustain. Prod. Consum. 2018, 13, 1–15. [Google Scholar] [CrossRef]
- Ni, Y.M.; Chen, Z.Y.; Fu, Y.; Liu, Y.; Zhu, W.L.; Liu, Z.M. Selective conversion of CO2 and H2 into aromatics. Nat. Commun. 2018, 9, 3457. [Google Scholar] [CrossRef]
- Gao, P.; Li, S.G.; Bu, X.N.; Dang, S.S.; Liu, Z.Y.; Wang, H.; Zhong, L.S.; Qiu, M.H.; Yang, C.G.; Cai, J.; et al. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017, 9, 1019–1024. [Google Scholar] [CrossRef]
- Mirzakhani, S.; Yin, B.H.; Masteri-Farahani, M.; Yip, A.C.K. Heterogeneous catalytic systems for carbon dioxide hydrogenation to value-added chemicals. Chempluschem 2023, 88, e202300157. [Google Scholar] [CrossRef]
- Song, C.S. Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing. Catal. Today 2006, 115, 2–32. [Google Scholar] [CrossRef]
- Cheng, K.; Li, Y.B.; Kang, J.C.; Zhang, Q.H.; Wang, Y. Selectivity Control by Relay Catalysis in CO and CO2 Hydrogenation to Multicarbon Compounds. Acc. Chem. Res. 2024, 57, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Sanz, I.; Korili, S.A.; Gil, A. Catalytic valorization of CO2 by hydrogenation: Current status and future trends. Catal. Rev.-Sci. Eng. 2023, 65, 698–772. [Google Scholar] [CrossRef]
- Mandal, S.C.; Das, A.; Roy, D.; Das, S.; Nair, A.S.; Pathak, B. Developments of the heterogeneous and homogeneous CO2 hydrogenation to value-added C2+-based hydrocarbons and oxygenated products. Coord. Chem. Rev. 2022, 471, 214737. [Google Scholar] [CrossRef]
- Ateka, A.; Rodriguez-Vega, P.; Erena, J.; Aguayo, A.T.; Bilbao, J. A review on the valorization of CO2. Focusing on the thermodynamics and catalyst design studies of the direct synthesis of dimethyl ether. Fuel Process. Technol. 2022, 233, 107310. [Google Scholar] [CrossRef]
- Wang, L.X.; Wang, L.; Xiao, F.S. Tuning product selectivity in CO2 hydrogenation over metal-based catalysts. Chem. Sci. 2021, 12, 14660–14673. [Google Scholar] [CrossRef] [PubMed]
- Atsbha, T.A.; Yoon, T.; Seongho, P.; Lee, C.J. A review on the catalytic conversion of CO2 using H2 for synthesis of CO, methanol, and hydrocarbons. J. CO2 Util. 2021, 44, 101413. [Google Scholar] [CrossRef]
- Chen, G.B.; Waterhouse, G.I.N.; Shi, R.; Zhao, J.Q.; Li, Z.H.; Wu, L.Z.; Tung, C.H.; Zhang, T.R. From Solar Energy to Fuels: Recent Advances in Light-Driven C1 Chemistry. Angew. Chem.-Int. Ed. 2019, 58, 17528–17551. [Google Scholar] [CrossRef]
- Dalena, F.; Senatore, A.; Basile, M.; Knani, S.; Basile, A.; Iulianelli, A. Advances in Methanol Production and Utilization, with Particular Emphasis toward Hydrogen Generation via Membrane Reactor Technology. Membranes 2018, 8, 98. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.P.; Ma, X.B.; Gong, J.L. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Z.; Hao, C.N.; Liu, H.M.; Zhang, R.Q.; Li, Y.Q.; Guo, J.W.; Vilancuo, C.C.; Guo, J.P. Photocatalytic CO2 Conversion to Ethanol: A Concise Review. Catalysis 2022, 12, 1549. [Google Scholar] [CrossRef]
- Shah, M.A.; Shibiru, A.L.; Kumar, V.; Srivastava, V.C. Carbon dioxide conversion to value-added products and fuels: Opportunities and challenges: A critical review. Int. J. Green Energy 2023, 16, 1654. [Google Scholar] [CrossRef]
- Syuy, A.V.; Shtarev, D.S.; Kozlova, E.A.; Kurenkova, A.Y.; Zhurenok, A.V.; Shtareva, A.V.; Gurin, M.S.; Tselikov, G.I.; Tikhonowski, G.V.; Arsenin, A.; et al. Photocatalytic activity of tiNbC-modified TiO2 during hydrogen evolution and CO2 reduction. Appl. Sci. 2023, 13, 9410. [Google Scholar] [CrossRef]
- Montalvo, D.; Corro, G.; Banuelos, F.; Olivares-Xometl, O.; Arellanes, P.; Pal, U. Selective alcohols production through CO2 photoreduction using Co3O4/TiO2 photocatalyst exploiting synergetic interactions between Ti3+, Co2+ and Co3+. Appl. Catal. B Environ. Energy 2023, 330, 122652. [Google Scholar] [CrossRef]
- Ouyang, Y.X.; Shi, L.; Bai, X.W.; Ling, C.Y.; Li, Q.; Wang, J.L. Selectivity of Electrochemical CO2 Reduction toward Ethanol and Ethylene: The Key Role of Surface-Active Hydrogen. ACS Catal. 2023, 13, 15448–15456. [Google Scholar] [CrossRef]
- Su, D.J.; Xiang, S.Q.; Jiang, Y.M.; Li, Q.; Liu, X.H.; Zhang, W.; Zhao, L.B. Binary alloys for electrocatalytic CO2 conversion to hydrocarbons and alcohols. Appl. Surf. Sci. 2023, 635, 157734. [Google Scholar] [CrossRef]
- Xu, Z.H.; Han, Y.X.; Sun, J.Y.; Xu, M.; Zhao, W.L.; Wang, Q.F. Confinement and interface engineering of CuSiO3 nanotubes for enhancing CO2 electroreduction to C2+ products. Electrochim. Acta 2023, 470, 143291. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Y.H.; Chen, T.; Shi, G.Q.; Zhu, L.; Sun, Y.; Yu, M. Insight into the Electrochemical CO2-to-Ethanol Conversion Catalyzed by Cu2S Nanocrystal-Decorated Cu Nanosheets. ACS Appl. Mater. Interfaces 2023, 15, 18857–18866. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.Q.; Hu, Y.Y.; Lou, Y.J.; Zhou, M.X.; Li, Y.M.; Wang, Y.J.; Jiang, G.Y.; Xu, C.M. Research progress on the design, preparation and properties of catalysts for CO2 Hydrogenation to alcohols. Acta Chim. Sin. 2023, 81, 1081–1100. [Google Scholar] [CrossRef]
- Christensen, E.; Yanowitz, J.; Ratcliff, M.; McCormick, R.L. Renewable Oxygenate Blending Effects on Gasoline Properties. Energy Fuels 2011, 25, 4723–4733. [Google Scholar] [CrossRef]
- Liu, S.; He, Y.; Fu, W.; Chen, J.; Ren, J.; Liao, L.; Sun, R.; Tang, Z.; Mebrahtu, C.; Zeng, F. Hetero-site cobalt catalysts for higher alcohols synthesis by CO2 hydrogenation: A review. J. CO2 Util. 2023, 67, 102322. [Google Scholar] [CrossRef]
- Angelici, C.; Weckhuysen, B.M.; Bruijnincx, P.C.A. Chemocatalytic Conversion of Ethanol into Butadiene and Other Bulk Chemicals. ChemSusChem 2013, 6, 1595–1614. [Google Scholar] [CrossRef]
- Sheng, Y.; Polynski, M.V.; Eswaran, M.K.; Zhang, B.; Lim, A.M.H.; Zhang, L.; Jiang, J.; Liu, W.; Kozlov, S.M. A review of mechanistic insights into CO2 reduction to higher alcohols for rational catalyst design. Appl. Catal. B Environ. 2024, 343, 123550. [Google Scholar] [CrossRef]
- Bai, F.W.; Anderson, W.A.; Moo-Young, M. Ethanol fermentation technologies from sugar and starch feedstocks. Biotechnol. Adv. 2008, 26, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Mahnaz, F.; Dunlap, V.; Helmer, R.; Borkar, S.S.; Navar, R.; Yang, X.; Shetty, M. Selective Valorization of CO2 towards Valuable Hydrocarbons through Methanol-mediated Tandem Catalysis. Chemcatchem 2023, 15, e202300402. [Google Scholar] [CrossRef]
- Alberto, B. Carbon dioxide hydrogenation for sustainable energy storage. Int. J. Hydrogen Energy 2024, 58, 1386–1395. [Google Scholar]
- Hua, Z.; Yang, Y.; Liu, J. Direct hydrogenation of carbon dioxide to value-added aromatics. Coord. Chem. Rev. 2023, 478, 214982. [Google Scholar] [CrossRef]
- Nie, X.W.; Li, W.H.; Jiang, X.; Guo, X.W.; Song, C.S. Recent advances in catalytic CO2 hydrogenation to alcohols and hydrocarbons. Adv. Catal. 2019, 65, 121–233. [Google Scholar]
- Yi, Q.; Li, W.Y.; Feng, J.; Xie, K.C. Carbon cycle in advanced coal chemical engineering. Chem. Soc. Rev. 2015, 44, 5409–5445. [Google Scholar] [CrossRef]
- Stangeland, K.; Li, H.; Yu, Z. Thermodynamic Analysis of Chemical and Phase Equilibria in CO2 Hydrogenation to Methanol, Dimethyl Ether, and Higher Alcohols. Ind. Eng. Chem. Res. 2018, 57, 4081–4094. [Google Scholar] [CrossRef]
- Jia, C.; Gao, J.; Dai, Y.; Zhang, J.; Yang, Y. The thermodynamics analysis and experimental validation for complicated systems in CO2 hydrogenation process. J. Energy Chem. 2016, 25, 1027–1037. [Google Scholar] [CrossRef]
- Tatsumi, T.; Muramatsu, A.; Tominaga, H.O. Alcohol synthesis from CO2/H2 on silica-supported molybdenum catalysts. Chem. Lett. 1985, 14, 593–594. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Y.Q.; Lu, H.J.; Liu, T.K.; Hong, X.L.; Liu, G.L. Highly Dispersed K- and Pd-Comodified Fe Catalyst for CO2 Hydrogenation to Higher Alcohols. ACS Sustain. Chem. Eng. 2024, 12, 3322–3330. [Google Scholar] [CrossRef]
- Zhang, J.; Kang, X.S.; Yan, Y.C.; Ding, X.; He, L.; Li, Y.G. Cascade Electrocatalytic and Thermocatalytic Reduction of CO2 to Propionaldehydes. Angew. Chem.-Int. Ed. 2024, 63, e202315777. [Google Scholar] [CrossRef]
- Guo, S.Y.; Asset, T.; Atanassov, P. Catalytic Hybrid Electrocatalytic/Biocatalytic Cascades for Carbon Dioxide Reduction and Valorization. ACS Catal. 2021, 11, 5172–5188. [Google Scholar] [CrossRef]
- Lee, M.G.; Li, X.Y.; Ozden, A.; Wicks, J.; Ou, P.F.; Li, Y.H.; Dorakhan, R.; Lee, J.; Park, H.K.; Yang, J.W.; et al. Selective synthesis of butane from carbon monoxide using cascade electrolysis and thermocatalysis at ambient conditions. Nat. Catal. 2023, 6, 310–318. [Google Scholar] [CrossRef]
- Zhang, T.T.; Zhang, M.L.; Wu, L.H.; Guo, S.Y.; Liu, X.J.; Zhao, J.K.; Xue, W.Q.; Li, J.W.; Liu, C.X.; Li, X.; et al. Upcycling CO2 into energy-rich long-chain compounds via electrochemical and metabolic engineering. Nat. Catal. 2022, 5, 388–396. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Hong, X. Sulfate-Promoted Higher Alcohol Synthesis from CO2 Hydrogenation. ACS Sustain. Chem. Eng. 2022, 10, 8980–8987. [Google Scholar] [CrossRef]
- Zeng, F.; Mebrahtu, C.; Xi, X.; Liao, L.; Ren, J.; Xie, J.; Heeres, H.J.; Palkovits, R. Catalysts design for higher alcohols synthesis by CO2 hydrogenation: Trends and future perspectives. Appl. Catal. B Environ. 2021, 291, 120073. [Google Scholar] [CrossRef]
- Ye, R.-P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q.; et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 5698. [Google Scholar] [CrossRef] [PubMed]
- Sheerin, E.; Reddy, G.K.; Smirniotis, P. Evaluation of Rh/CexTi1−xO2 Catalysts for Synthesis of Oxygenates from Syngas Using XPS and TPR Techniques. Catal. Today 2016, 263, 75–83. [Google Scholar] [CrossRef]
- Huang, X.; Teschner, D.; Dimitrakopoulou, M.; Fedorov, A.; Frank, B.; Kraehnert, R.; Rosowski, F.; Kaiser, H.; Schunk, S.; Kuretschka, C. Atomic-Scale Observation of the Metal–Promoter Interaction in Rh-Based Syngas-Upgrading Catalysts. Angew. Chem. Int. Ed. 2019, 58, 8709–8713. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, J.; Qin, G.; Wang, L.; Zuidema, E.; Yang, Q.; Dang, S.; Yang, C.; Xiao, J.; Meng, X. Direct Conversion of Syngas to Ethanol within Zeolite Crystals. Chem 2020, 6, 646–657. [Google Scholar] [CrossRef]
- Zhu, C.; Xu, K.T.; Fang, Y.; Lv, Q.; Jiang, K.; Zhao, H.; Chen, Y.; Wang, P.; Yang, H.; Wu, L. Synergetic effect of Ce/Zr for ethanol synthesis from syngas over Rh-based catalyst. Fuel 2023, 334, 126770. [Google Scholar] [CrossRef]
- Inoue, T.; Iizuka, T.; Tanabe, K. Hydrogenation of carbon-dioxide and carbon-monoxide over supported rhodium catalysts under 10 bar pressure. Appl. Catal. 1989, 46, 1–9. [Google Scholar] [CrossRef]
- Bando, K.K.; Soga, K.; Kunimori, K.; Arakawa, H. Effect of Li additive on CO2 hydrogenation reactivity of zeolite supported Rh catalysts. Appl. Catal. A-Gen. 1998, 175, 67–81. [Google Scholar] [CrossRef]
- Kusama, H.; Okabe, K.; Sayama, K.; Arakawa, H. Ethanol synthesis by catalytic hydrogenation of CO2 over Rh-Fe/SiO2 catalysts. Energy 1997, 22, 343–348. [Google Scholar] [CrossRef]
- Wang, G.; Luo, R.; Yang, C.; Song, J.; Xiong, C.; Tian, H.; Zhao, Z.-J.; Mu, R.; Gong, J. Active sites in CO2 hydrogenation over confined VOx-Rh catalysts. Sci. China Chem. 2019, 62, 1710–1719. [Google Scholar] [CrossRef]
- Yang, C.; Mu, R.; Wang, G.; Song, J.; Tian, H.; Zhao, Z.-J.; Gong, J. Hydroxyl-mediated ethanol selectivity of CO2 hydrogenation. Chem. Sci. 2019, 10, 3161–3167. [Google Scholar] [CrossRef]
- Zheng, K.; Li, Y.; Liu, B.; Jiang, F.; Xu, Y.; Liu, X. Ti-doped CeO2 Stabilized Single-Atom Rhodium Catalyst for Selective and Stable CO2 Hydrogenation to Ethanol. Angew. Chem.-Int. Edit. 2022, 61, e202210991. [Google Scholar] [CrossRef]
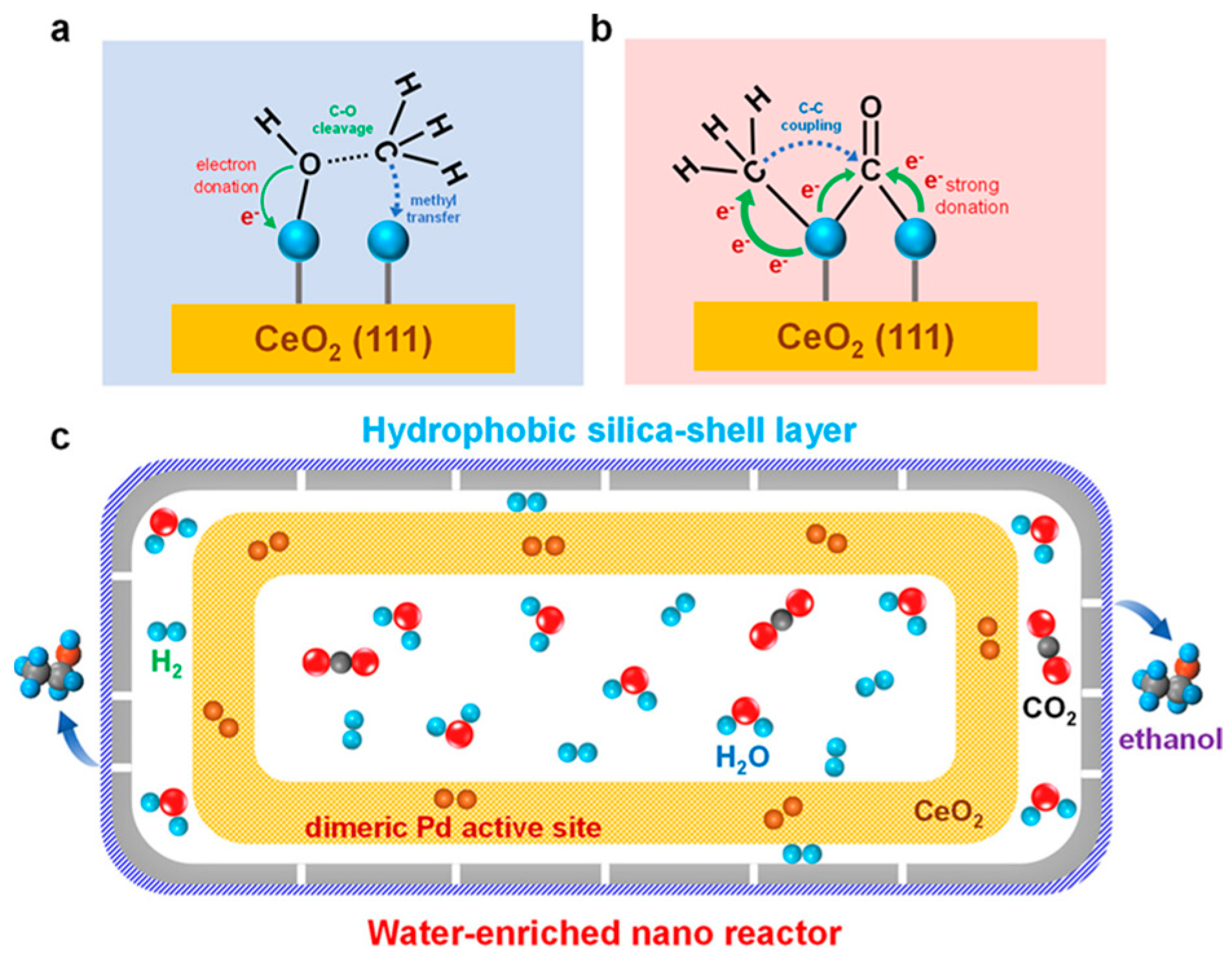
- Chen, J.; Zha, Y.; Liu, B.; Li, Y.; Xu, Y.; Liu, X. Rationally Designed Water Enriched Nano Reactor for Stable CO2 Hydrogenation with Near 100% Ethanol Selectivity over Diatomic Palladium Active Sites. ACS Catal. 2023, 13, 7110–7121. [Google Scholar] [CrossRef]
- Ouyang, B.; Xiong, S.; Zhang, Y.; Liu, B.; Li, J. The Study of Morphology Effect of Pt/Co3O4 Catalysts for Higher Alcohol Synthesis from CO2 Hydrogenation. Appl. Catal. A-Gen. 2017, 543, 189–195. [Google Scholar] [CrossRef]
- He, Z.; Qian, Q.; Zhang, Z.; Meng, Q.; Zhou, H.; Jiang, Z.; Han, B. Synthesis of Higher Alcohols from CO2 Hydrogenation over a PtRu/Fe2O3 Catalyst under Supercritical Condition. Philos. Trans. R. Soc. A 2015, 373, 20150006. [Google Scholar] [CrossRef]
- Xu, S.S.; Chansai, S.; Xu, S.J.; Stere, C.E.; Jiao, Y.L.; Yang, S.H.; Hardacre, C.; Fan, X.L. CO Poisoning of Ru Catalysts in CO2 Hydrogenation under Thermal and Plasma Conditions: A Combined Kinetic and Diffuse Reflectance Infrared Fourier Transform Spectroscopy−Mass Spectrometry Study. ACS Catal. 2020, 10, 12828–12840. [Google Scholar] [CrossRef]
- Guo, S.Y.; Liu, Y.C.; Murphy, E.; Ly, A.; Xu, M.J.; Matanovic, I.; Pan, X.Q.; Atanassov, P. Robust palladium hydride catalyst for electrocatalytic formate formation with high CO tolerance. Appl. Catal. B-Environ. 2022, 316, 121659. [Google Scholar] [CrossRef]
- Wang, P.; Chen, S.; Bai, Y.; Gao, X.; Li, X.; Sun, K.; Xie, H.; Yang, G.; Han, Y.; Tan, Y. Effect of the Promoter and Support on Cobalt-Based Catalysts for Higher Alcohols Synthesis through CO Hydrogenation. Fuel 2017, 195, 69–81. [Google Scholar] [CrossRef]
- Li, W.; Nie, X.; Jiang, X.; Zhang, A.; Ding, F.; Liu, M.; Liu, Z.; Guo, X.; Song, C. ZrO2 Support Imparts Superior Activity and Stability of Co Catalysts for CO2 Methanation. Appl. Catal. B-Environ. 2018, 220, 397–408. [Google Scholar] [CrossRef]
- Gnanamani, M.K.; Shafer, W.D.; Sparks, D.E.; Davis, B.H. Fischer–Tropsch Synthesis: Effect of CO2 Containing Syngas over Pt Promoted Co/γ-Al2O3 and K-Promoted Fe Catalysts. Catal. Commun. 2011, 12, 936–939. [Google Scholar] [CrossRef]
- Niu, J.; Liu, H.; Jin, Y.; Fan, B.; Qi, W.; Ran, J. Comprehensive Review of Cu-Based CO2 Hydrogenation to CH3OH: Insights from Experimental Work and Theoretical Analysis. Int. J. Hydrogen Energy 2022, 47, 9183–9200. [Google Scholar] [CrossRef]
- Gnanamani, M.K.; Jacobs, G.; Hamdeh, H.H.; Shafer, W.D.; Liu, F.; Hopps, S.D.; Thomas, G.A.; Davis, B.H. Hydrogenation of Carbon Dioxide over Co–Fe Bimetallic Catalysts. ACS Catal. 2016, 6, 913–927. [Google Scholar] [CrossRef]
- Liu, S.; Yang, C.; Zha, S.; Sharapa, D.; Studt, F.; Zhao, Z.J.; Gong, J. Moderate Surface Segregation Promotes Selective Ethanol Production in CO2 Hydrogenation Reaction over CoCu Catalysts. Angew. Chem. Int. Ed. 2022, 61, e202109027. [Google Scholar] [CrossRef]
- Okabe, K.; Yamada, H.; Hanaoka, T.; Matsuzaki, T.; Arakawa, H.; Abe, Y. CO2 hydrogenation to alcohols over highly dispersed Co/SiO2 catalysts derived from acetate. Chem. Lett. 2001, 30, 904–905. [Google Scholar] [CrossRef]
- Gnanamani, M.K.; Jacobs, G.; Keogh, R.A.; Shafer, W.D.; Sparks, D.E.; Hopps, S.D.; Thomas, G.A.; Davis, B.H. Fischer-Tropsch synthesis: Effect of pretreatment conditions of cobalt on activity and selectivity for hydrogenation of carbon dioxide. Appl. Catal. A-Gen. 2015, 499, 39–46. [Google Scholar] [CrossRef]
- Liu, B.; Ouyang, B.; Zhang, Y.; Lv, K.; Li, Q.; Ding, Y.; Li, J. Effects of mesoporous structure and Pt promoter on the activity of Co-based catalysts in low-temperature CO2 hydrogenation for higher alcohol synthesis. J. Catal. 2018, 366, 91–97. [Google Scholar] [CrossRef]
- An, K.; Zhang, S.; Wang, J.; Liu, Q.; Zhang, Z.; Liu, Y. A highly selective catalyst of Co/La4Ga2O9 for CO2 hydrogenation to ethanol. J. Energy Chem. 2021, 56, 486–495. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, Z.; Liu, X.; Shao, Z.; Xia, L.; Zhong, L.; Wang, H.; Sun, Y. Tuning the interaction between Na and Co2C to promote selective CO2 hydrogenation to ethanol. Appl. Catal. B-Environ. 2021, 293, 120207. [Google Scholar]
- Liu, S.; Zhou, H.; Zhang, L.; Ma, Z.; Wang, Y. Activated Carbon-Supported Mo-Co-K Sulfide Catalysts for Synthesizing Higher Alcohols from CO2. Chem. Eng. Technol. 2019, 42, 962–970. [Google Scholar] [CrossRef]
- Zhang, H.; Han, H.; Xiao, L.; Wu, W. Highly Selective Synthesis of Ethanol via CO2 Hydrogenation over CoMoCx Catalysts. Chemcatchem 2021, 13, 3333–3339. [Google Scholar] [CrossRef]
- Liao, F.; Huang, Y.; Ge, J.; Zheng, W.; Tedsree, K.; Collier, P.; Hong, X.; Tsang, S.C. Morphology-Dependent Interactions of ZnO with Cu Nanoparticles at the Materials’ Interface in Selective Hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2011, 50, 2162–2165. [Google Scholar] [CrossRef]
- Kattel, S.; Ramírez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Catalysis active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef]
- Larmier, K.; Liao, W.-C.; Tada, S.; Lam, E.; Verel, R.; Bansode, A.; Urakawa, A.; Comas-Vives, A.; Copéret, C. CO2-to-Methanol Hydrogenation on Zirconia-Supported Copper Nanoparticles: Reaction Intermediates and the Role of the Metal–Support Interface. Angew. Chem. Int. Ed. 2017, 56, 2318–2323. [Google Scholar] [CrossRef]
- Hong, Z.; Wang, J.R.; Gao, Z.H.; Huang, W. Tuning CuO crystallite size by different solvents for higher alcohols synthesis from syngas over CuZnAl catalyst. Int. J. Hydrogen Energy 2024, 56, 1032–1037. [Google Scholar] [CrossRef]
- Tian, M.; Tian, X.; Ma, E.J.; Bai, K.Y.; Zuo, Z.J.; Huang, W. Investigation of the role of oxygen vacancies in CuZn catalysts for the formation of higher alcohols from syngas. Fuel 2024, 360, 130595. [Google Scholar] [CrossRef]
- Anton, J.; Nebel, J.; Song, H.; Froese, C.; Weide, P.; Ruland, H.; Muhler, M.; Kaluza, S. Structure–Activity Relationships of Co-Modified Cu/ZnO/Al2O3 Catalysts Applied in the Synthesis of Higher Alcohols from Synthesis Gas. Appl. Catal. A-Gen. 2015, 505, 326–333. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, R.; Wang, B. Insight into the Preference Mechanism for CC Chain Formation of C2 Oxygenates and the Effect of Promoters in Syngas Conversion over Cu-Based Catalysts. Appl. Catal. A-Gen. 2013, 466, 77–89. [Google Scholar] [CrossRef]
- Gupta, M.; Smith, M.L.; Spivey, J.J. Heterogeneous Catalytic Conversion of Dry Syngas to Ethanol and Higher Alcohols on Cu-Based Catalysts. ACS Catal. 2011, 1, 641–656. [Google Scholar] [CrossRef]
- Luk, H.T.; Mondelli, C.; Ferre, D.C.; Stewart, J.A.; Perez-Ramirez, J. Status and prospects in higher alcohols synthesis from syngas. Chem. Soc. Rev. 2017, 46, 1358–1426. [Google Scholar] [CrossRef]
- Ding, L.; Shi, T.; Gu, J.; Cui, Y.; Zhang, Z.; Yang, C.; Chen, T.; Lin, M.; Wang, P.; Xue, N.; et al. CO2 Hydrogenation to Ethanol over Cu@Na-Beta. Chem 2020, 6, 2673–2689. [Google Scholar] [CrossRef]
- Iltsiou, D.; Mielby, J.; Kegnaes, S. Direct Conversion of CO2 into Alcohols Using Cu-Based Zeolite Catalysts. ChemPlusChem 2023, 89, e202300313. [Google Scholar] [CrossRef]
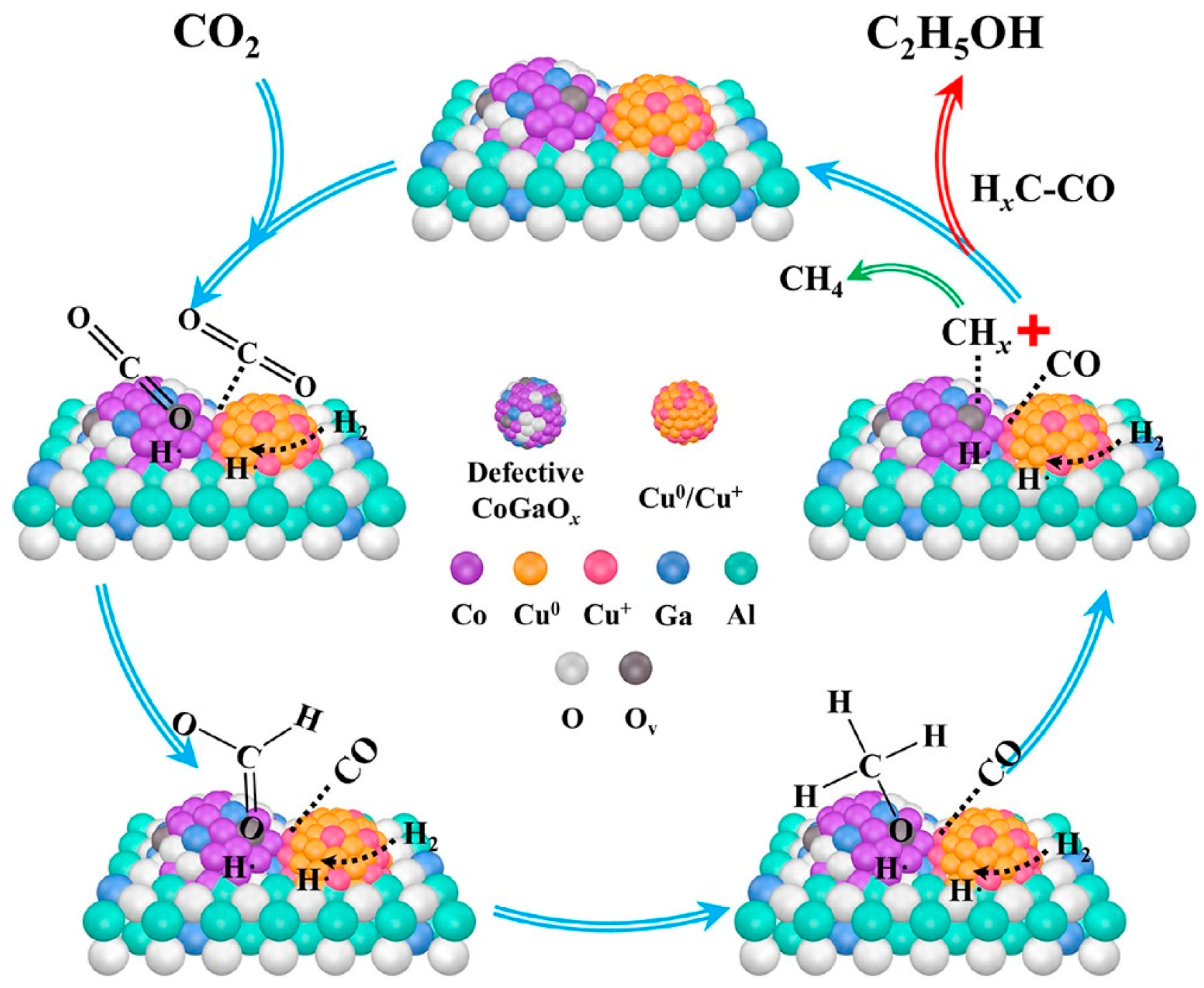
- Zhang, G.; Fan, G.; Zheng, L.; Li, F. Ga-Promoted CuCo-Based Catalysts for Efficient CO2 Hydrogenation to Ethanol: The Key Synergistic Role of Cu-CoGaOx Interfacial Sites. ACS Appl. Mater. Interfaces 2022, 14, 35569–35580. [Google Scholar] [CrossRef]
- Irshad, M.; Chun, H.-J.; Khan, M.K.; Jo, H.; Kim, S.K.; Kim, J. Synthesis of n-butanol-rich C3+ alcohols by direct CO2 hydrogenation over a stable Cu-Co tandem catalyst. Appl. Catal. B-Environ. 2024, 340, 123201. [Google Scholar]
- Xu, D.; Ding, M.; Hong, X.; Liu, G.; Tsang, S.C.E. Selective C2+ Alcohol Synthesis from Direct CO2 Hydrogenation over a Cs-Promoted Cu-Fe-Zn Catalyst. ACS Catal. 2020, 10, 5250–5260. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, S.; Geng, R.; Wang, P.; Dong, M.; Wang, J.; Fan, W. Hydrogenation of CO2 to higher alcohols on an efficient Cr-modified CuFe catalyst. Appl. Catal. B-Environ. 2023, 337, 123013. [Google Scholar]
- Si, Z.; Wang, L.; Han, Y.; Yu, J.; Ge, Q.; Zeng, C.; Sun, J. Synthesis of Alkene and Ethanol in CO2 Hydrogenation on a Highly Active Sputtering CuNaFe Catalyst. ACS Sustain. Chem. Eng. 2022, 10, 14972–14979. [Google Scholar] [CrossRef]
- He, Z.; Qian, Q.; Ma, J.; Meng, Q.; Zhou, H.; Song, J.; Liu, Z.; Han, B. Water-Enhanced Synthesis of Higher Alcohols from CO2 Hydrogenation over a Pt/Co3O4 Catalyst under Milder Conditions. Angew. Chem.-Int. Ed. 2016, 55, 737–741. [Google Scholar] [CrossRef]
- Chang, Q.; Li, J.; Suo, H.; Qing, M.; Wang, H.; Zhang, C.; Wen, X.; Xiang, H.; Yang, Y.; Li, Y. Unravelling the formation of Fe2SiO4 on Fischer-Tropsch Fe/SiO2 catalyst. Catal. Today 2024, 431, 114605. [Google Scholar] [CrossRef]
- Raghav, H.; Pendem, C.; Tripathi, S.; Kumar, S.; Sarkar, B. Enhanced light olefin production from CO2 over potassium promoted Fe-Co bimetallic ZrO2 supported catalysts. Fuel 2024, 368, 131645. [Google Scholar] [CrossRef]
- Jung, W.; Rhim, G.B.; Kim, K.Y.; Youn, M.H.; Chun, D.H.; Lee, J. Comprehensive analysis of syngas-derived Fischer–Tropsch synthesis using iron-based catalysts with varied acidities. Chem. Eng. J. 2024, 484, 149408. [Google Scholar] [CrossRef]
- Wu, K.; Zhang, Z.; Shan, R. Encapsulating Fischer-Tropsch synthesis catalyst with porous graphite-carbon enables ultrahigh activity for syngas to α-olefins. Appl. Catal. B Environ. Energy 2024, 353, 124067. [Google Scholar] [CrossRef]
- Ngo, X.D.; Phung, T.L.H.; Nguyen, N.H.; Nguyen, T.A.; Vinh, N.T.; Quy, N.V.; Le, A.T. A Physical Insight of Phase-Dependent Electron Transfer Kinetic Behaviors and Electrocatalytic Activity of an Iron Oxide-Based Sensing Nanoplatform towards Chloramphenicol. J. Electrochem. Soc. 2024, 171, 056503. [Google Scholar] [CrossRef]
- Aitbekova, A.; Goodman, E.D.; Wu, L.; Boubnov, A.; Hoffman, A.S.; Genc, A.; Cheng, H.; Casalena, L.; Bare, S.R.; Cargnello, M. Engineering of Ruthenium-Iron Oxide Colloidal Heterostructures: Improved Yields in CO2 Hydrogenation to Hydrocarbons. Angew. Chem.-Int. Ed. 2019, 58, 17451–17457. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, M.; Rodemerck, U.; Schneider, M.; Broering, M.; Baabe, D.; Kondratenko, E.V. Unexpectedly efficient CO2 hydrogenation to higher hydrocarbons over non-doped Fe2O3. Appl. Catal. B-Environ. 2017, 204, 119–126. [Google Scholar] [CrossRef]
- Jiang, J.; Wen, C.; Tian, Z.; Wang, Y.; Zhai, Y.; Chen, L.; Li, Y.; Liu, Q.; Wang, C.; Ma, L. Manganese-Promoted Fe3O4 Microsphere for Efficient Conversion of CO2 to Light Olefins. Ind. Eng. Chem. Res. 2020, 59, 2155–2162. [Google Scholar] [CrossRef]
- Kasipandi, S.; Bae, J.W. Recent Advances in Direct Synthesis of Value-Added Aromatic Chemicals from Syngas by Cascade Reactions over Bifunctional Catalysts. Adv. Mater. 2019, 31, e1803390. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Wu, B.; Yu, Y.; Liu, N.; Niu, Q.; Li, C.; Wei, J.; Ge, Q. Regulating the electronic property of iron catalysts for higher alcohols synthesis from CO2 hydrogenation. Appl. Catal. B Environ. Energy 2024, 355, 124159. [Google Scholar] [CrossRef]
- Yao, R.; Wei, J.; Ge, Q.; Xu, J.; Han, Y.; Ma, Q.; Xu, H.; Sun, J. Monometallic iron catalysts with synergistic Na and S for higher alcohols synthesis via CO2 hydrogenation. Appl. Catal. B-Environ. 2021, 298, 120556. [Google Scholar] [CrossRef]
- Lu, F.; Chen, X.; Wang, W.; Zhang, Y. Adjusting the CO2 hydrogenation pathway via the synergic effects of iron carbides and iron oxides. Catal. Sci. Technol. 2021, 11, 7694–7703. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, G.; Zhu, J.; Wang, M.; Ding, F.; Song, C.; Guo, X. Boosting the production of higher alcohols from CO2 and H2 over Mn-and K-modified iron carbide. Ind. Eng. Chem. Res. 2022, 21, 61. [Google Scholar] [CrossRef]
- Xi, X.; Zeng, F.; Zhang, H. CO2 Hydrogenation to Higher Alcohols over K-Promoted Bimetallic Fe-In Catalysts on a Ce-ZrO2 Support. ACS Sustain. Chem. Eng. 2021, 9, 6235–6249. [Google Scholar] [CrossRef]
- Goud, D.; Churipard, S.R.; Bagchi, D.; Singh, A.K.; Riyaz, M.; Vinod, C.P.; Peter, S.C. Strain-Enhanced Phase Transformation of Iron Oxide for Higher Alcohol Production from CO2. ACS Catal. 2022, 12, 11118–11128. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Zhou, Y.; Zhang, X.X.; Wang, M.R.; Liu, T.K.; Wei, J.X.; Zhang, G.H.; Hong, X.L.; Liu, G.L. PdFe Alloy-Fe5C2 interfaces for efficient CO2 hydrogenation to higher alcohols. Appl. Catal. B Environ. Energy 2024, 345, 123691. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; He, R.; Li, M.; Zhang, J.; Cao, F.; Liu, J.; Lin, S.; Gao, X.; Yang, G.; et al. Carbon-Based Electron Buffer Layer on ZnOx-Fe5C2-Fe3O4 Boosts Ethanol Synthesis from CO2 Hydrogenation. Angew. Chem.-Int. Ed. 2023, 62, e202311786. [Google Scholar] [CrossRef]
- Chernyak, S.A.; Corda, M.; Marinova, M.; Safonova, O.V.; Kondratenko, V.A.; Kondratenko, E.V.; Kolyagin, Y.G.; Cheng, K.; Ordomsky, V.V.; Khodakov, A.Y. Decisive influence of SAPO-34 zeolite on light olefin selectivity in methanol-meditated CO2 hdrogenation over metal oxide-zeolite catalysts. ACS Catal. 2023, 13, 14627–14638. [Google Scholar] [CrossRef]
- Chen, S.; Wang, J.; Feng, Z.; Jiang, Y.; Hu, H.; Qu, Y.; Tang, S.; Li, Z.; Liu, J.; Wang, J. Hydrogenation of CO2 to Light Olefins over ZnZrOx /SSZ-13. Angew. Chem. Int. Ed. 2024, 63, e202316874. [Google Scholar] [CrossRef]
- Ma, K.; Zhao, S.; Dou, M.; Ma, X.; Dai, C. Enhancing the stability of methanol-to-olefins reaction catalyzed by SAPO-34 zeolite in the presence of CO2 and oxygen-vacancy-rich ZnCeZrOx. ACS Catal. 2024, 14, 594–607. [Google Scholar] [CrossRef]
- Zhang, Z.; Yin, H.; Yu, G.; He, S.; Kang, J.; Liu, Z.; Cheng, K.; Zhang, Q.; Wang, Y. Selective Hydrogenation of CO2 and CO into Olefins over Sodium-and Zinc-Promoted Iron Carbide Catalysts. J. Catal. 2021, 395, 350–361. [Google Scholar] [CrossRef]
- Li, Z.; Qu, Y.; Wang, J.; Liu, H.; Li, M.; Miao, S.; Li, C. Highly selective conversion of carbon dioxide to aromatics over tandem catalysts. Joule 2019, 3, 570–583. [Google Scholar] [CrossRef]
- Wei, J.; Yao, R.; Han, Y.; Ge, Q.; Sun, J. Towards the development of the emerging process of CO2 heterogenous hydrogenation into high-value unsaturated heavy hydrocarbons. Chem. Soc. Rev. 2021, 50, 10764–10805. [Google Scholar] [CrossRef]
- Al-Qadri, A.A.; Nasser, G.A.; Adamu, H.; Muraza, O.; Saleh, T.A. CO2 Utilization in Syngas Conversion to Dimethyl Ether and Aromatics: Roles and Challenges of Zeolites-Based Catalysts. J. Energy Chem. 2023, 79, 418–449. [Google Scholar] [CrossRef]
- Wen, C.; Xu, X.; Song, X.; Lu, L.; Zhuang, X.; Jin, K.; Jiang, Q.; Zhang, X.; Chen, L.; Wang, C.; et al. Selective CO2 hydrogenation to light aromatics over the Cu-modified Fe-based/ZSM-5 catalyst system. Energy Fuel 2023, 37, 518–528. [Google Scholar] [CrossRef]
- Liu, B.; Wang, Y.; Xie, Y.; Xiao, L.; Wang, W.; Wu, W. yIn2O3 ZnZrOx/hierarchical ZSM-5 tandem catalysts for CO2 hydrogenation to aromatics rich in tetramethylbenzene. ACS Sustain. Resour. Manag. 2023, 11, 17340–17354. [Google Scholar] [CrossRef]
- Uslamin, E.; Kosinov, N.; Filonenko, G.; Mezari, B.; Pidko, E.; Hensen, E. Co-Aromatization of Furan and Methanol over ZSM-5—A Pathway to Bio-Aromatics. ACS Catal. 2019, 9, 8547–8554. [Google Scholar] [CrossRef]
- Gao, W.; Guo, L.; Wu, Q.; Wang, C.; Guo, X.; He, Y.; Zhang, P.; Yang, G.; Liu, G.; Wu, J. Capsule-like Zeolite Catalyst Fabricated by Solvent-Free Strategy for Para-Xylene Formation from CO2 Hydrogenation. Appl. Catal. B-Environ. 2022, 303, 120906. [Google Scholar]
- Zhang, L.; Gao, W.; Wang, F.; Wang, C.; Liang, J.; Guo, X.; He, Y.; Yang, G.; Tsubaki, N. Highly Selective Synthesis of Light Aromatics from CO2 by Chromium-Doped ZrO2 Aerogels in Tandem with HZSM-5@SiO2 Catalyst. Appl. Catal. B-Environ. 2023, 328, 122535. [Google Scholar] [CrossRef]
- Cheng, K.; Zhou, W.; Kang, J.; He, S.; Wang, Y. Bifunctional Catalysts for One-Step Conversion of Syngas into Aromatics with Excellent Selectivity and Stability. Chem 2017, 3, 334–347. [Google Scholar] [CrossRef]
- Rudolph, M.; Isbrücker, P.; Schomäcker, R. Bifunctional Catalysts for the Conversion of CO2 into Value-Added Products—Distance as a Design Parameter for New Catalysts. Catal. Sci. Technol. 2023, 13, 3469–3482. [Google Scholar] [CrossRef]
- Kang, J.; He, S.; Zhou, W.; Shen, Z.; Li, Y.; Chen, M.; Zhang, Q.; Wang, Y. Single-pass transformation of syngas into ethanol with high selectivity by triple tandem catalysis. Nat. Commun. 2020, 11, 827. [Google Scholar] [CrossRef]
- Yamamoto, T.; Inui, T. Highly effective synthesis of ethanol from CO2 on Fe, Cu-based novel catalysts. In Advances in Chemical Conversions for Mitigating Carbon Dioxide; Inui, T., Anpo, M., Izui, K., Yanagida, S., Yamaguchi, T., Eds.; Elsevier Science: Amsterdam, The Netherlands, 1998; Volume 114, pp. 513–516. [Google Scholar]
- Guo, H.; Li, S.; Peng, F.; Zhang, H.; Xiong, L.; Huang, C.; Wang, C.; Chen, X. Roles Investigation of Promoters in K/Cu-Zn Catalyst and Higher Alcohols Synthesis from CO2 Hydrogenation over a Novel Two-Stage Bed Catalyst Combination System. Catal. Lett. 2015, 145, 620–630. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, K.; Zhang, B.; Peng, X.; Gao, X.; Yang, G.; Hu, H.; Wu, M.; Tsubaki, N. Direct Conversion of CO2 to Ethanol Boosted by Intimacy-Sensitive Multifunctional Catalysts. ACS Catal. 2021, 11, 11742–11753. [Google Scholar] [CrossRef]
- Xu, D.; Yang, H.; Hong, X.; Liu, G.; Tsang, S.C.E. Tandem Catalysis of Direct CO2 Hydrogenation to Higher Alcohols. ACS Catal. 2021, 11, 8978–8984. [Google Scholar] [CrossRef]
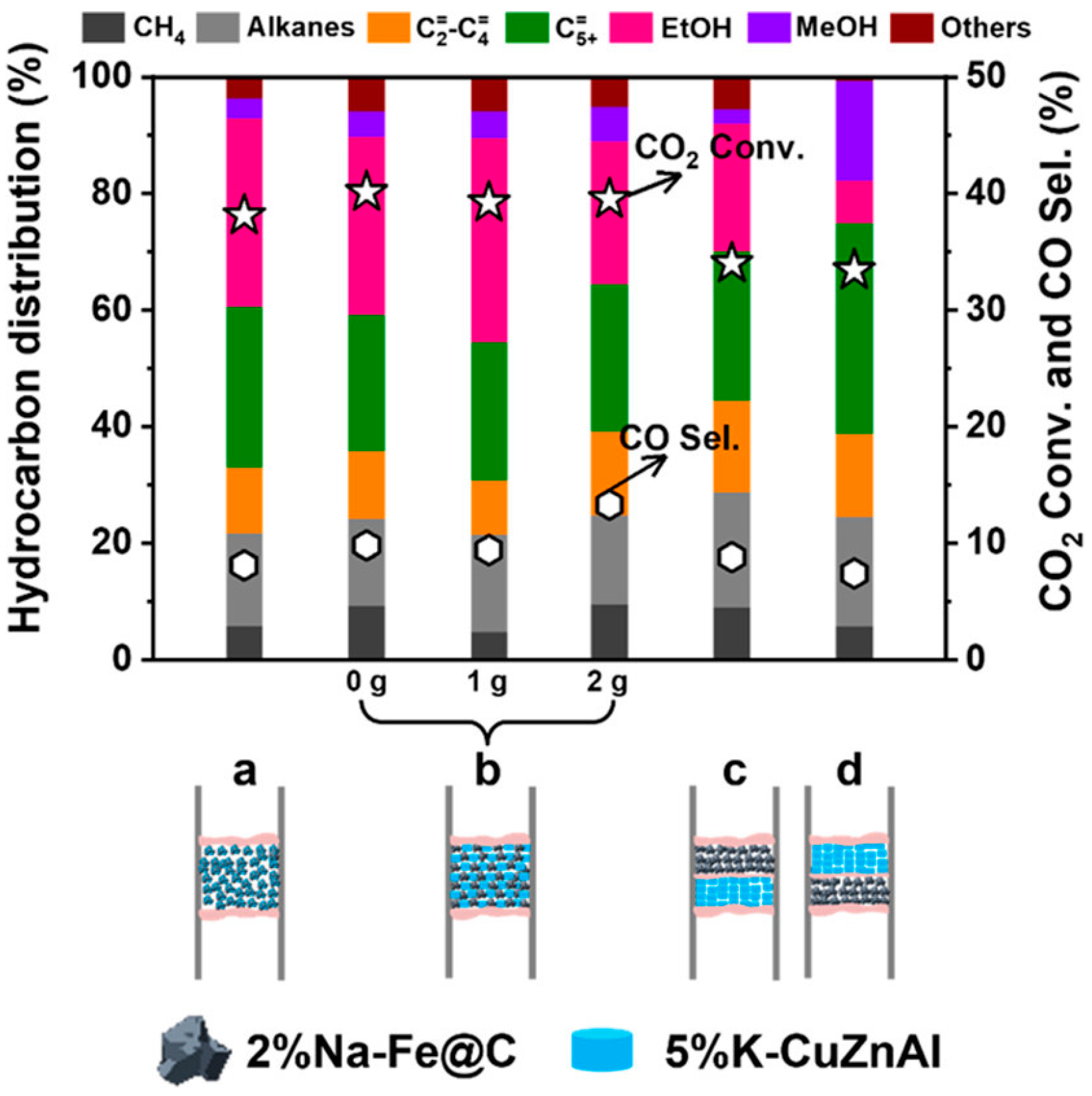
- Huang, J.; Zhang, G.; Wang, M.; Zhu, J.; Ding, F.; Song, C.; Guo, X. The synthesis of higher alcohols from CO2 hydrogenation over Mn-Cu-K modified Fe5C2 and CuZnAlZr tandem catalysts. Front. Energy Res. 2023, 10, 995800. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, D.; Zhang, X.; Hong, X.; Liu, G. Selective C2+ alcohol synthesis by CO2 hydrogenation via a reaction-coupling strategy. Catal. Sci. Technol. 2022, 12, 1539–1550. [Google Scholar] [CrossRef]
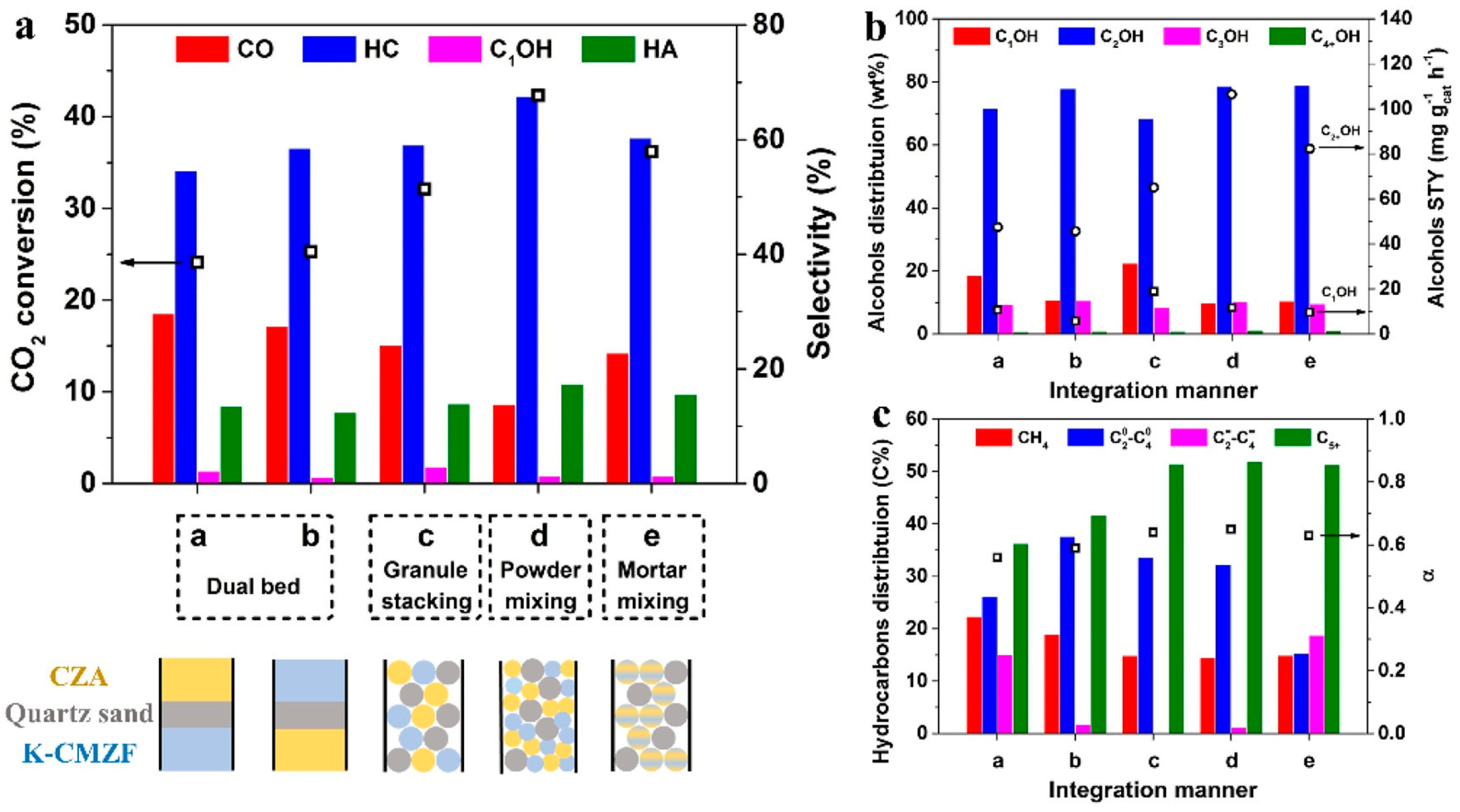
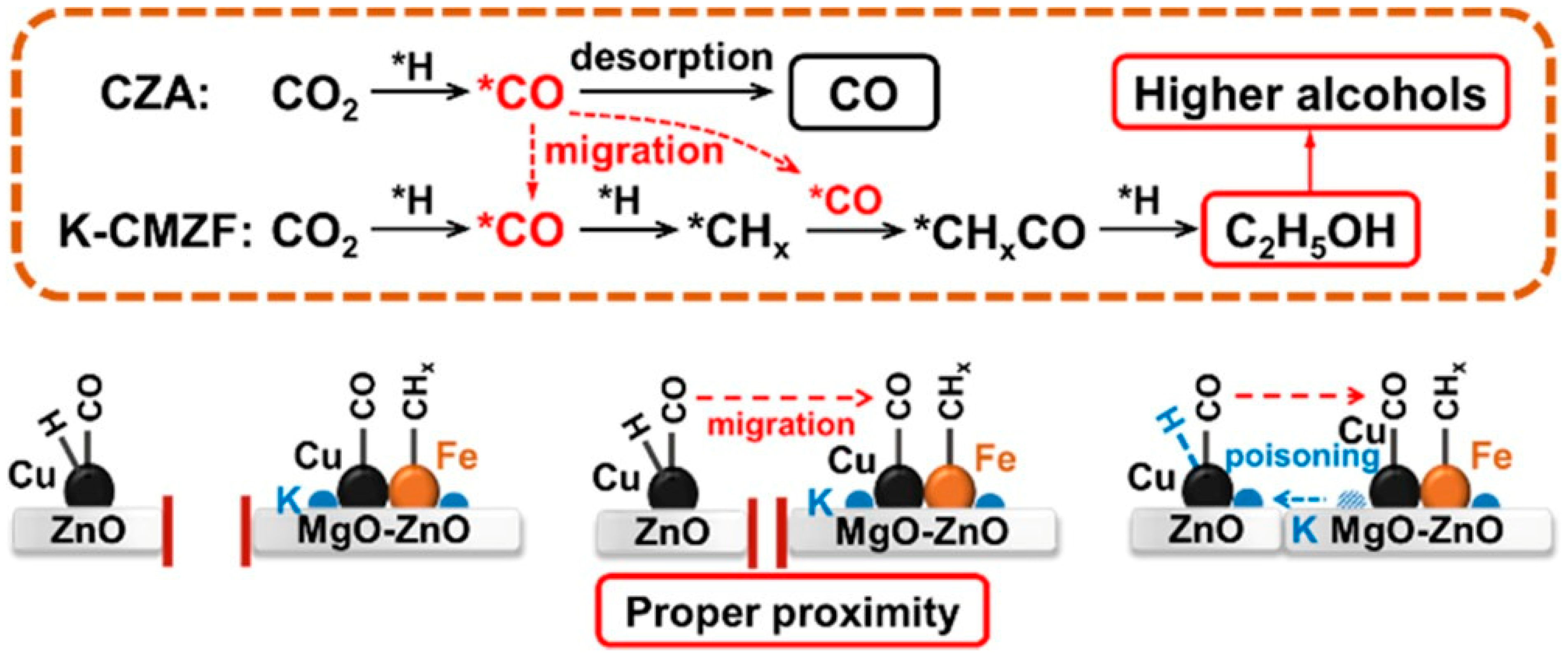
- Zhang, Q.; Wang, S.; Shi, X.R.; Dong, M.; Chen, J.G.; Zhang, J.; Wang, J.G.; Fan, W.B. Conversion of CO2 to higher alcohols on K-CuZnAl/Zr-CuFe composite. Appl. Catal. B Environ. Energy 2024, 346, 123748. [Google Scholar] [CrossRef]
- Zhang, S.; Huang, C.; Shao, Z.; Zhou, H.; Chen, J.; Li, L.; Lu, J.; Liu, X.; Luo, H.; Xia, L.; et al. Revealing and Regulating the Complex Reaction Mechanism of CO2 Hydrogenation to Higher Alcohols on Multifunctional Tandem Catalysts. ACS Catal. 2023, 13, 3055–3065. [Google Scholar] [CrossRef]
- Wang, X.; Ramirez, P.J.; Liao, W.; Rodriguez, J.A.; Liu, P. Cesium-Induced Active Sites for C-C Coupling and Ethanol Synthesis from CO2 Hydrogenation on Cu/ZnO(000(1)over-bar) Surfaces. J. Am. Chem. Soc. 2021, 143, 13103–13112. [Google Scholar] [CrossRef]
- Cui, M.; Qian, Q.L.; He, Z.H.; Zhang, Z.F.; Ma, J.; Wu, T.B.; Yang, G.Y.; Han, B.X. Bromide promoted hydrogenation of CO2 to higher alcohols using Ru-Co homogeneous catalyst. Chem. Sci. 2016, 7, 5200–5205. [Google Scholar] [CrossRef]
- Graciani, J.; Grinter, D.C.; Ramirez, P.J.; Palomino, R.M.; Xu, F.; Waluyo, I.; Stacchiola, D.; Sanz, J.F.; Senanayake, S.D.; Rodriguze, J.A. Conversion of CO2 to Methanol and Ethanol on Pt/CeOx/TiO2(110): Enabling Role of Water in C-C Bond Formation. ACS Catal. 2023, 12, 15097–15109. [Google Scholar] [CrossRef]
- Chao, S.; Kim, C.; Kim, J. Techno-economic assessment and early-stage screening of CO2 direct hydrogenation catalysts for methanol production using knowledge-based surrogate modeling. Energy Convers. Manag. 2021, 244, 114477. [Google Scholar] [CrossRef]
- Dutta, A.; Hensley, J.; Bain, R.; Magrini, K.; Tan, E.; Apanel, G.; Barton, D.; Groenendijk, P.; Ferrari, D.; Jablonski, W.; et al. Technoeconomic analysis for the production of mixed alcohols via indirect gasification of biomass based on demonstration experiments. Ind. Eng. Chem. Res. 2014, 53, 12149–12159. [Google Scholar] [CrossRef]
- Dutta, A.; Bain, R.L.; Biddy, M.J. Techno-economics of the production of mixed alcohols from lignocellulosic biomass via high-temperature gasification. Environ. Prog. Sustain. Energy 2010, 29, 163–174. [Google Scholar] [CrossRef]
- Liu, T.; Xu, D.; Song, M.; Hong, X.; Liu, G. K-ZrO2 Interfaces Boost CO2 Hydrogenation to Higher Alcohols. ACS Catal. 2023, 13, 4667–4674. [Google Scholar] [CrossRef]



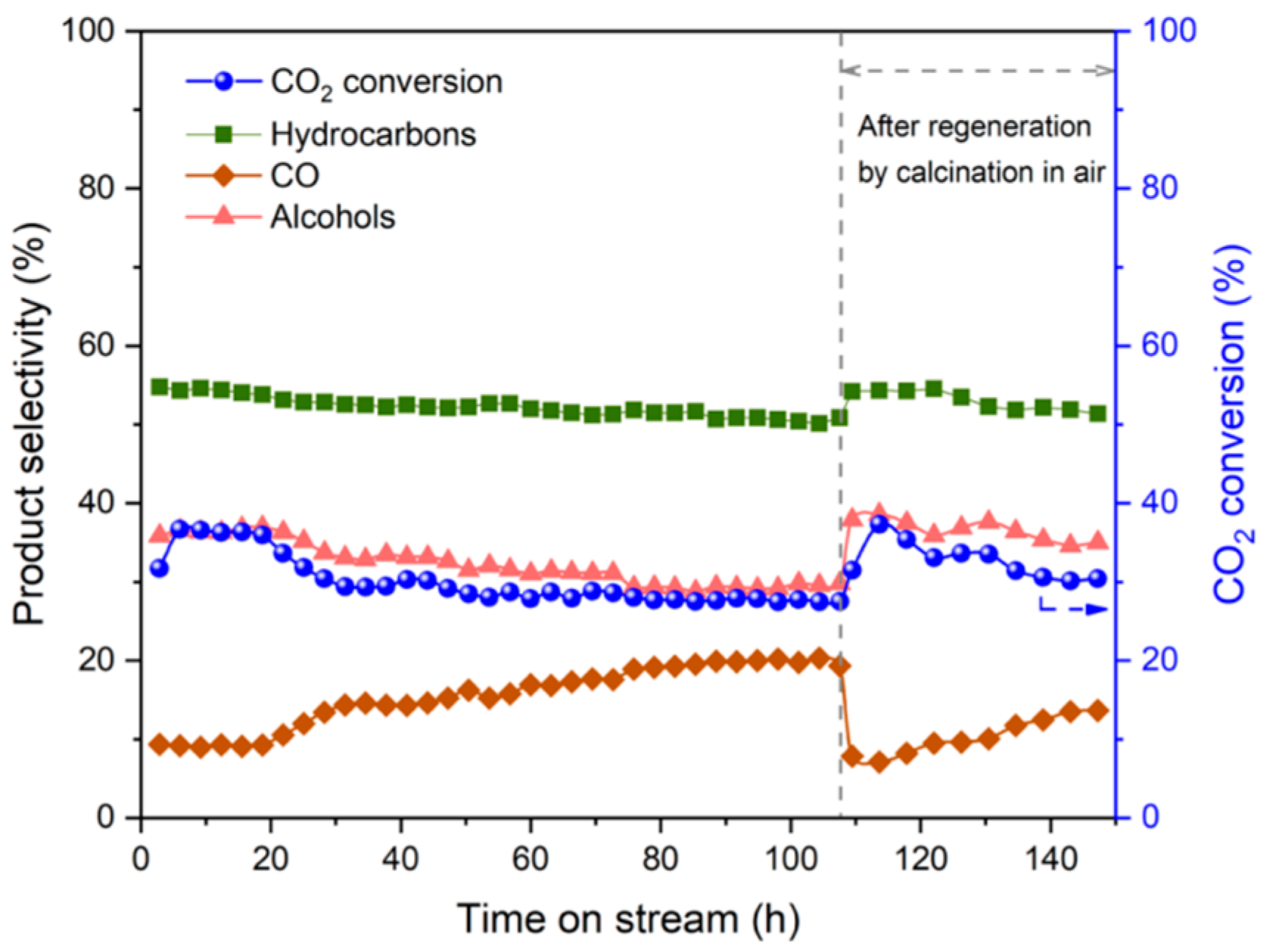
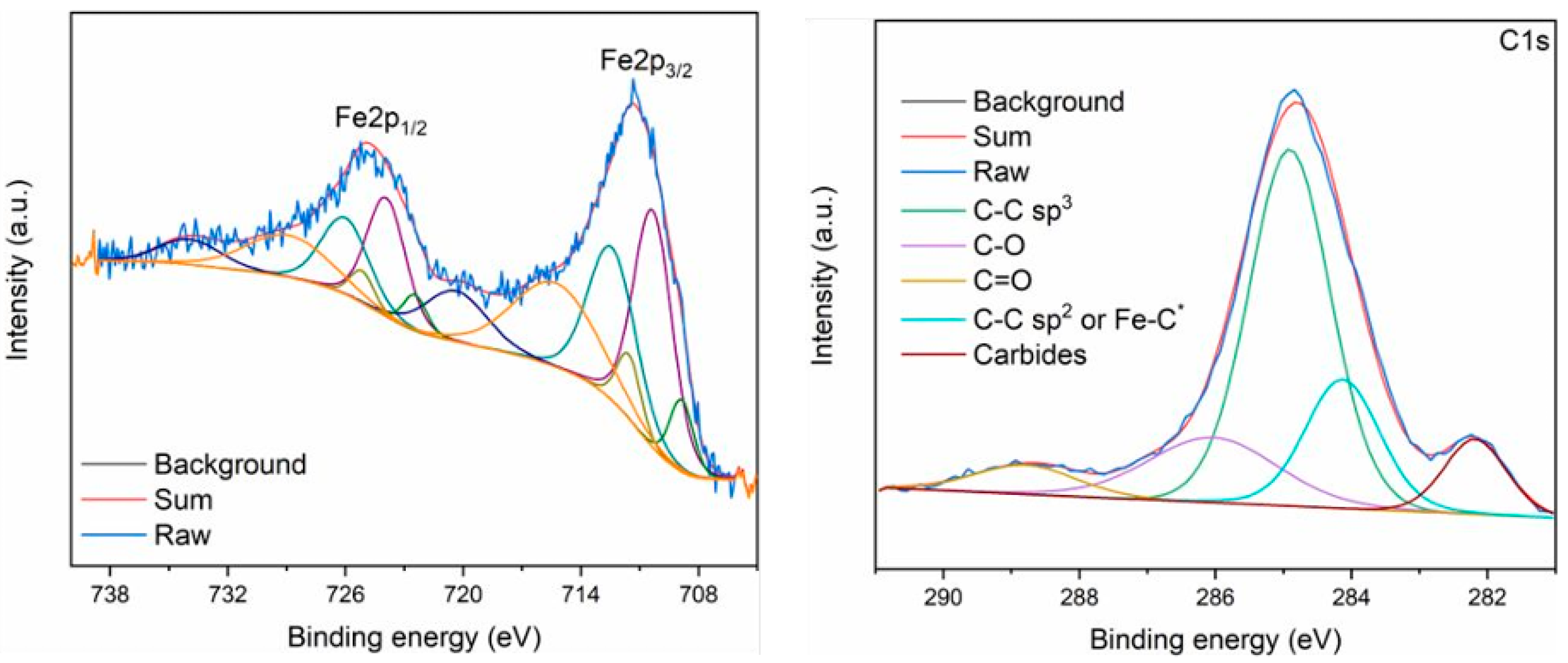
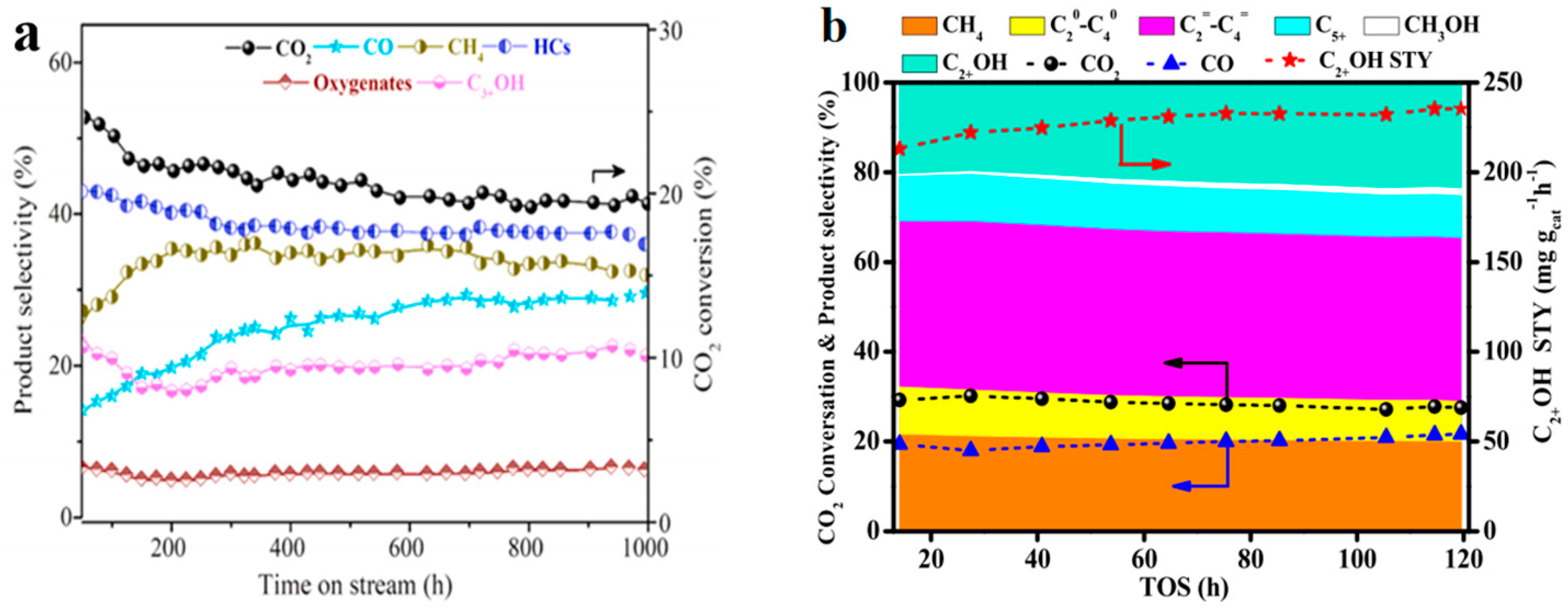
| Reaction Equation | ΔG298K (kJ/mol) | ΔH298K (kJ/mol) | K298K |
|---|---|---|---|
| CO2 + H2 ↔ CO + H2O | 28.6 | 41.1 | 9.67 × 10−6 |
| CO2 + 4H2 ↔ CH4 + 2H2O | −113.5 | −165.0 | 7.79 × 1019 |
| n CO2 + (3n + 1) H2 ↔ CnH2n+2 + 2n H2O | / | / | / |
| n CO2 + 3n H2 ↔ CnH2n + 2n H2O | / | / | / |
| CO2 +3H2 ↔ CH3OH + H2O | 3.5 | −49.3 | 2.45 × 10−1 |
| 2CO2 + 6H2 ↔ C2H5OH +3H2O | −32.4 | −86.7 | 4.70 × 105 |
| n CO2 + 3n H2 ↔ CnH2n+1OH + (2n − 1) H2O | / | / | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Liu, J.; Chen, X.; Gu, S.; Wei, Y.; Wang, L.; Wan, H.; Guan, G. Development of Multifunctional Catalysts for the Direct Hydrogenation of Carbon Dioxide to Higher Alcohols. Molecules 2024, 29, 2666. https://doi.org/10.3390/molecules29112666
Chen Y, Liu J, Chen X, Gu S, Wei Y, Wang L, Wan H, Guan G. Development of Multifunctional Catalysts for the Direct Hydrogenation of Carbon Dioxide to Higher Alcohols. Molecules. 2024; 29(11):2666. https://doi.org/10.3390/molecules29112666
Chicago/Turabian StyleChen, Yun, Jinzhao Liu, Xinyu Chen, Siyao Gu, Yibin Wei, Lei Wang, Hui Wan, and Guofeng Guan. 2024. "Development of Multifunctional Catalysts for the Direct Hydrogenation of Carbon Dioxide to Higher Alcohols" Molecules 29, no. 11: 2666. https://doi.org/10.3390/molecules29112666
APA StyleChen, Y., Liu, J., Chen, X., Gu, S., Wei, Y., Wang, L., Wan, H., & Guan, G. (2024). Development of Multifunctional Catalysts for the Direct Hydrogenation of Carbon Dioxide to Higher Alcohols. Molecules, 29(11), 2666. https://doi.org/10.3390/molecules29112666