

2-Aryladenine Derivatives as a Potent Scaffold for Adenosine Receptor Antagonists: The 6-Morpholino Derivatives

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results and Discussion
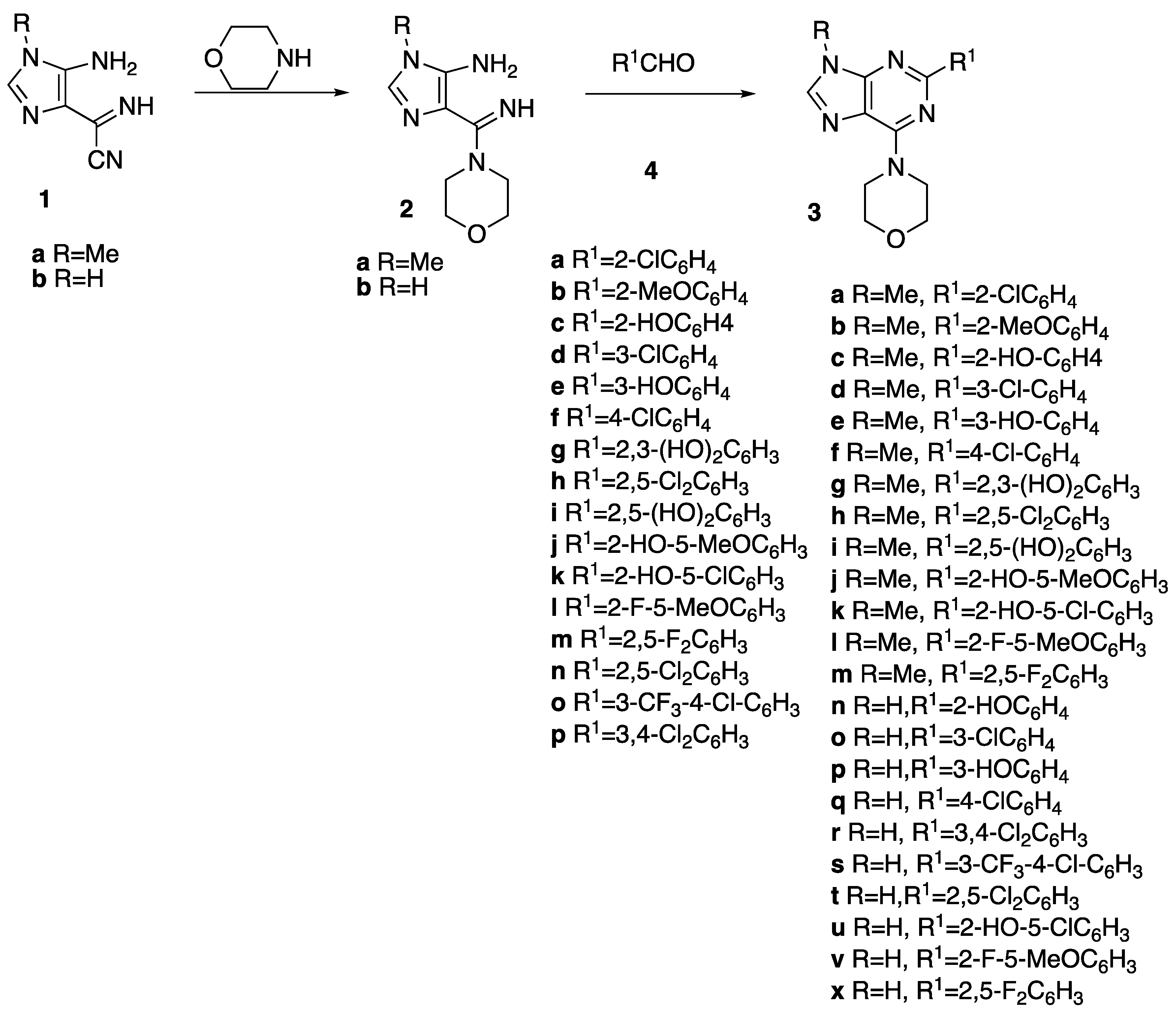
2.1. Chemistry
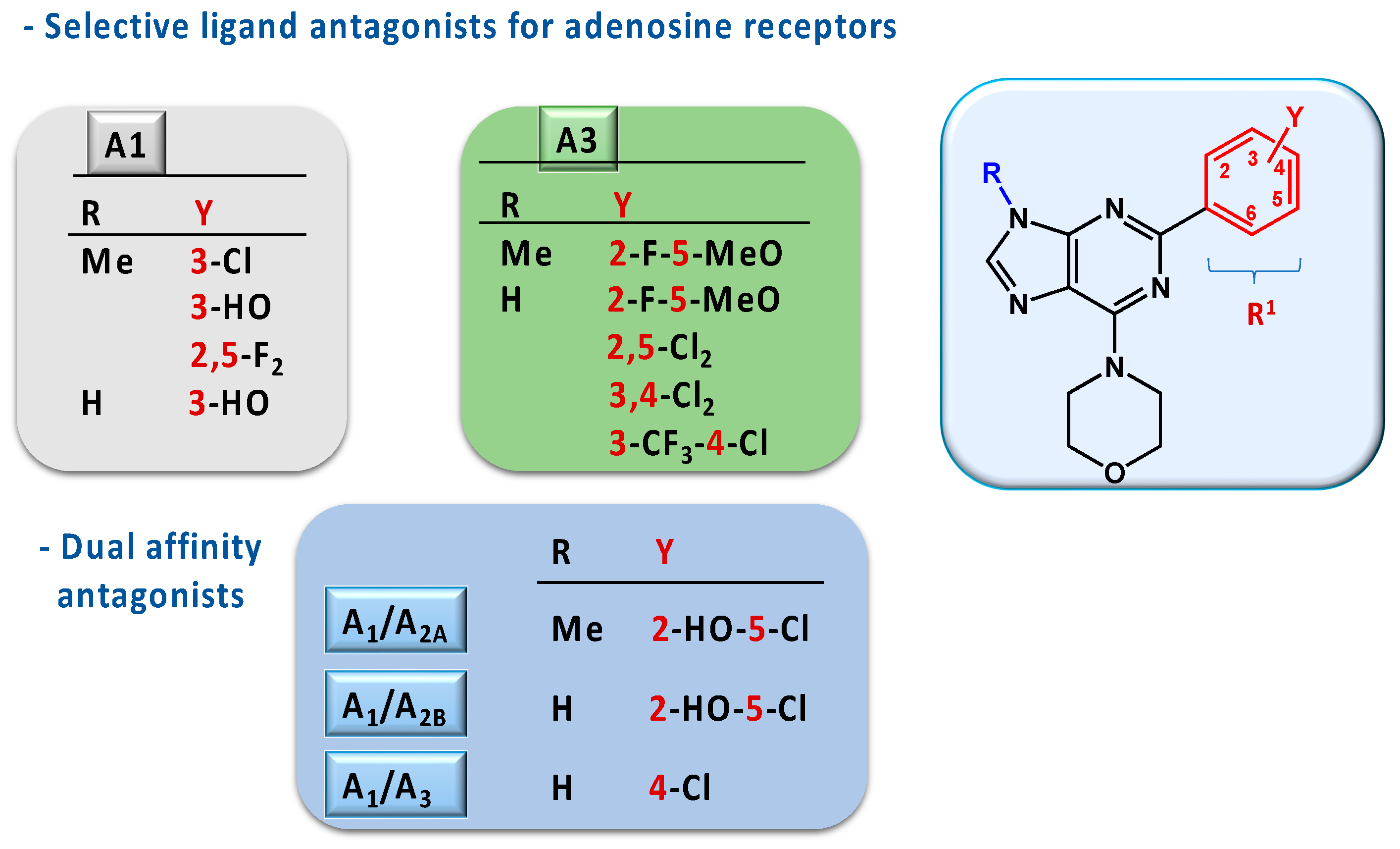
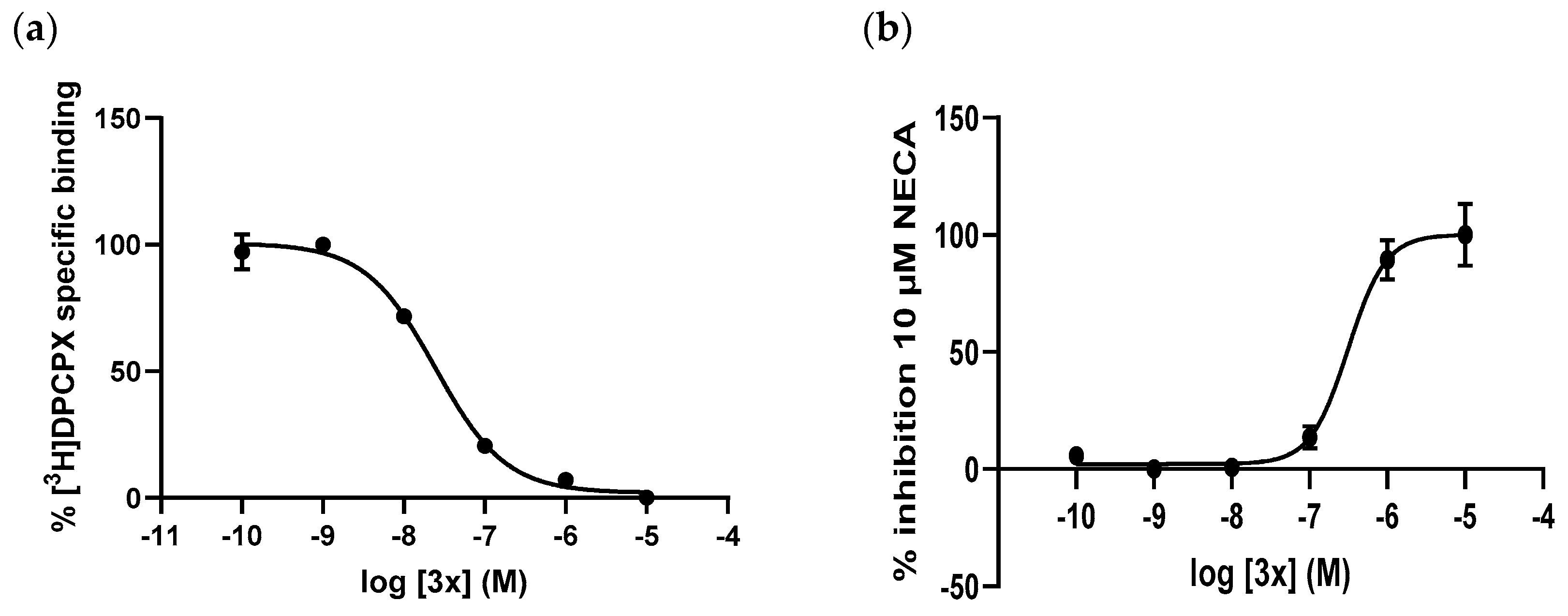
2.2. Pharmacology
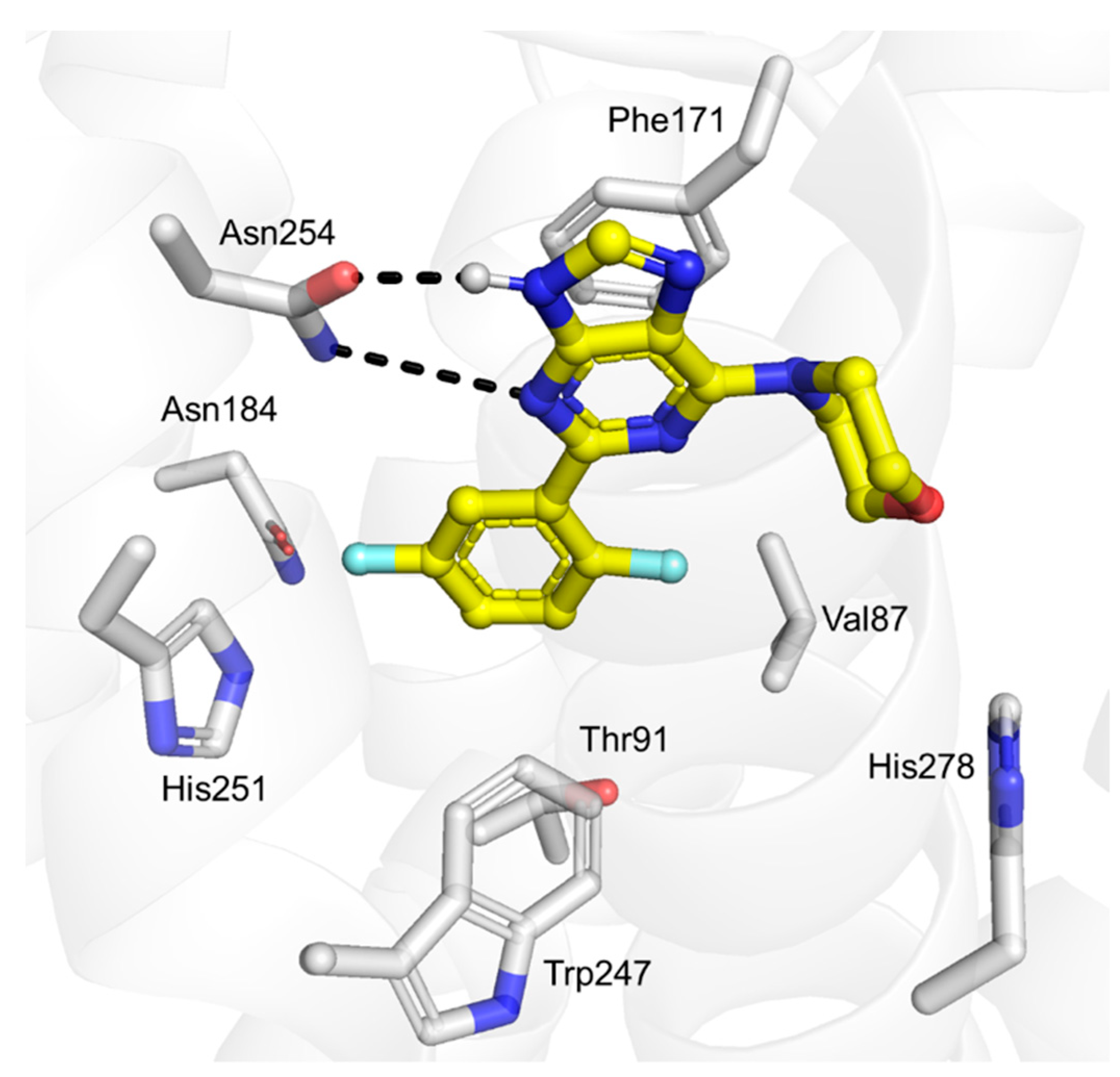
2.3. Docking Studies
3. Experimental Protocols
3.1. Chemistry
3.2. General Procedure for the Synthesis of 3a–x
3.2.1. 4-(2-(2-Chlorophenyl)-9-methyl-9H-purin-6-yl)morpholine 3a
3.2.2. 4-(2-(2-Methoxyphenyl)-9-methyl-9H-purin-6-yl)morpholine 3b
3.2.3. 2-(9-Methyl-6-morpholino-9H-purin-2-yl)phenol 3c
3.2.4. 4-(2-(3-Chlorophenyl)-9-methyl-9H-purin-6-yl)morpholine 3d
3.2.5. 3-(9-Methyl-6-morpholino-9H-purin-2-yl)phenol 3e
3.2.6. 4-(2-(4-Chlorophenyl)-9-methyl-9H-purin-6-yl)morpholine 3f
3.2.7. 3-(9-Methyl-6-morpholino-9H-purin-2-yl)benzene-1,2-diol 3g
3.2.8. 4-(2-(2,5-Dichlorophenyl)-9-methyl-9H-purin-6-yl)morpholine 3h
3.2.9. 2-(9-Methyl-6-morpholino-9H-purin-2-yl)benzene-1,4-diol 3i
3.2.10. 4-Methoxy-2-(9-methyl-6-morpholino-9H-purin-2-yl)phenol 3j
3.2.11. 4-Chloro-2-(9-methyl-6-morpholino-9H-purin-2-yl)phenol 3k
3.2.12. 4-(2-(2-Fluoro-5-methoxyphenyl)-9-methyl-9H-purin-6-yl)morpholine 3l
3.2.13. 4-(2-(2,5-Difluorophenyl)-9-methyl-9H-purin-6-yl)morpholine 3m
3.2.14. 2-(6-Morpholino-9H-purin-2-yl)phenol 3n
3.2.15. 4-(2-(3-Chlorophenyl)-9H-purin-6-yl)morpholine 3o
3.2.16. 3-(6-Morpholino-9H-purin-2-yl)phenol 3p
3.2.17. 4-(2-(4-Chlorophenyl)-9H-purin-6-yl)morpholine 3q
3.2.18. 4-(2-(3,4-Dichlorophenyl)-9H-purin-6-yl)morpholine 3r
3.2.19. 4-(2-(4-Chloro-3-(trifluoromethyl)phenyl)-9H-purin-6-yl)morpholine 3s
3.2.20. 4-(2-(2,5-Dichlorophenyl)-9H-purin-6-yl)morpholine 3t
3.2.21. 5-Chloro-2-(6-morpholino-9H-purin-2-yl)phenol 3u
3.2.22. 4-(2-(2-Fluoro-5-methoxyphenyl)-9H-purin-6-yl)morpholine 3v
3.2.23. 4-(2-(2,5-Difluorophenyl)-9H-purin-6-yl)morpholine 3x
3.3. Radioligand Binding Assays
3.3.1. Human A1 Receptor
3.3.2. Human A2A Receptor
3.3.3. Human A2B Receptor
3.3.4. Human A3 Receptor
3.4. Functional Studies
3.4.1. Human A1 Receptor
3.4.2. Human A3 Receptor
3.5. Data Analysis
3.6. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Batra, R.; Jain, V.; Sharma, P. Adenosine: A partially discovered medicinal agent. Future J. Pharm. Sci. 2021, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, S.; Mittal, A.; Kumar, P.; Alhayani, D.M.; Al-Aboudi, A. Therapeutic potential of agonists and antagonists of A1, A2a, A2b and A3 adenosine receptors. Curr. Pharm. Des. 2019, 25, 2892–2905. [Google Scholar] [CrossRef] [PubMed]
- Effendi, W.I.; Nagano, T.; Kobayashi, K.; Nishimura, Y. Focusing on adenosine receptors as a potential targeted therapy in human diseases. Cells 2020, 9, 785. [Google Scholar] [CrossRef] [PubMed]
- Ijzerman, A.P.; Jacobson, K.A.; Müller, C.E.; Cronstein, B.N.; Cunha, R.A. International union of basic and clinical pharmacology. CXII: Adenosine receptors: A further updates. Pharmacol. Rev. 2022, 74, 340–372. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.E.; Jacobson, K.A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta Biomembr. 2011, 1808, 1290–1308. [Google Scholar] [CrossRef] [PubMed]
- Borah, P.; Deka, S.; Mailavaram, R.P.; Deb, P.K. P1 receptor agonists/antagonists in clinical trials-potential drug candidates of the future. Curr. Pharm. Des. 2019, 25, 2792–2807. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-G.; Tosh, D.K.; Jain, S.; Yu, J.; Suresh, R.R.; Jacobson, K.A. A1 adenosine receptor agonists, antagonists, and allosteric modulators. In The Adenosine Receptors; Borea, P., Varani, K., Gessi, S., Merighi, S., Vincenzi, F., Eds.; Springer International Publishing: Cham, Germany, 2018; pp. 59–89. [Google Scholar]
- Stockwell, J.; Jakova, E.; Cayabyab, F. Adenosine A1 and A2A Receptors in the brain: Current research and their role in neurodegeneration. Molecules 2017, 22, 676. [Google Scholar] [CrossRef] [PubMed]
- Pak, E.S.; Cha, J.J.; Cha, D.R.; Kanasaki, K.; Ha, H. Adenosine receptors as emerging therapeutic targets for diabetic kidney disease. Kidney Res. Clin. Pract. 2022, 41, S74–S88. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Barbon, D.; Brienza, N.S.; Bigorra Rodríguez, T.; Mateus Medina, É.F.; Gich Saladich, I.; Puntes Rodríguez, M.; Antonijoan Arbos, R.M.; Quirce Gancedo, S.; Castro Palomino Laria, N.; Castro Palomino Laria, J. PBF-680, an oral A1 adenosine receptor antagonist, inhibits the late allergic response (LAR) in mild-to-moderate atopic asthmatics: A Phase-IIa trial. Eur. Respir. J. 2020, 56, 4784. [Google Scholar] [CrossRef]
- Nguyen, A.T.N.; Tran, Q.L.; Baltos, J.-A.; McNeill, S.M.; Nguyen, D.T.N.; May, L.T. Small molecule allosteric modulation of the adenosine A1 receptor. Front. Endocrinol. 2023, 14, 1184360. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.B.; Fresco, P.; Diniz, C.; Goncalves, J. Adenosine receptor ligands on cancer therapy: A review of patent literature. Recent Pat. Anticancer. Drug Discov. 2018, 13, 40–69. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, D.M.; Son, W.C.; Moon, B.G. A selective adenosine A3 receptor antagonist, HL3501, has therapeutic potential in preclinical liver and renal fibrosis models. Vivo 2022, 36, 2186–2193. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Romagnoli, R.; Saponaro, G.; Baraldi, S.; Tabrizi, M.A.; Preti, D. A3 adenosine receptor antagonists: History and future perspectives. In A3 Adenosine Receptors from Cell Biology to Pharmacology and Therapeutics; Borea, P.A., Ed.; Springer Science+Business Media B.V.: Dordrecht, The Netherlands, 2010; pp. 121–147. [Google Scholar]
- Spinaci, A.; Buccioni, M.; Dal Ben, D.; Maggi, F.; Marucci, G.; Francucci, B.; Santoni, G.; Lambertucci, C.; Volpini, R. A3 adenosine receptor antagonists with nucleoside structures and their anticancer activity. Pharmaceuticals 2022, 15, 164. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Merighi, S.; Varani, K.; Borea, P.A.; Baraldi, S.; Aghazadeh Tabrizi, M.; Romagnoli, R.; Baraldi, P.G.; Ciancetta, A.; Tosh, D.K.; et al. A3 adenosine receptors as modulators of inflammation: From medicinal chemistry to therapy. Med. Res. Rev. 2018, 38, 1031–1072. [Google Scholar] [CrossRef]
- Spinozzi, E.; Baldassarri, C.; Acquaticci, L.; Del Bello, F.; Grifantini, M.; Cappellacci, L.; Riccardo, P. Adenosine receptors as promising targets for the management of ocular diseases. Med. Chem. Res. 2021, 30, 353–370. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Yang, J.; Kim, J.Y.; Lee, J.M.; Son, W.C.; Moon, B.G. HL3501, a novel selective A3 adenosine receptor antagonist, lowers intraocular pressure (IOP) in animal glaucoma models. Transl. Vis. Sci. Technol. 2022, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Barkan, K.; Lagarias, P.; Stampelou, M.; Stamatis, D.; Hoare, S.; Safitri, D.; Klotz, K.N.; Vrontaki, E.; Kolocouris, A.; Ladds, G. Pharmacological characterisation of novel adenosine A3 receptor antagonists. Sci. Rep. 2020, 10, 20781. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, P.; Carradori, S.; Campestre, C.; Poce, G. Novel therapies for glaucoma: A patent review (2013–2019). Expert Opin. Ther. Pat. 2019, 29, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, S.; Kim, D.; Ahn, K.H.; Lee, G.B.; Kim, D.; Hwang, H.S. Compounds antagonizing A3 adenosine receptor, method for preparing them, and medical-use thereof. WO2017123058A1, 20 July 2017. [Google Scholar]
- Petrelli, R.; Torquati, I.; Kachler, S.; Luongo, L.; Maione, S.; Franchetti, P.; Grifantini, M.; Novellino, E.; Lavecchia, A.; Klotz, K.N.; et al. 5′-C-ethyl-tetrazolyl-N 6-substituted adenosine and 2-chloro-adenosine derivatives as highly potent dual acting A1 adenosine receptor agonists and A3 adenosine receptor antagonists. J. Med. Chem. 2015, 58, 2560–2566. [Google Scholar] [CrossRef]
- Abdelrahman, A.; Yerande, S.G.; Namasivayam, V.; Klapschinski, T.A.; Alnouri, M.W.; El-Tayeb, A.; Müller, C.E. Substituted 4-phenylthiazoles: Development of potent and selective A1, A3 and dual A1/A3 adenosine receptor antagonists. Eur. J. Med. Chem. 2020, 186, 111879. [Google Scholar] [CrossRef]
- Massie, B.M.; O’Connor, C.M.; Metra, M.; Ponikowski, P.; Teerlink, J.R.; Cotter, G.; Weatherley, B.D.; Cleland, J.G.F.; Givertz, M.M.; Voors, A.; et al. Rolofylline, an adenosine A1-receptor antagonist, in acute heart failure. N. Engl. J. Med. 2010, 363, 1419–1428. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Iragui, V.J.; Mohr, J.P.; Carson, P.E.; Hauptman, P.J.; Lovett, D.H.; Miller, A.B.; Piña, I.L.; Thomson, S.; Varosy, P.D.; et al. The safety of an adenosine A1-receptor antagonist, rolofylline, in patients with acute heart failure and renal impairment. Drug Saf. 2012, 35, 233–244. [Google Scholar] [CrossRef]
- Mitrovic, V.; Seferovic, P.; Dodic, S.; Krotin, M.; Neskovic, A.; Dickstein, K.; de Voogd, H.; Bocker, C.; Ziegler, D.; Godes, M.; et al. Cardio-renal effects of the A1 adenosine receptor antagonist SLV320 in patients with heart failure. Circ. Hear. Fail. 2009, 2, 523–531. [Google Scholar] [CrossRef]
- Areias, F.; Correia, C.; Rocha, A.; Brea, J.; Castro, M.; Loza, M.I.; Proença, M.F.; Carvalho, M.A. 2-Aryladenine derivatives as a potent scaffold for A1, A3 and dual A1/A3 adenosine receptor antagonists: Synthesis and structure-activity relationships. Bioorganic Med. Chem. 2019, 27, 3551–3558. [Google Scholar] [CrossRef]
- Alves, M.J.; Booth, B.L.; Proença, M.F.J.R.P. Synthesis of 5-amino-4-(cyanoformimidoyl)-1H-imidazole: A reactive intermediate for the synthesis of 6-carbamoyl-1,2-dihydropurines and 6-carbamoylpurines. J. Chem. Soc. Perkin Trans. 1 1990, 1, 1705–1712. [Google Scholar] [CrossRef]
- Correia, C.; Carvalho, M.A.; Proença, M.F. Synthesis and in vitro activity of 6-amino-2,9-diarylpurines for Mycobacterium tuberculosis. Tetrahedron 2009, 65, 6903–6911. [Google Scholar] [CrossRef]
- Jacobson, K.A. Introduction to adenosine receptors as therapeutic targets. In Adenosine Receptors in Health and Disease; Wilson, C.N., Mustafa, S.J., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 1–24. [Google Scholar]
- Giorgi, I.; Nieri, P. Adenosine A 1 modulators: A patent update (2008 to present). Expert Opin. Ther. Pat. 2013, 23, 1109–1121. [Google Scholar] [CrossRef]
- Shook, B.C.; Rassnick, S.; Osborne, M.C.; Davis, S.; Westover, L.; Boulet, J.; Hall, D.; Rupert, K.C.; Heintzelman, G.R.; Hansen, K.; et al. In vivo characterization of a dual adenosine A2A/A1 receptor antagonist in animal models of Parkinson’s disease. J. Med. Chem. 2010, 53, 8104–8115. [Google Scholar] [CrossRef]
- Jung, J.; Lee, Y.; Moon, A.-N.; Ann, J.; Jeong, J.J.; Do, N.; Lee, J. Discovery of Novel Dual Adenosine A2A and A1 receptor antagonists with 1H-Pyrazolo[3,4-d]pyrimidin-6-amine core scaffold as anti-Parkinson’s disease agents. Pharmaceuticals 2022, 15, 922. [Google Scholar] [CrossRef]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867–877. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Gao, Z.; Matricon, P.; Eddy, M.T.; Carlsson, J. Adenosine A 2A receptor antagonists: From caffeine to selective non-xanthines. Br. J. Pharmacol. 2022, 179, 3496–3511. [Google Scholar] [CrossRef]
- Leff, P.; Dougall, I.G. Further concerns over Cheng-Prusoff analysis. Trends Pharmacol. Sci. 1993, 14, 110–112. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | R1 | A1 | A2A | A2B | A3 | |
|---|---|---|---|---|---|---|
| a | Me | 2-ClC6H4 | 36 ± 1% | 11 ± 2% | 22 ± 5% | 14 ± 2% |
| b | Me | 2-MeOC6H4 | 22 ± 1% | 12 ± 5% | 12 ± 2% | 36 ± 5% |
| c | Me | 2-HOC6H4 | 6.51 ± 0.11 | 6.47 ± 0.05 | 5.70 ± 0.07 | 6 ± 5% |
| d | Me | 3-ClC6H4 | 6.80 ± 0.07 | 15 ± 4% | 2 ± 2% | 25 ± 4% |
| e | Me | 3-HOC6H4 | 6.16 ± 0.21 | 37 ± 1% | 34 ± 3% | 52 ± 4% |
| f | Me | 4-ClC6H4 | 37 ± 4% | 42 ± 5% | 15 ± 4% | 31 ± 5% |
| g | Me | 2,3-(HO)2C6H3 | 33 ± 0.3% | 25 ± 2% | 22 ± 1.5% | 42 ± 3% |
| h | Me | 2,5-Cl2C6H3 | 41 ± 2% | 2 ± 3% | 22 ± 3% | 8 ± 1% |
| i | Me | 2,5-(HO)2C6H3 | 6.36 ± 0.15 | 6.25 ± 0.12 | 6.32 ± 0.01 | 53 ± 5% |
| j | Me | 2-HO-5-MeOC6H3 | 6.28 ± 0.17 | 5.91 ± 0.11 | 5.78 ± 0.03 | 19 ± 11% |
| k | Me | 2-HO-5-ClC6H3 | 6.91 ± 0.08 | 6.15 ± 0.03 | 55 ± 1% | 53 ± 4% |
| l | Me | 2-F-5-MeOC6H3 | 61 ± 4% | 42 ± 1% | 27 ± 2% | 5.28 ± 0.11 |
| m | Me | 2,5-F2C6H3 | 6.28 ± 0.13 | 37 ± 2% | 30 ± 4% | 25 ± 3% |
| n | H | 2-HOC6H4 | 7.19 ± 0.10 | 6.97 ± 0.07 | 6.86 ± 0.10 | 6.59 ± 0.06 |
| o | H | 3-ClC6H4 | 7.75 ± 0.14 | 7.33 ± 0.07 | 7.12 ± 0.13 | 7.40 ± 0.14 |
| p | H | 3-HOC6H4 | 7.49 ± 0.06 | 6.26 ± 0.12 | 60 ± 6% | 6.33 ± 0.08 |
| q | H | 4-ClC6H4 | 7.02 ± 0.07 | 56 ± 3% | 50 ± 4% | 7.47 ± 0.05 |
| r | H | 3,4-Cl2C6H3 | 29 ± 1% | 22 ± 5% | 35 ± 3% | 7.15 ± 0.08 |
| s | H | 3-CF3-4-ClC6H3 | 43 ± 5% | 13 ± 5% | 8 ± 5% | 6.02 ± 0.04 |
| t | H | 2,5-Cl2C6H3 | 5.97 ± 0.11 | 44 ± 2% | 26 ± 1% | 7.16 ± 0.17 |
| u | H | 2-HO-5-ClC6H3 | 7.05 ± 0.09 | 35 ± 1% | 6.21 ± 0.08 | 37 ± 1% |
| v | H | 2-F-5-MeOC6H3 | 38 ± 2% | 45 ± 4% | 6.40 ± 0.16 | 7.83 ± 0.16 |
| x | H | 2,5-F2C6H3 | 8.23 ± 0.06 | 7.15 ± 0.05 | 7.44 ± 0.11 | 7.63 ± 0.12 |
| Compounds | Potency (pKB) | |
|---|---|---|
| A1 | A3 | |
| 3v | N.D. | 8.24 ± 0.11 |
| 3x | 8.25 ± 0.16 | N.D. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Areias, F.; Correia, C.; Rocha, A.; Teixeira, S.; Castro, M.; Brea, J.; Hu, H.; Carlsson, J.; Loza, M.I.; Proença, M.F.; et al. 2-Aryladenine Derivatives as a Potent Scaffold for Adenosine Receptor Antagonists: The 6-Morpholino Derivatives. Molecules 2024, 29, 2543. https://doi.org/10.3390/molecules29112543
Areias F, Correia C, Rocha A, Teixeira S, Castro M, Brea J, Hu H, Carlsson J, Loza MI, Proença MF, et al. 2-Aryladenine Derivatives as a Potent Scaffold for Adenosine Receptor Antagonists: The 6-Morpholino Derivatives. Molecules. 2024; 29(11):2543. https://doi.org/10.3390/molecules29112543
Chicago/Turabian StyleAreias, Filipe, Carla Correia, Ashly Rocha, Sofia Teixeira, Marián Castro, Jose Brea, Huabin Hu, Jens Carlsson, Maria I. Loza, M. Fernanda Proença, and et al. 2024. "2-Aryladenine Derivatives as a Potent Scaffold for Adenosine Receptor Antagonists: The 6-Morpholino Derivatives" Molecules 29, no. 11: 2543. https://doi.org/10.3390/molecules29112543
APA StyleAreias, F., Correia, C., Rocha, A., Teixeira, S., Castro, M., Brea, J., Hu, H., Carlsson, J., Loza, M. I., Proença, M. F., & Carvalho, M. A. (2024). 2-Aryladenine Derivatives as a Potent Scaffold for Adenosine Receptor Antagonists: The 6-Morpholino Derivatives. Molecules, 29(11), 2543. https://doi.org/10.3390/molecules29112543