
Monofluorophos–Metal Complexes: Ripe for Future Discoveries in Homogeneous Catalysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
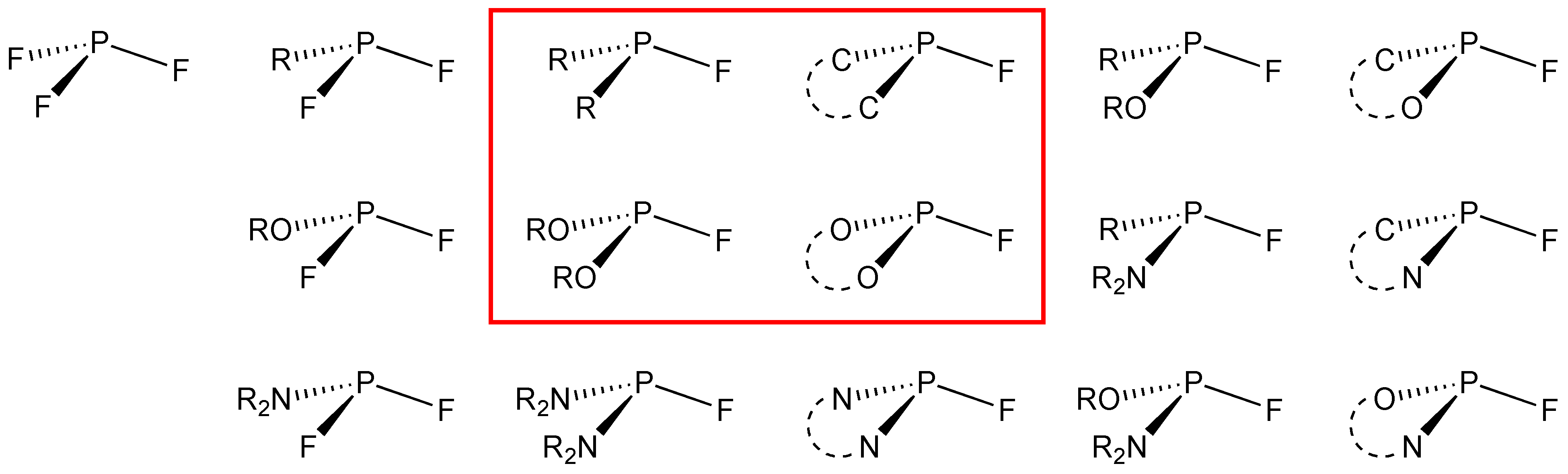
2. Monofluorophosphites
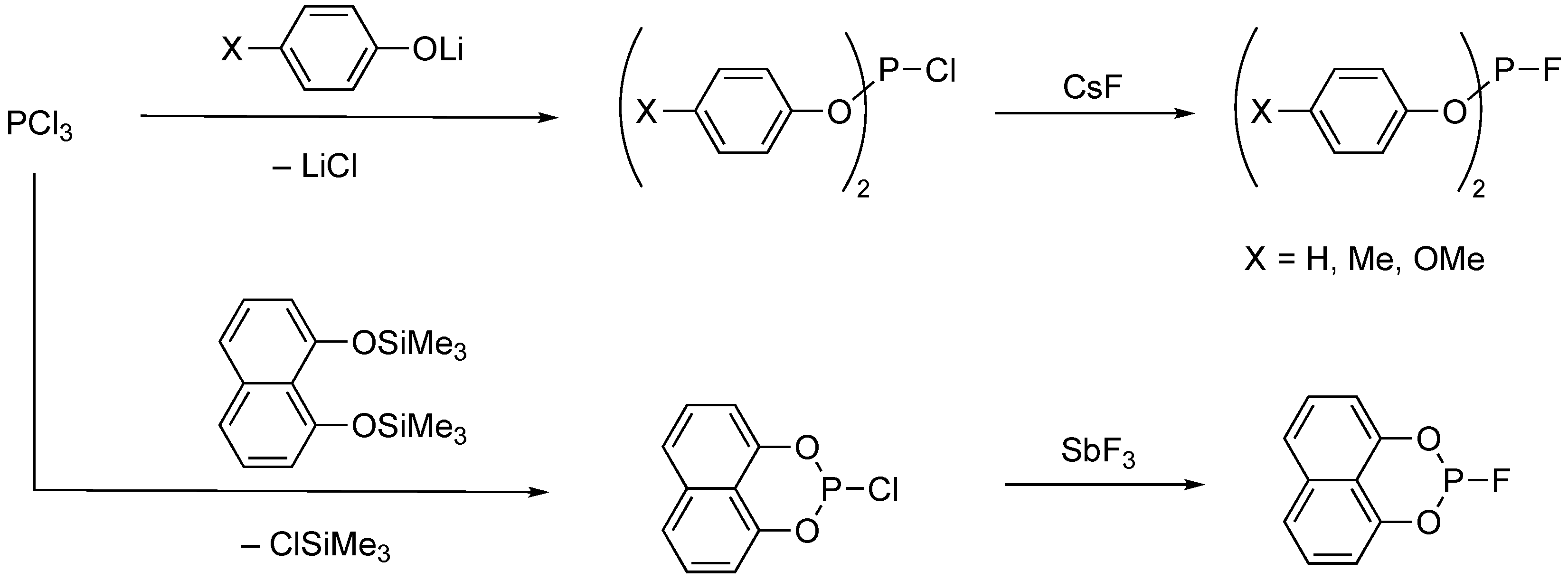
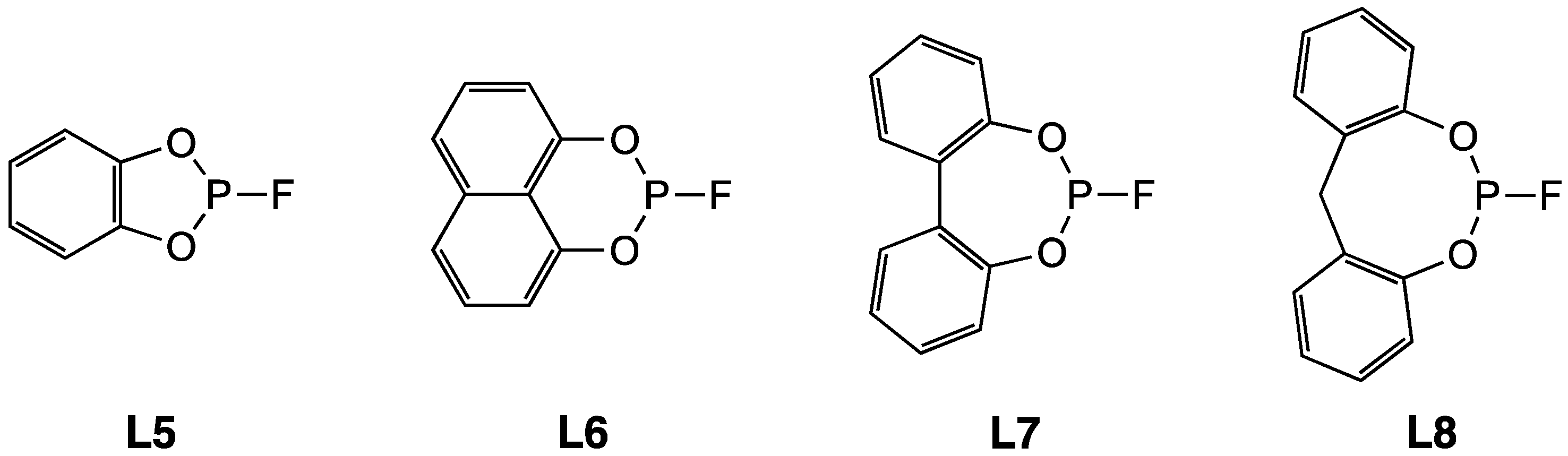
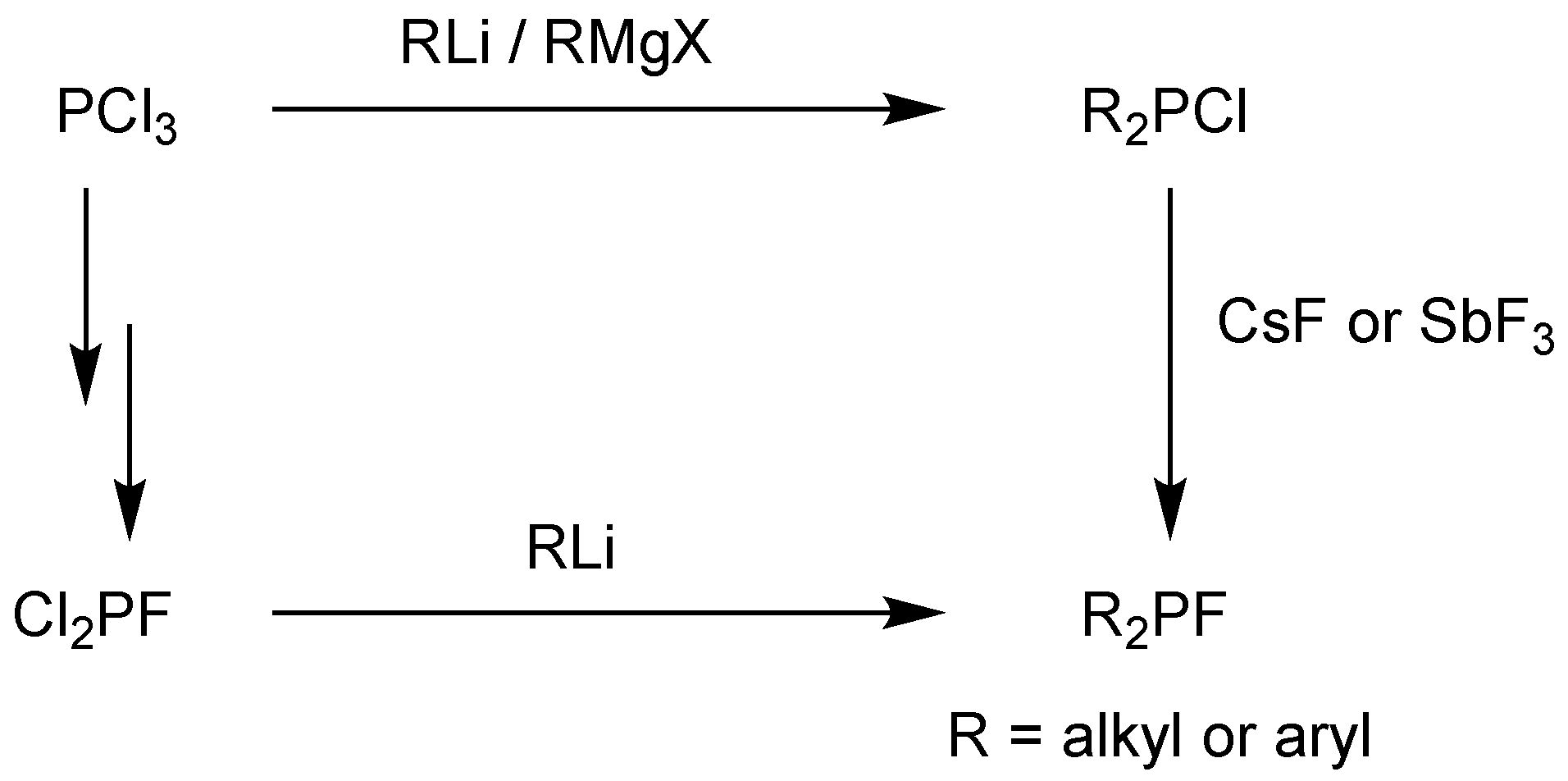
2.1. Synthesis and Hydrolytic Stability of Monofluorophosphites

2.2. Coordination Chemistry of Monofluorophosphites
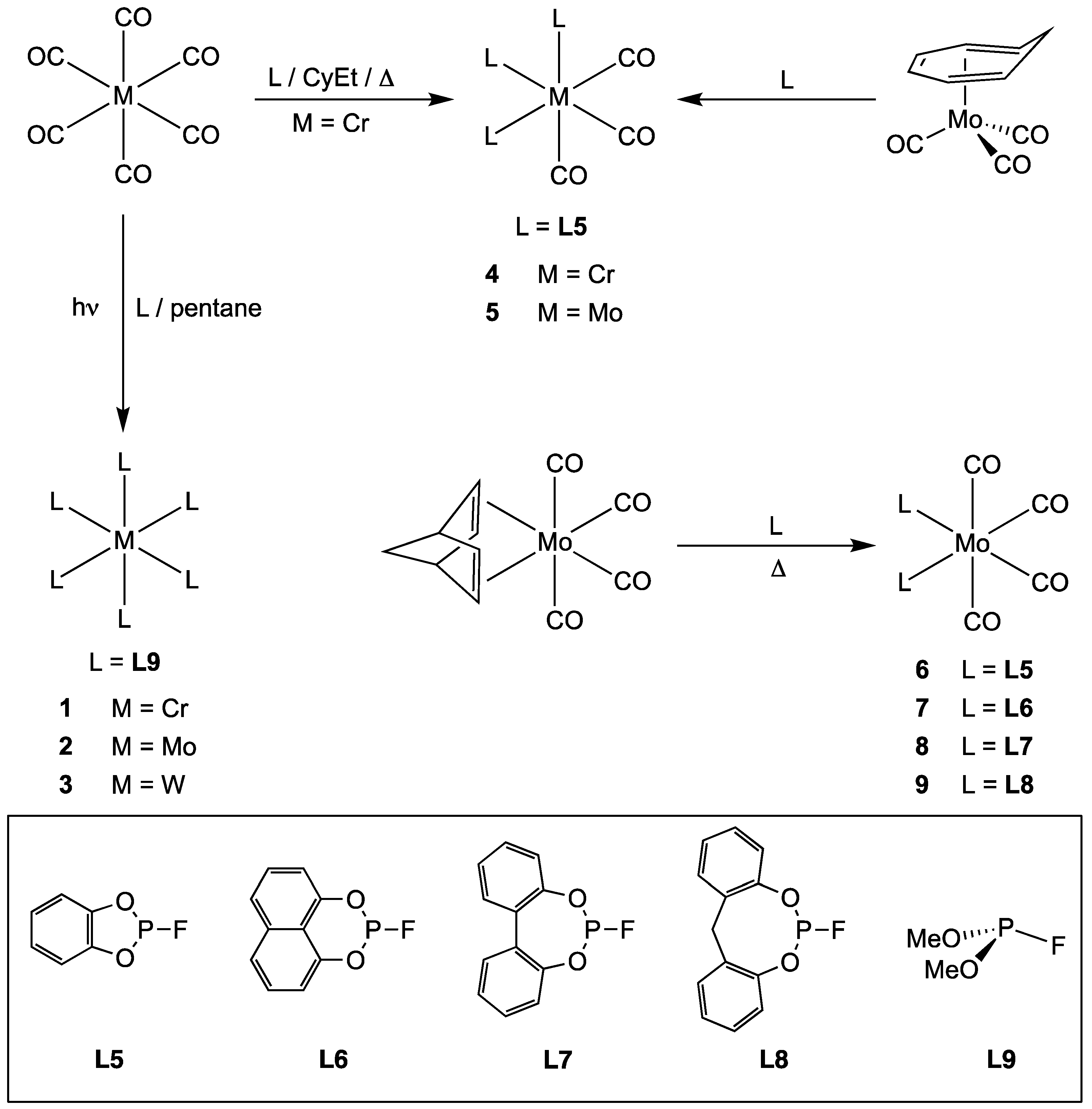
2.2.1. Group 6 Metal Complexes of Monofluorophosphites
2.2.2. Group 8 Metal Complexes of Monofluorophosphites
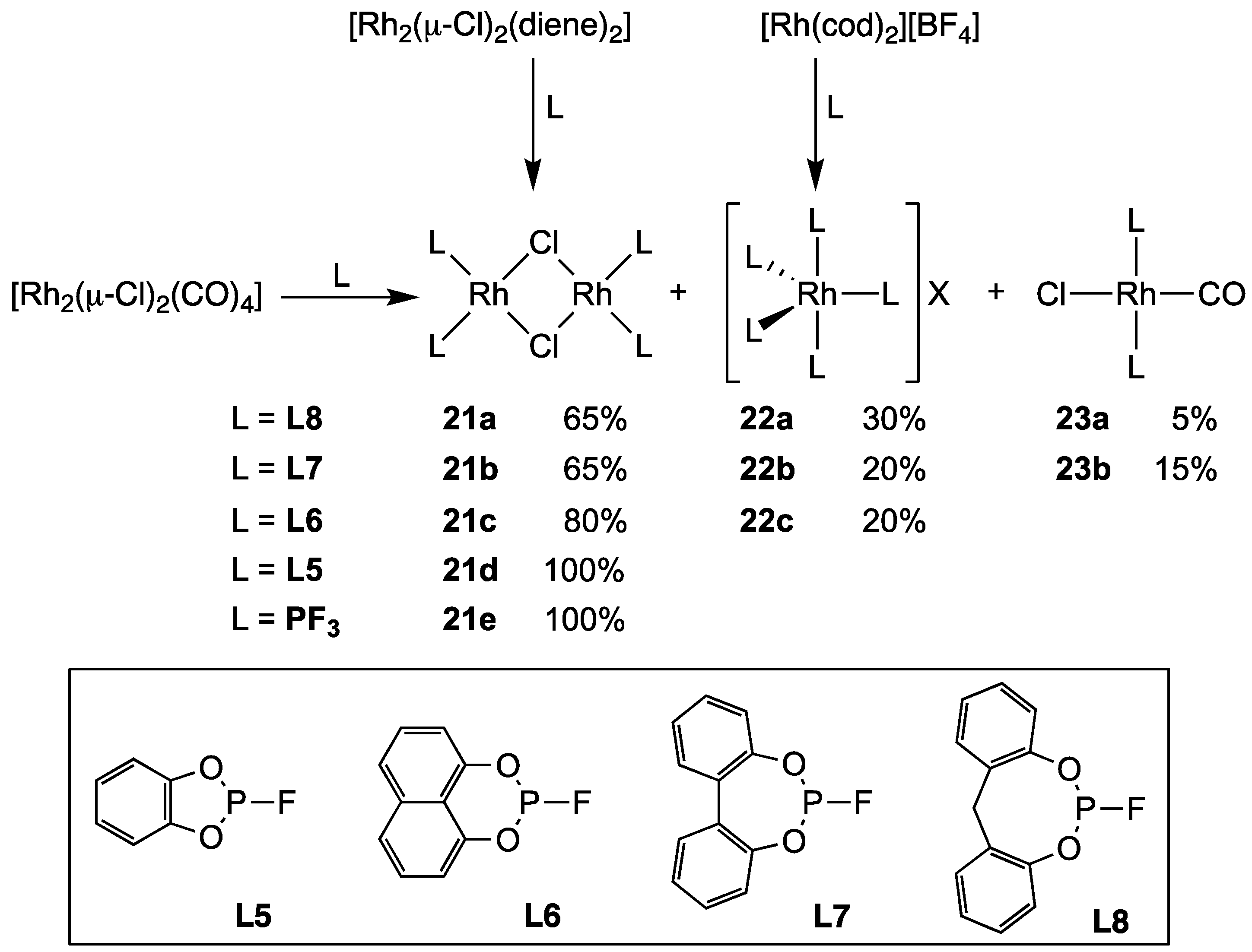
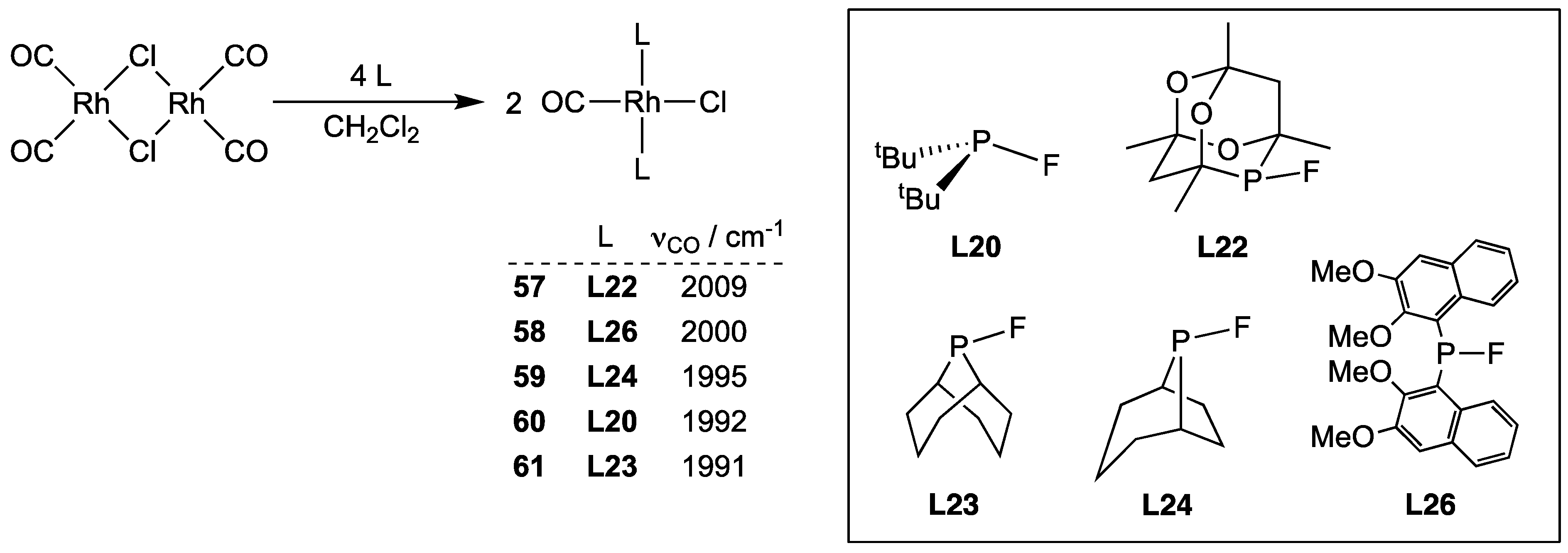
2.2.3. Group 9 Metal Complexes of Monofluorophosphites
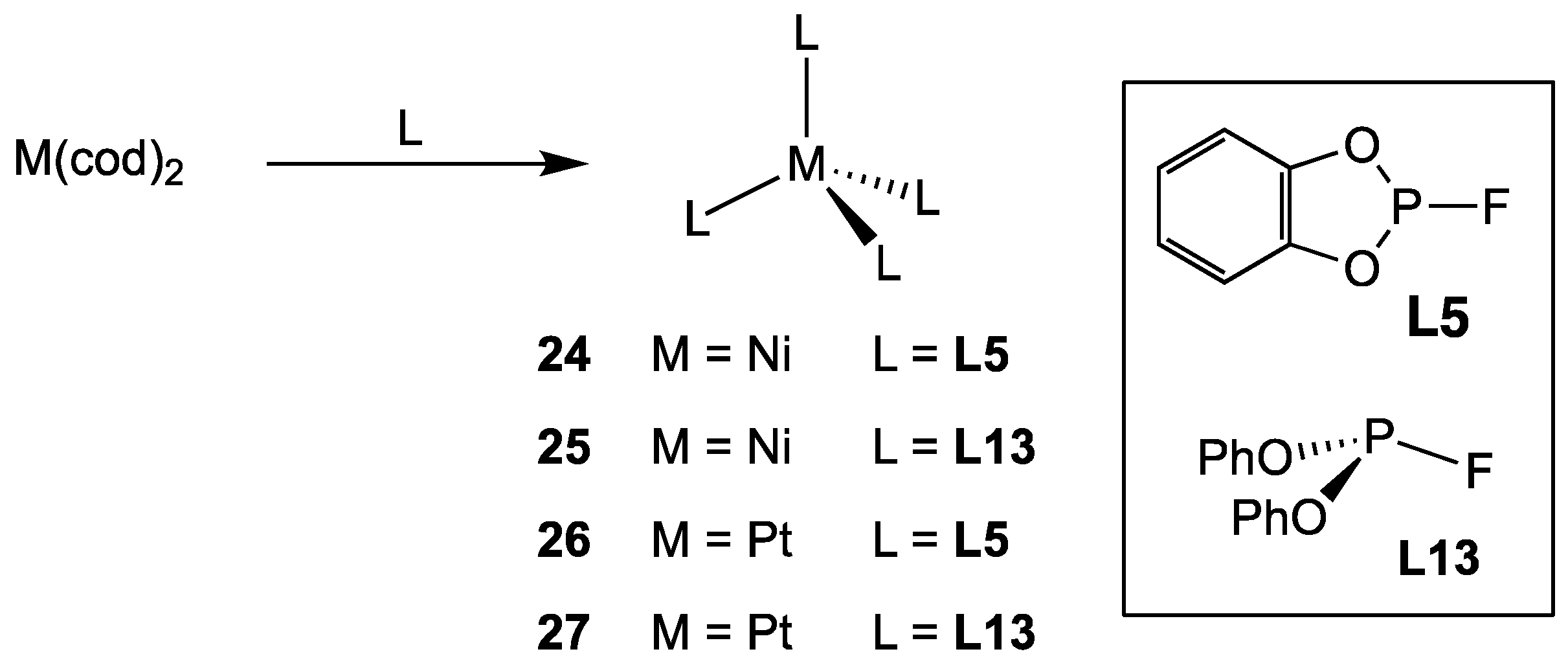
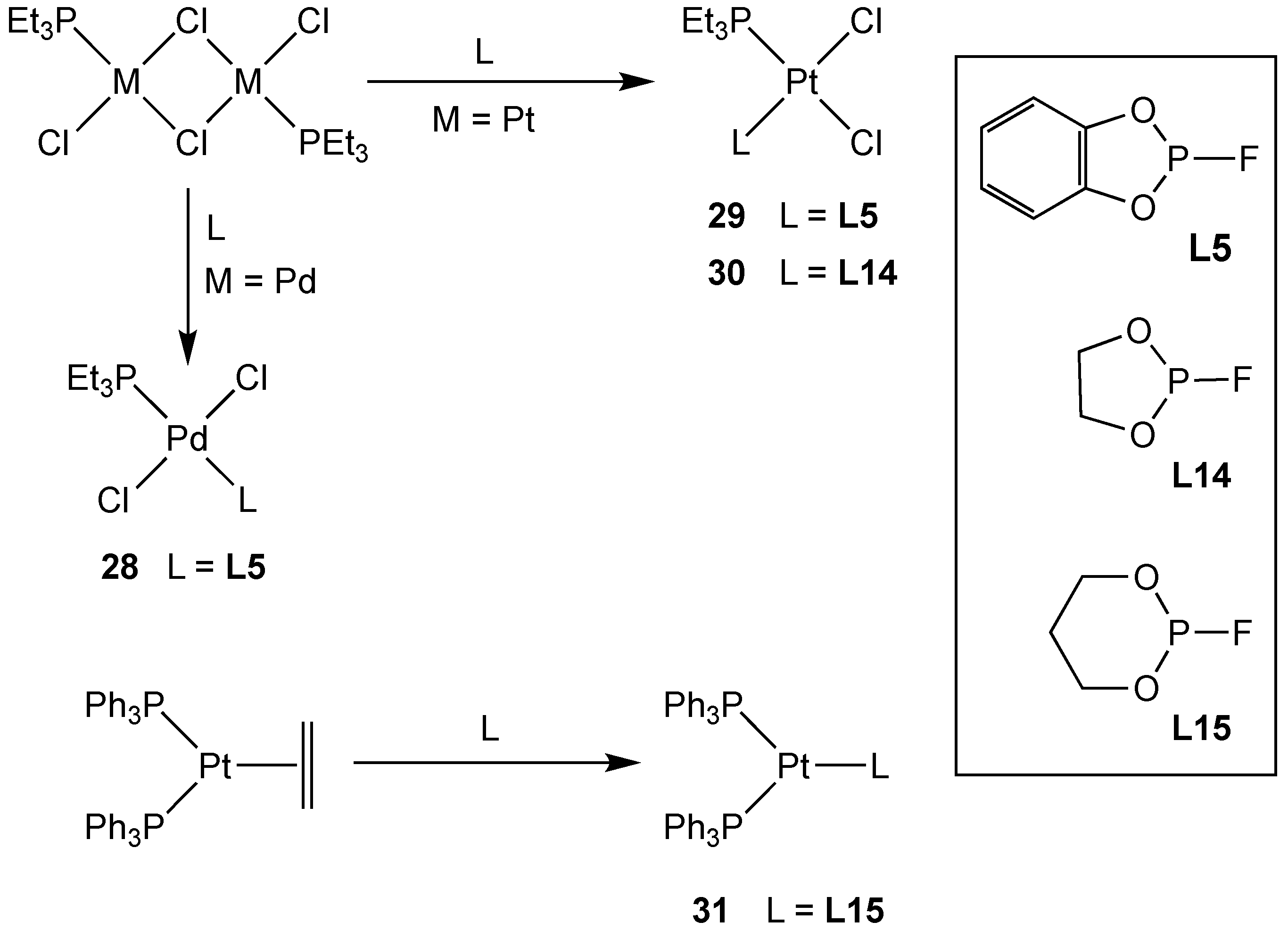
2.2.4. Group 10 Metal Complexes of Monofluorophosphites
2.3. Catalysis with Complexes of Monofluorophosphites
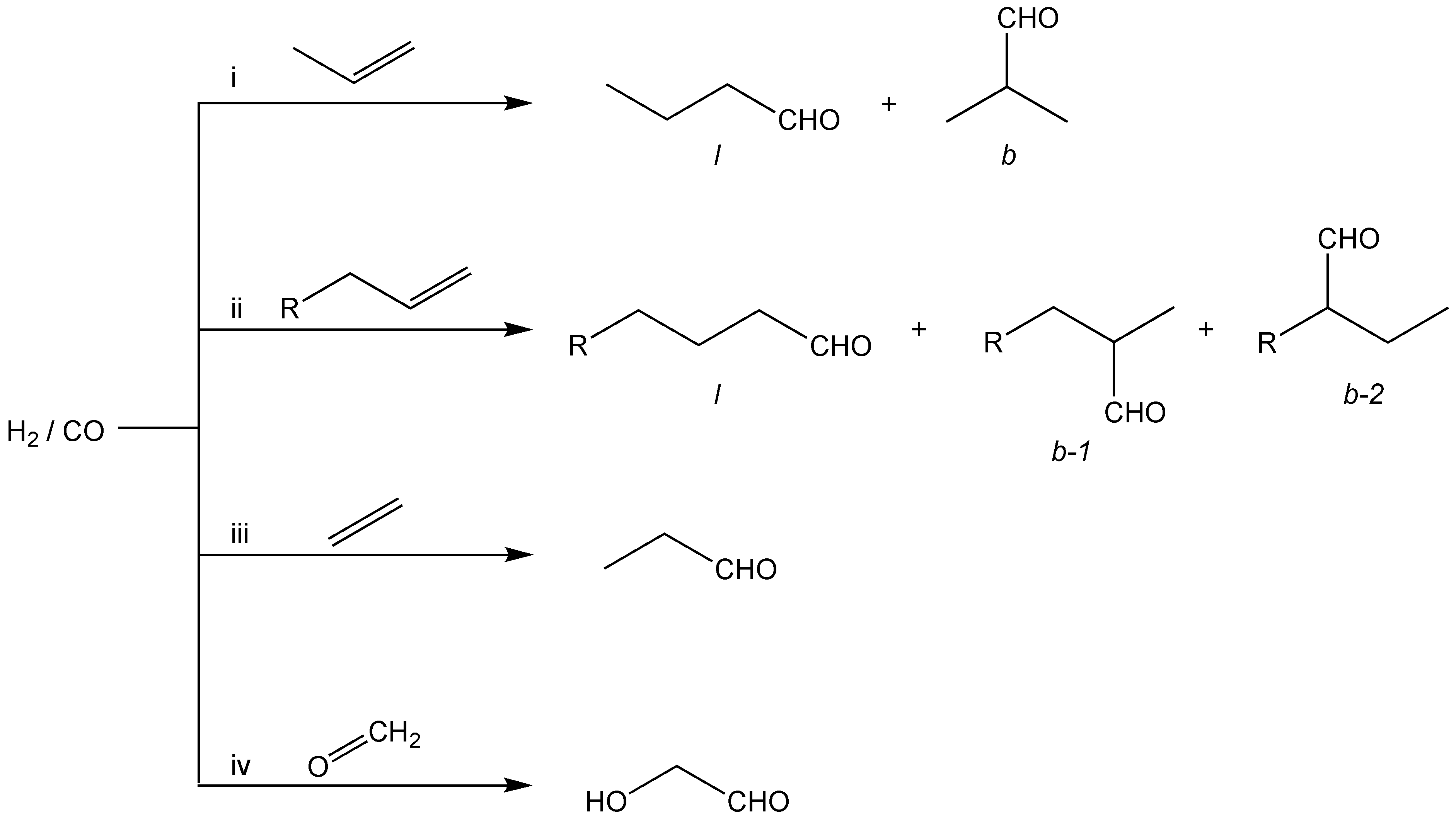
2.3.1. Hydroformylation Catalysis with Rhodium Complexes of Monofluorophosphites
2.3.2. Other Catalytic Reactions with Monofluorophosphite Ligands
3. Monofluorophosphines
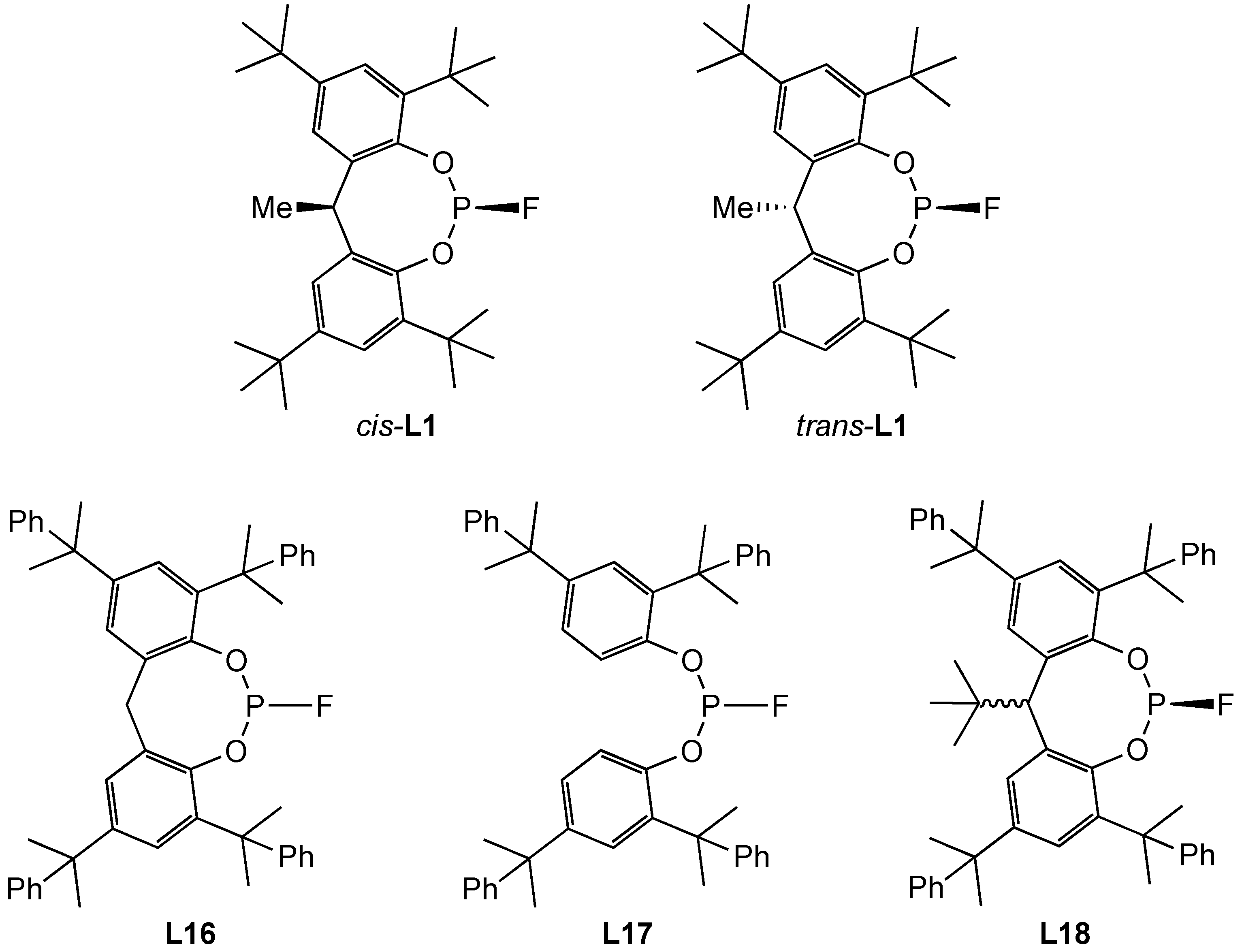
3.1. Synthesis and Stability of Monofluorophosphines
- (1)
- (2)
- (3)
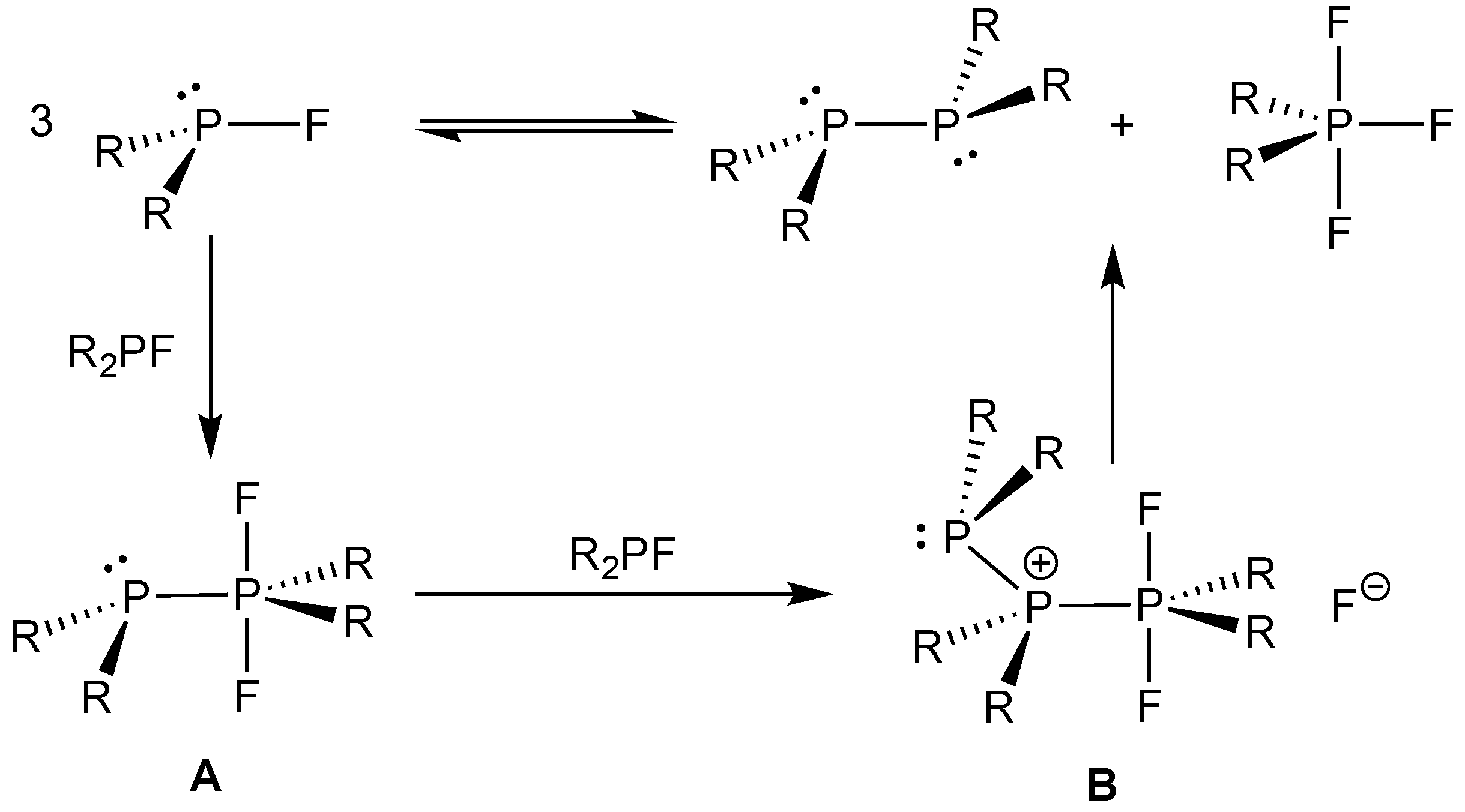
- Cyclic monofluorophosphines with constrained C–P–C bonds are more stable with respect to disproportionation than acyclic analogues [61].
3.2. Coordination Chemistry of Monofluorophosphines
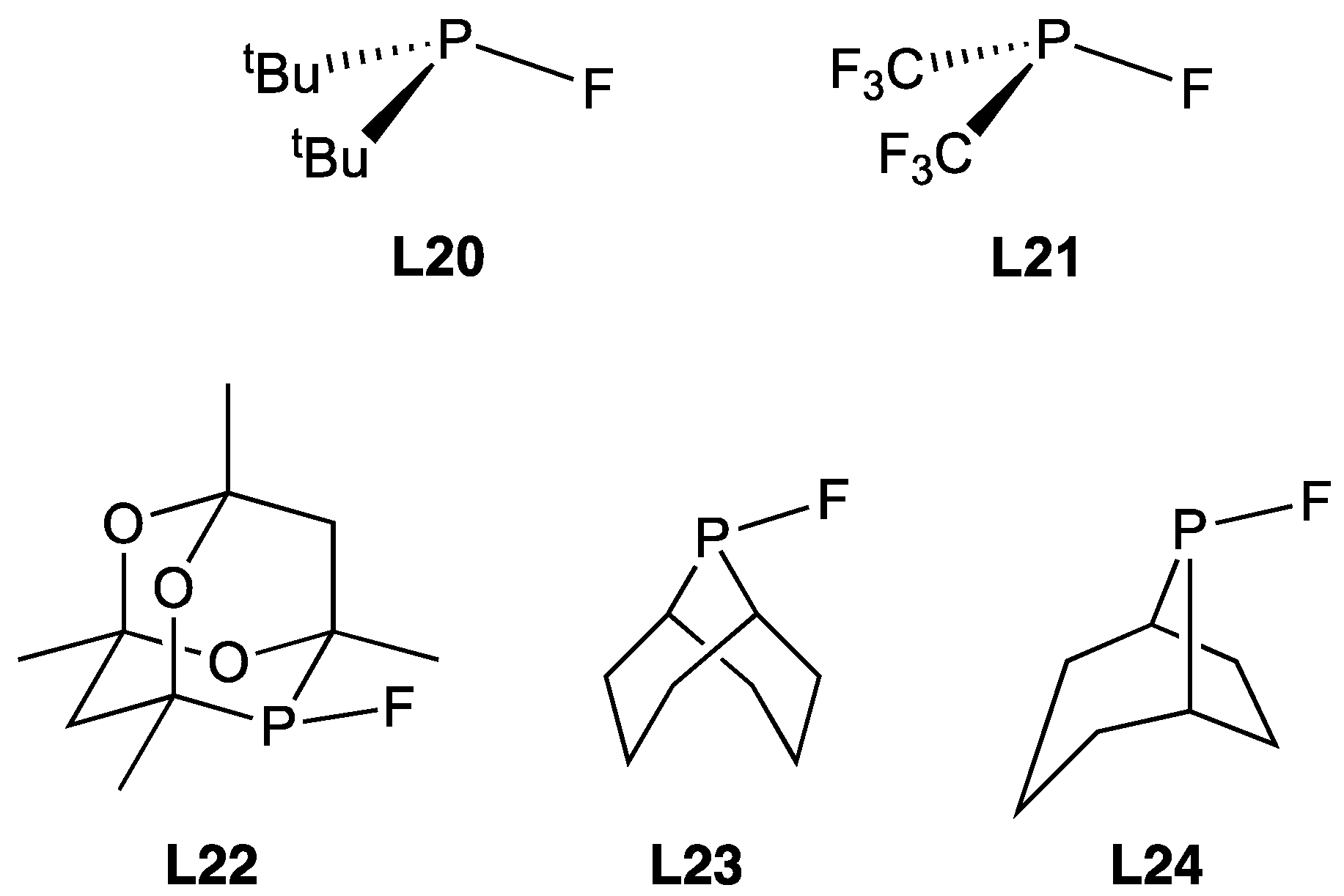
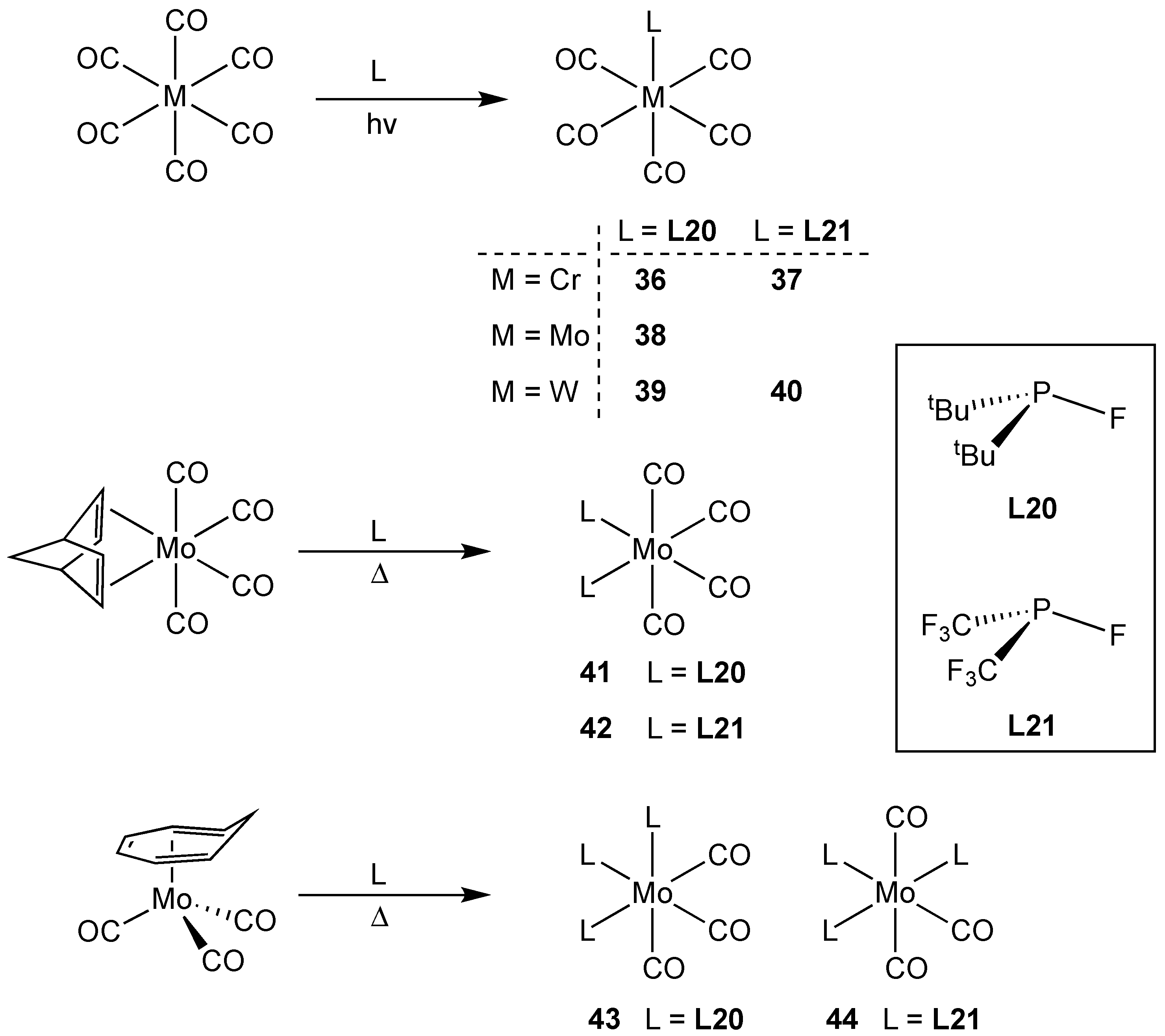
3.2.1. Group 6 Metal Complexes of Monofluorophosphines
3.2.2. Group 7 Metal Complexes of Monofluorophosphines
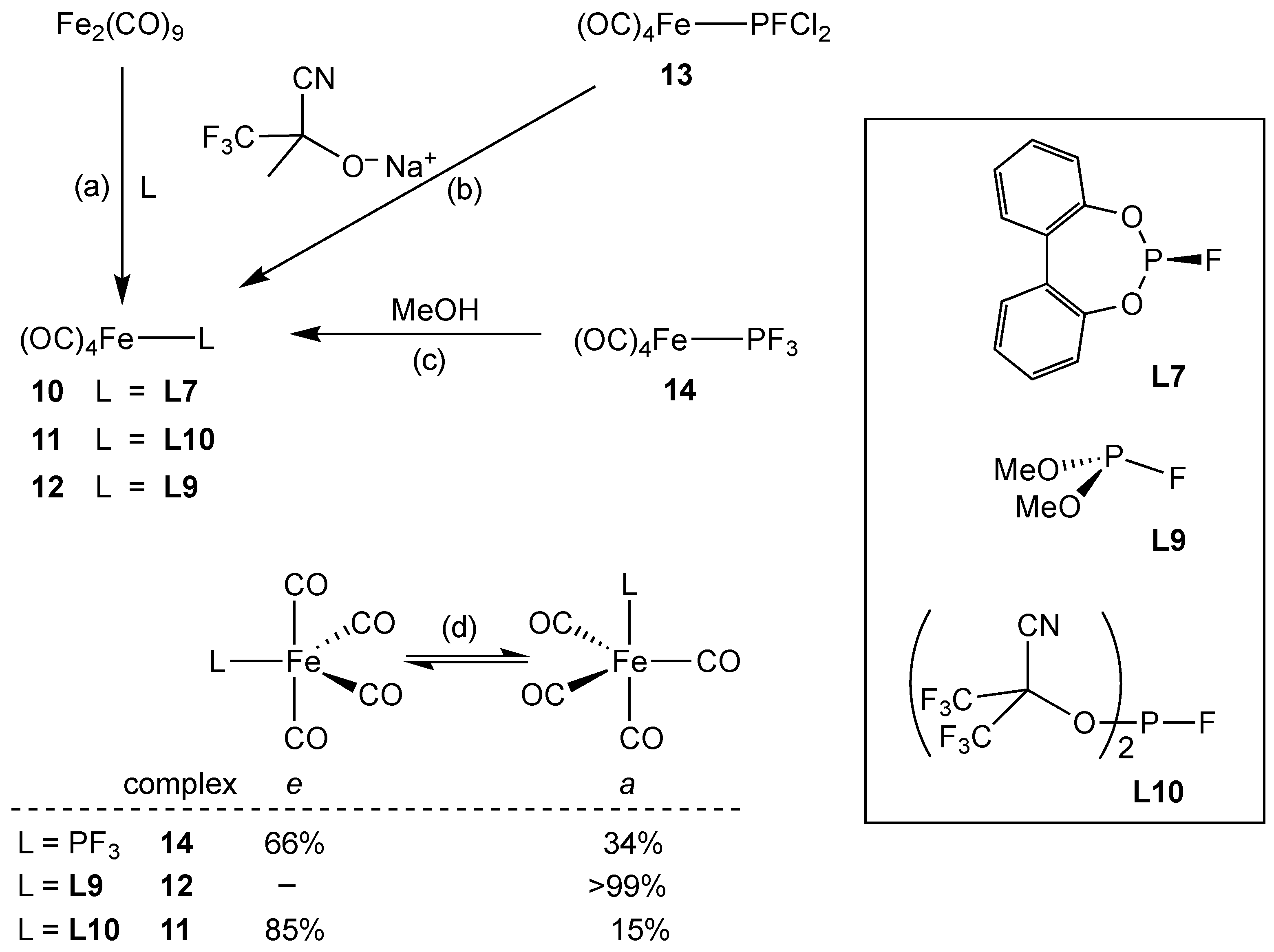
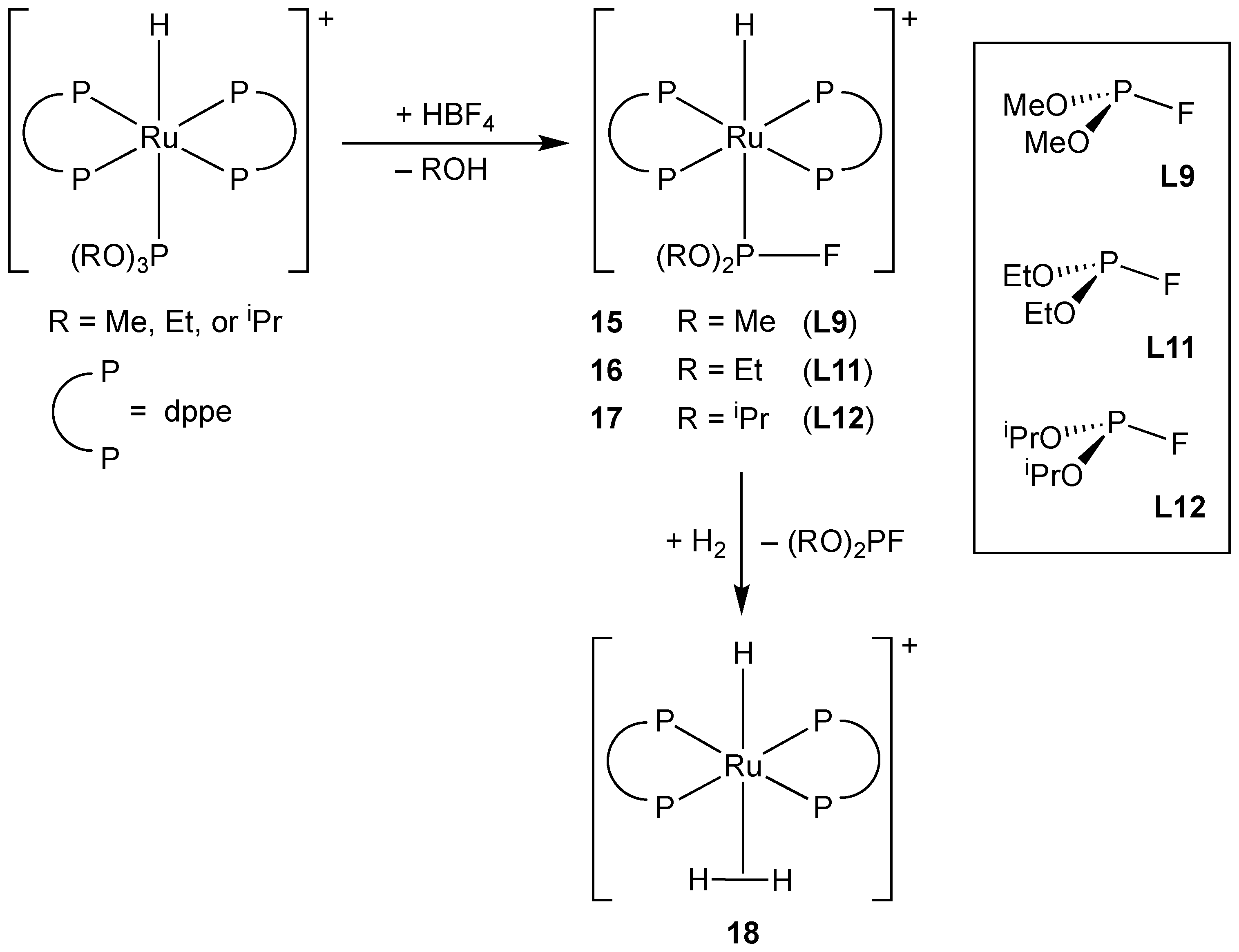
3.2.3. Group 8 Metal Complexes of Monofluorophosphines
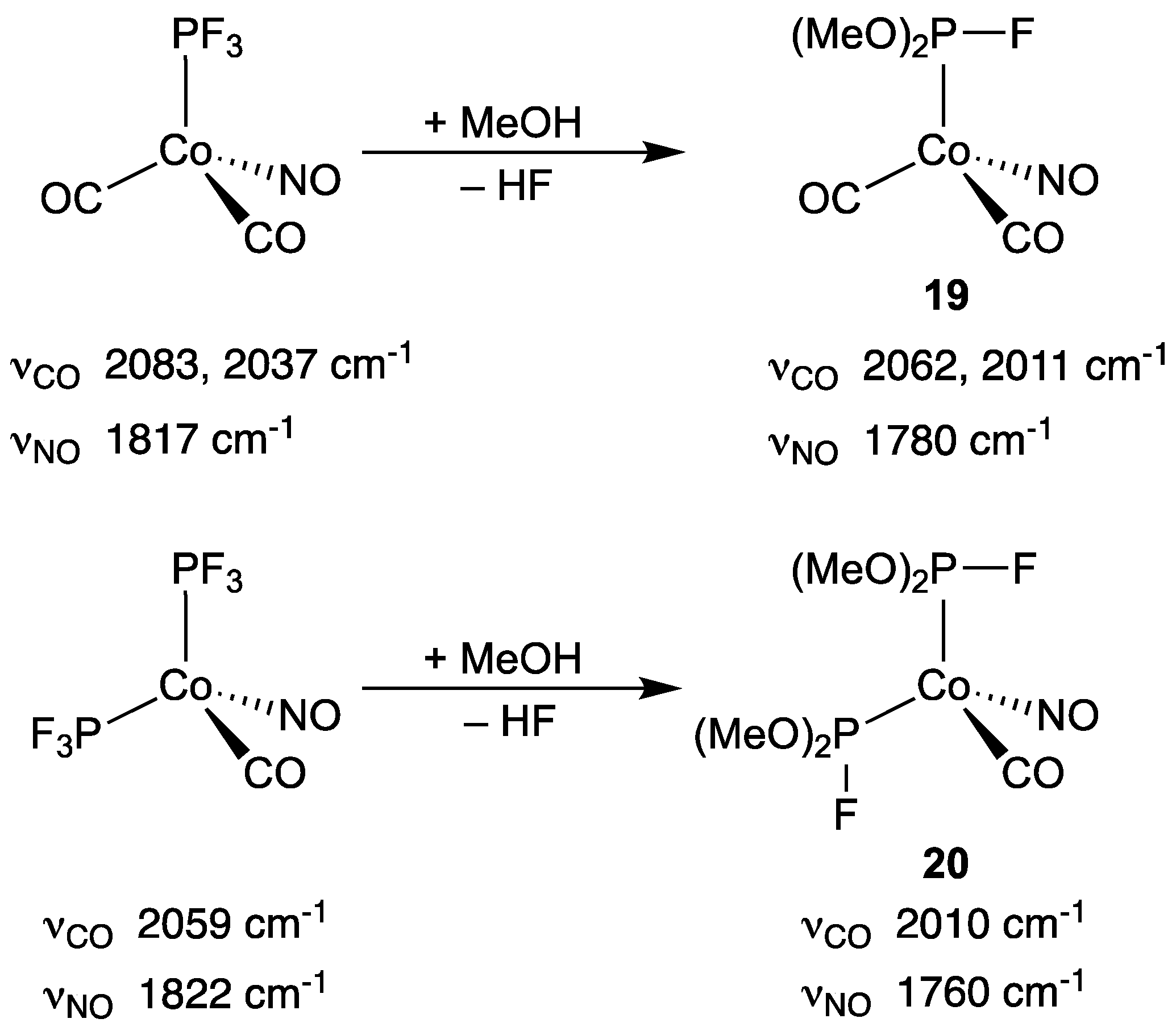
3.2.4. Group 9 Metal Complexes of Monofluorophosphines
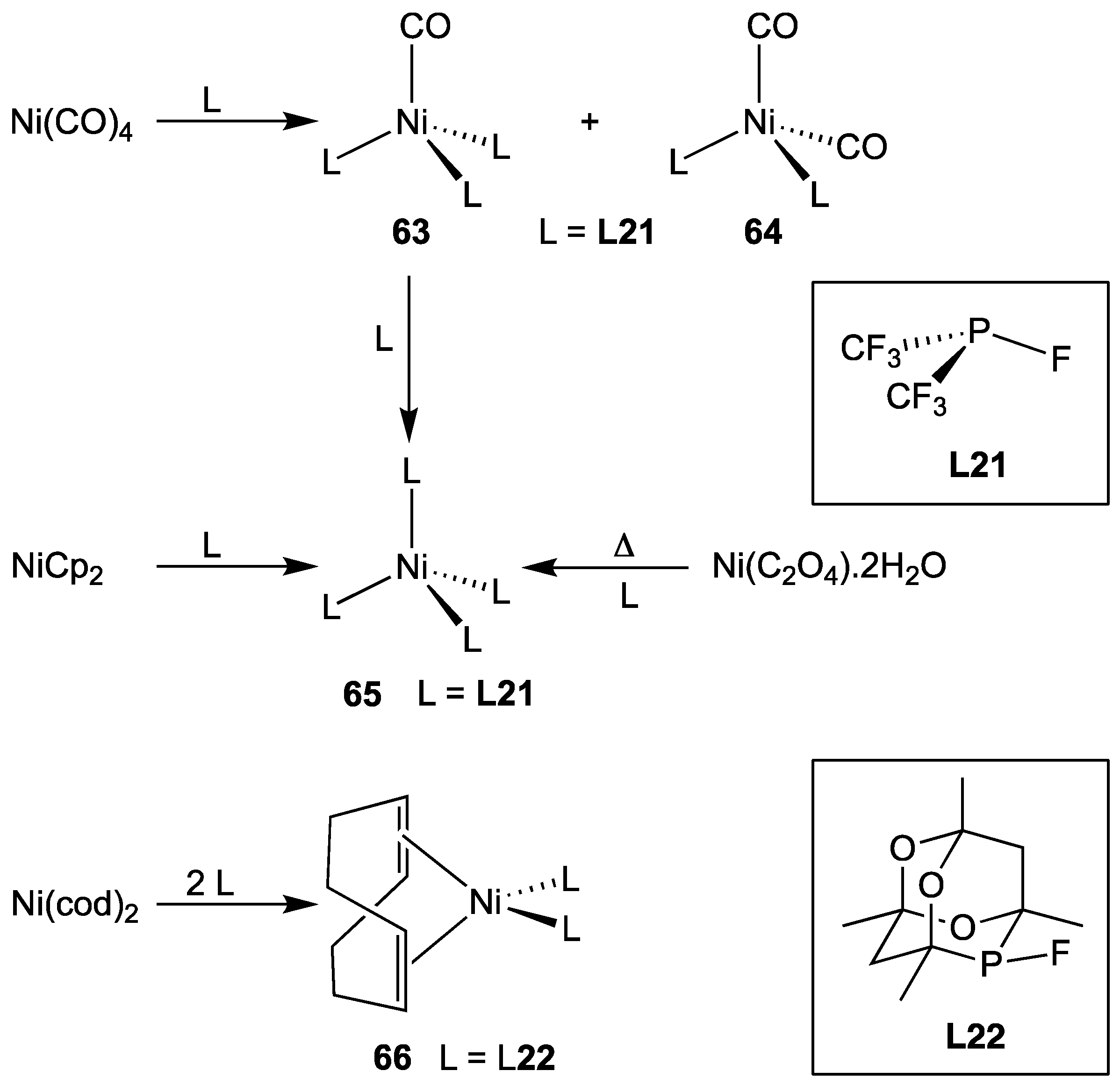
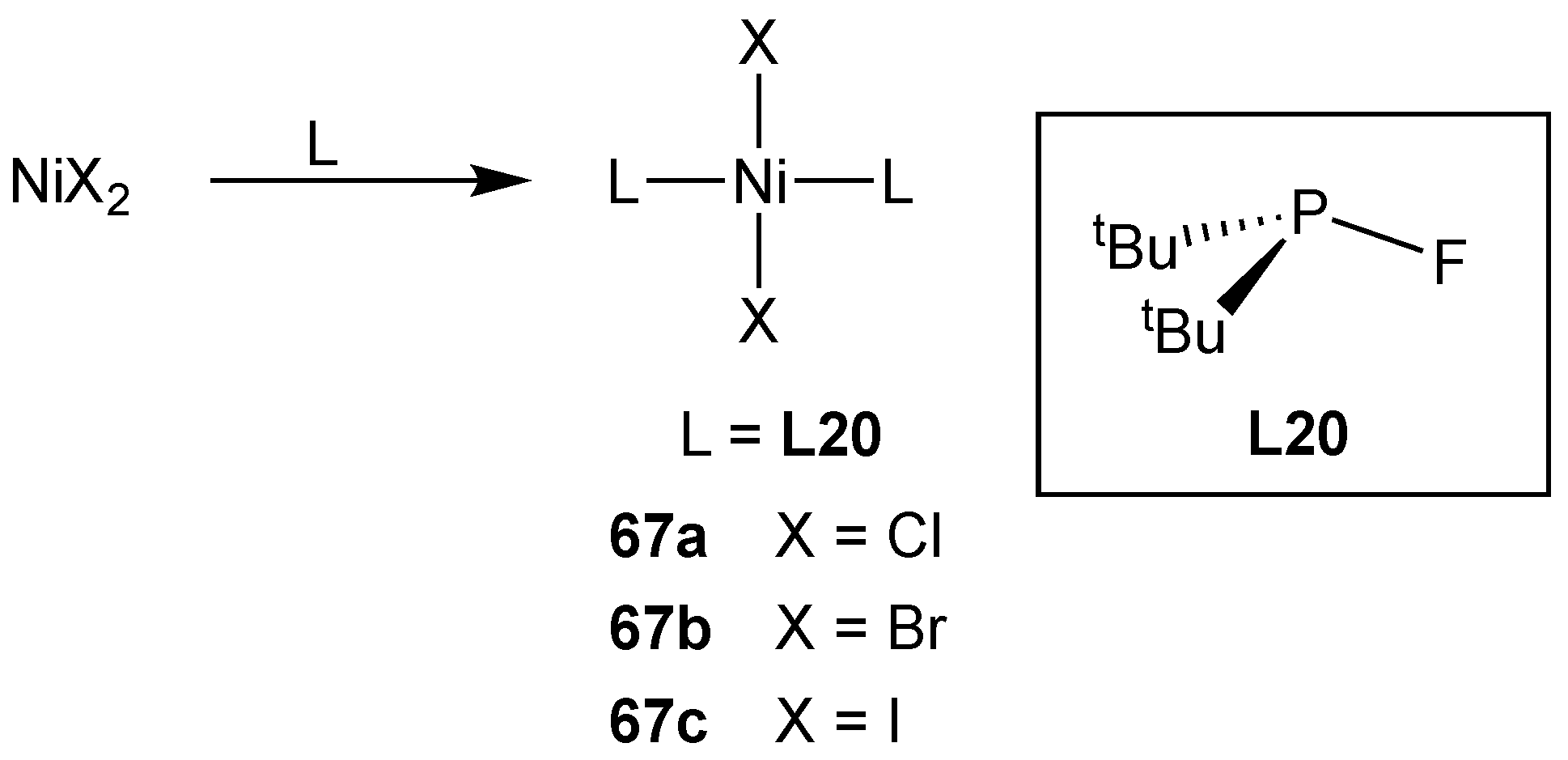
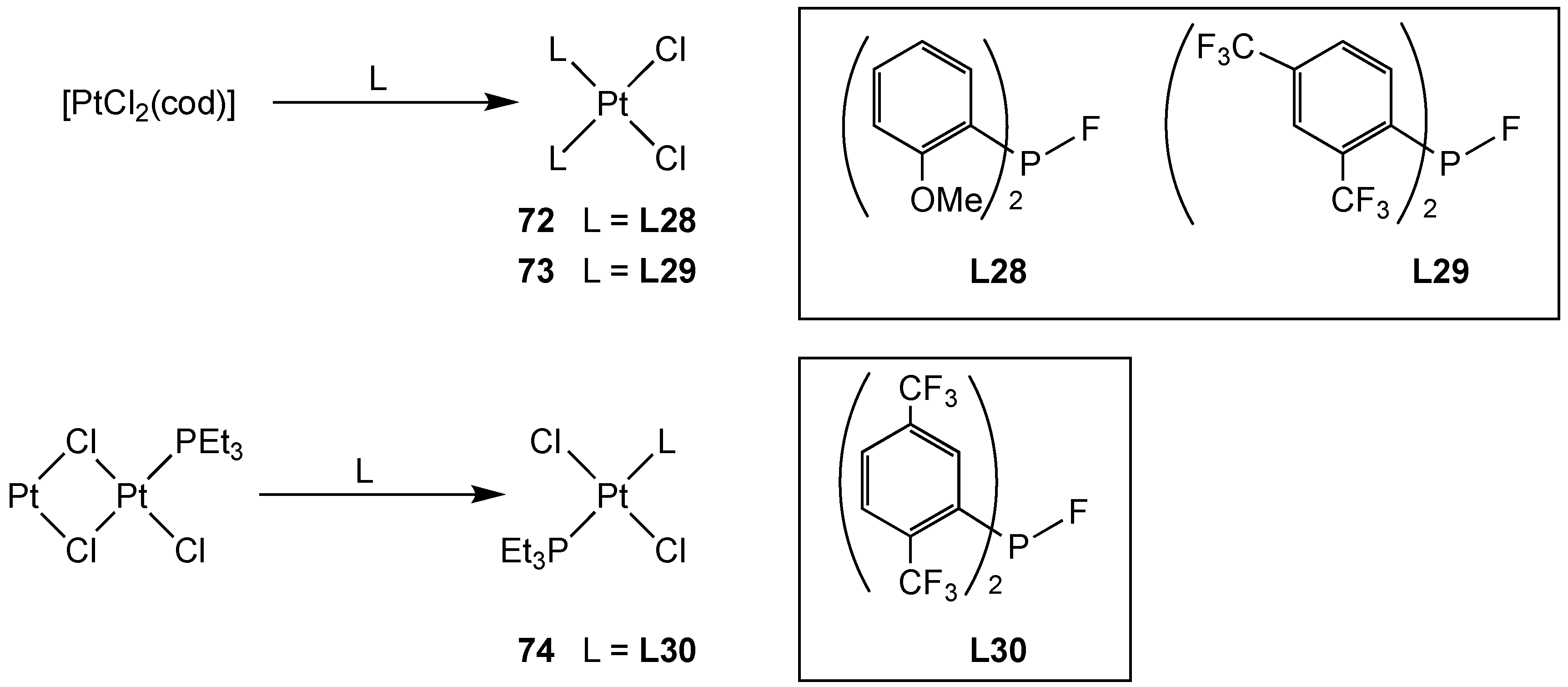
3.2.5. Group 10 Metal Complexes of Monofluorophosphines
3.3. Catalysis with Complexes of Monofluorophosphines
3.3.1. Hydroformylation Catalysis with Rhodium Complexes of Monofluorophosphines
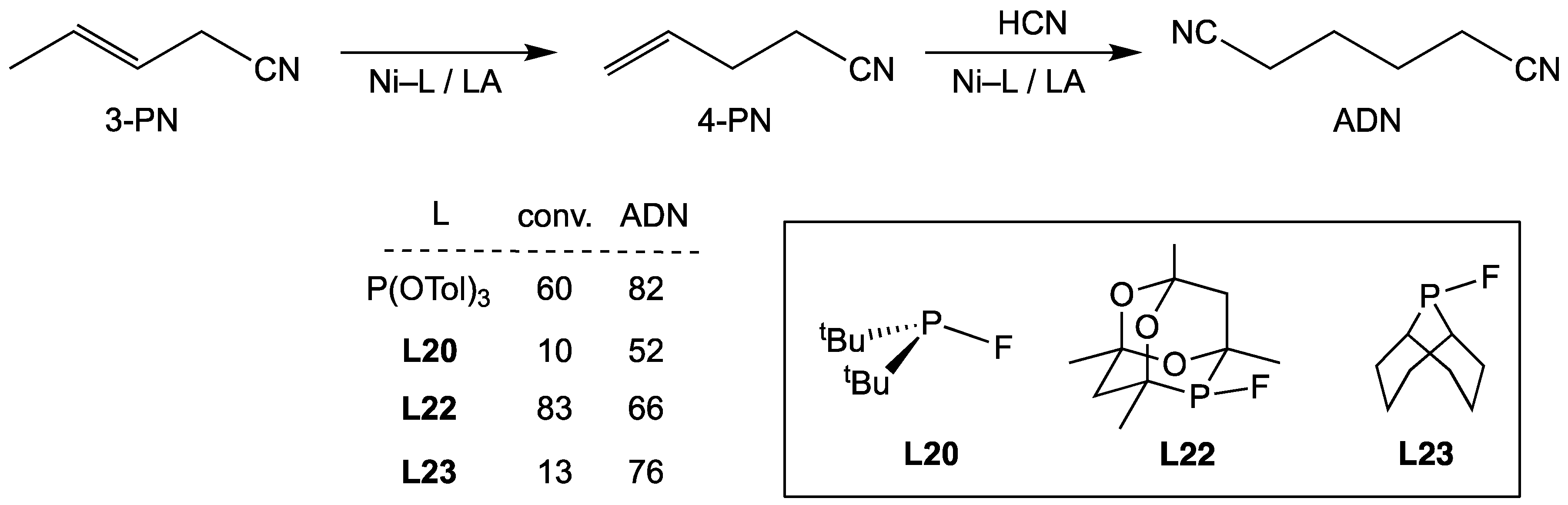
3.3.2. Hydrocyanation Catalysis with Nickel Complexes of Monofluorophosphines
4. Conclusions and Prospective Applications of Monofluorophos Ligands in Coordination Chemistry and Catalysis
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Puckette, T.A.; Struck, G.E. Hydroformylation Process Using Novel Phosphite-Metal Catalyst System. US 5840647, 24 November 1998. [Google Scholar]
- Burton, L.P.J. Antioxidant Aromatic Fluorophosphites. European Patent EP0280938B1, 15 June 1994. [Google Scholar]
- Kaprinidis, N.; Chandrika, G.; Zingg, J. Flame Retardant Compositions. World Patent WO 2004/031286 A1, 15 April 2004. [Google Scholar]
- Chemjobber blog. Fluorine: The T rex of the periodic table. Chemistry World. 30 July 2019. Available online: https://www.chemistryworld.com/opinion/fluorine-the-t-rex-of-the-periodic-table/3010748.article (accessed on 16 May 2024).
- Hansch, C.; Leo, A.; Taft, R.W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Tricas, H.; Diebolt, O.; van Leeuwen, P.W.N.M. Bulky Monophosphite Ligands for Ethene Hydroformylation. J. Catal. 2013, 298, 198–205. [Google Scholar] [CrossRef]
- Billig, E.; Abatjoglou, A.G.; Bryant, D.R. Transition Metal Complex Catalysed Processes. US 4769498, 6 September 1988. [Google Scholar]
- van Leeuwen, P.W.N.M.; Claver, C. (Eds.) Rhodium Catalyzed Hydroformylation; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar]
- Börner, A.; Franke, R. Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis; Wiley-VCH GmbH: Weinheim, Germany, 2016. [Google Scholar]
- Chakrabortty, S.; Almasalma, A.A.; de Vries, J.G. Recent Developments in Asymmetric Hydroformylation. Catal. Sci. Technol. 2021, 11, 5388–5411. [Google Scholar] [CrossRef]
- Tazawa, T.; Phanopoulos, A.; Nozaki, K. Enantioselective Hydroformylation; Wiley Online Library: Hoboken, NJ, USA, 2021. [Google Scholar]
- Nixon, J.F. Trifluorophosphine Complexes of Transition Metals. Adv. Inorg. Chem. Radiochem. 1985, 29, 41–141. [Google Scholar]
- Vargas Garcia, J.R.; Goto, T. Chemical Vapor Deposition of Iridium, Platinum, Rhodium and Palladium. Mater. Trans. 2003, 44, 1717–1728. [Google Scholar] [CrossRef]
- Tran, P.D.; Doppelt, P. Gold CVD Using Trifluorophosphine Gold(I) Chloride Precursor and Its Toluene Solutions. J. Electrochem. Soc. 2007, 154, D520–D525. [Google Scholar] [CrossRef]
- Utke, L.; Swiderek, P.; Höflich, K.; Madajska, K.; Jurczyk, J.; Martinović, P.; Szymańska, I.B. Coordination and Organometallic Precursors of Group 10 and 11: Focused Electron Beam Induced Deposition of Metals and Insight Gained from Chemical Vapour Deposition, Atomic Layer Deposition, and Fundamental Surface and Gas Phase Studies. Coord. Chem. Rev. 2022, 458, 213851. [Google Scholar] [CrossRef]
- Carpenter, A.E.; Singleton, D.G.; Kheir, S.A. Hydroformylation Catalysts Comprising Fluorophosphine Ligands and Precursors Thereof. World Patent WO 2021/202225 A1, 7 October 2021. [Google Scholar]
- Kloprogge, T.; Ponce, C.P.; Loomis, T. The Periodic Table: Nature’s Building Blocks: An Introduction to the Naturally Occurring Elements, Their Origins and Their Uses; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Heuer, L. Fluorophosphine Complexes of the Platinum Group Metals. Platin. Met. Rev. 1991, 35, 86–93. [Google Scholar] [CrossRef]
- Miles-Hobbs, A.M.; Hunt, E.; Pringle, P.G.; Sparkes, H.A. Ring Size Effects in Cyclic Fluorophosphites: Ligands That Span the Bonding Space between Phosphites and PF3. Dalton Trans. 2019, 48, 9712–9724. [Google Scholar] [CrossRef]
- Meyer, T.G.; Fischer, A.; Jones, P.G.; Schmutzler, R. Darstellung und Einkristall-Röntgenstrukturanalyse Einiger Fluorphosphite Und Phosphitester. Z. Naturforsch. B 1993, 48, 659–671. [Google Scholar] [CrossRef]
- Albers, W.; Krüger, W.; Storzer, W.; Schmutzler, R. Improved Synthesis of Halo-Phosphorus(III) Fluorides. Synth. React. Inorg. Met.-Org. Chem. 1985, 15, 187–195. [Google Scholar] [CrossRef]
- Quin, L.D. A Guide to Organophosphorus Chemistry; Wiley-Interscience: New York, NY, USA, 2000. [Google Scholar]
- Tolleson, G.S.; Puckette, T.A. Hydroformylation Process Using Chlorophosphite-Metal Catalyst System. EP 1133356 B1, 17 March 2004. [Google Scholar]
- Trillo, R.B.; Neudörfl, J.M.; Goldfuss, B. An unusually stable chlorophosphite: What makes BIFOP–Cl so robust against hydrolysis? Bellstein J. Org. Chem. 2015, 11, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Puckette, T.A. Hydroformylation Catalysis at Eastman Chemical: Generations of Catalysts. Top. Catal. 2012, 55, 421–425. [Google Scholar] [CrossRef]
- Schmutzler, R. Complexes of Organophosphorus Fluorides with Zerovalent Transition Metals. US 3242171, 22 March 1966. [Google Scholar]
- Mathieu, R.; Poilblanc, R. New Penta- and Hexasubstituted Derivatives of Group VIb Metal Hexacarbonyls. Inorg. Chem. 1972, 11, 1858–1861. [Google Scholar] [CrossRef]
- Bauer, D.P.; Ruff, J.K. Novel Iron Tetracarbonyl Fluorophosphine Complexes. Inorg. Chem. 1983, 22, 1686–1689. [Google Scholar] [CrossRef]
- Udovich, C.A.; Clark, R.J.; Haas, H. Stereochemical Nonrigidity in Iron Carbonyl Fluorophosphine Compounds. Inorg. Chem. 1969, 8, 1066–1072. [Google Scholar] [CrossRef]
- Mathew, N.; Jagirdar, B.R. Influence of the Cone Angles and the π-Acceptor Properties of Phosphorus-Containing Ligands in the Chemistry of Dihydrogen Complexes of Ruthenium. Organometallics 2000, 19, 4506–4517. [Google Scholar] [CrossRef]
- Clark, R.J.; Morgan, K.A. Methanol Solvolysis of Metal Trifluorophosphine Complexes. Inorg. Chim. Acta 1968, 2, 93–96. [Google Scholar] [CrossRef]
- Nixon, J.F.; Swain, J.R. Trifluorophosphine Complexes of Rhodium(1): Syntheses and Ligand-exchange Studies. J. Chem. Soc. Dalton Trans. 1972, 10, 1044–1048. [Google Scholar] [CrossRef]
- Hitchcock, P.B.; Morton, S.; Nixon, J.F. Fluorophosphine Complexes of Rhodium(I) and Iridium(I): Towards the Design of Systems with Extended Metal-Metal Interactions. The Crystal Structure of [{IrCl(PF3)2}2]. J. Chem. Soc. Dalton Trans. 1985, 7, 1295–1301. [Google Scholar] [CrossRef]
- Reddy, G.S.; Schmutzler, R. Phosphorus-Fluorine Chemistry. XVIII. Nuclear Magnetic Resonance Studies on Coordination Compounds Involving Fluorine-Containing Phosphine Ligands. Inorg. Chem. 1967, 6, 823–830. [Google Scholar] [CrossRef]
- Crocker, C.; Goodfellow, R.J. Heteronuclear INDOR Spectra of Some Tetrakis(Fluorophosphine)-Nickel(0) and -Platinum(0) Complexes Having the [AX]4 (Td) Spin System. J. Chem. Soc. Dalton Trans. 1977, 17, 1687–1689. [Google Scholar] [CrossRef]
- Lynden-Bell, R.M. The [AX]4 Nuclear Spin System with Tetrahedral Symmetry. Mol. Phys. 1968, 15, 523–531. [Google Scholar] [CrossRef]
- Matos, R.M.; da Costa, R.F.F.; Knupp, V.F.; Silva, J.A.D.; Passos, B.F.T. Syntheses and 31P NMR Studies of Transition Metal Complexes Containing Derivatives of Dioxaphospholane and Dioxaphosphorinane. J. Braz. Chem. Soc. 2000, 11, 311–316. [Google Scholar] [CrossRef]
- Puckette, T.A. Halophosphite Ligands for the Rhodium Catalyzed Low-Pressure Hydroformylation Reaction. In Catalysis of Organic Reactions; Schmidt, S.R., Ed.; CRC Press: Boca Raton, FL, USA, 2006; Chapter 4. [Google Scholar]
- Tau, K.D. Production of 2-Methylbutanal. US 4605781, 12 August 1986. [Google Scholar]
- Klender, G.J.; Gatto, V.J.; Jones, K.R.; Calhoun, C.W. Further Developments in the Study of Fluorophosphonite Stabilizers. In Polymer Preprints; American Chemical Society: Washington, DC, USA, 1993; Volume 34, pp. 156–157. [Google Scholar]
- Puckette, T.A.; Tolleson, G.S.; Devon, T.J.; Stavinoha, J.L. Epoxide Stabilization of Fluorophosphite-Metal Catalyst System in a Hydroformylation Process. WO 02/098825 A2, 12 December 2002. [Google Scholar]
- Puckette, T.A.; Tolleson, G.S. Stabilization of Fluorophosphite-Containing Catalysts. US 6831035 B2, 14 December 2004. [Google Scholar]
- Puckette, T.A.; Shan, X.; Rogers, J.L.; Green, B.E. Hydroformylation Catalyst. US 9550179 B1, 24 January 2017. [Google Scholar]
- Puckette, T.A.; Shan, X.; Rogers, J.L.; Green, B.E. Hydroformylation Catalyst Containing Isomerically Enriched Halophosphite. WO 2017/044277 A1, 16 March 2017. [Google Scholar]
- Zuidema, E.; Daura-Oller, E.; Carbó, J.J.; Bo, C.; van Leeuwen, P.W.N.M. Electronic Ligand Effects on the Regioselectivity of the Rhodium-Diphosphine-Catalyzed Hydroformylation of Propene. Organometallics 2007, 26, 2234–2242. [Google Scholar] [CrossRef]
- Liu, Y.-S.; Rodgers, J.L. Fluorophosphite Containing Catalysts for Hydroformylation Processes. US 7872156 B2, 18 January 2011. [Google Scholar]
- Ibrahim, M.Y.S.; Bennett, J.A.; Mason, D.; Rodgers, J.; Abolhasani, M. Flexible Homogeneous Hydroformylation: On-Demand Tuning of Aldehyde Branching with a Cyclic Fluorophosphite Ligand. J. Catal. 2022, 409, 105–117. [Google Scholar] [CrossRef]
- Puckette, T.A. Acetylene Tolerant Hydroformylation Catalysts. US 2010/0069679 A1, 18 March 2010. [Google Scholar]
- Puckette, T.A. Process for the Preparation of Glycolaldehyde. US 7301054 B1, 27 November 2007. [Google Scholar]
- Trillo, R.B.; Leven, M.; Neudörfl, J.M.; Goldfuss, B. Electronegativity Governs Enantioselectivity: Alkyl-Aryl Cross-Coupling with Fenchol-Based Palladium-Phosphorus Halide Catalysts. Adv. Synth. Catal. 2012, 354, 1451–1465. [Google Scholar] [CrossRef]
- Brüllingen, E.; Neudörfl, J.M.; Goldfuss, B. Enantioselective Cu-Catalyzed 1,4-Additions of Organozinc and Grignard Reagents to Enones: Exceptional Performance of the Hydrido-Phosphite-Ligand BIFOP-H. New J. Chem. 2019, 43, 4787–4799. [Google Scholar] [CrossRef]
- Meyer, T.G.; Jones, P.G.; Schmutzler, R. Darstellung Neuer Monofluorphosphine Und Einiger Ihrer Übergangsmetallkomplexe; Einkristall-Röntgenstrukturanalyse Eines Platin(II)-Komplexes. Z. Naturforsch. B 1993, 48, 875–885. [Google Scholar] [CrossRef]
- Seel, F.; Rudolph, K.; Gombler, W. Dimethylfluorophosphine. Angew. Chem. Int. Ed. 1967, 6, 708. [Google Scholar] [CrossRef]
- Seel, F.; Rudolph, K. Uber Dimethylfluorophosphin und Dimethyldifluorophosphoran. Z. Anorg. Allg. Chem. 1968, 363, 233–244. [Google Scholar] [CrossRef]
- Riesel, L.; Haenel, J.; Ohms, G. Zur Disproportionierung der Phenylfluorphosphane (C6H5)2PF Und (C6H5)PF2. J. Fluor. Chem. 1988, 38, 335–340. [Google Scholar] [CrossRef]
- Haenel, J.; Ohms, G.; Riesel, L. Die Dimerisierung yon Di(n-buty1)fluorphosphan und seine Reaktion mit Benzaldehyd. Z. Anorg. Allg. Chem. 1992, 607, 161–163. [Google Scholar] [CrossRef]
- Brown, C.; Murray, M.; Schmutzler, R. Fluorodiphenylphosphine. J. Chem. Soc. C 1970, 878–881. [Google Scholar] [CrossRef]
- Schmutzler, R.; Stelzer, O.; Liebman, J.F. Catalytic and Autocatalytic Disproportionation Reactions of Fluorophosphines and Related Lower Valence Nonmetal Fluorides. J. Fluor. Chem. 1984, 25, 289–299. [Google Scholar] [CrossRef]
- Fild, M.; Schmutzler, R. Phosphorus-Fluorine Chemistry. Part XXIII. t-Butyl-Fluorophosphines and -Fluorophosphoranes and Their Derivatives. J. Chem. Soc. A 1970, 2359–2364. [Google Scholar] [CrossRef]
- Fild, M.; Schmutzler, R. Phosphorus-Fluorine Chemistry. Part XXI. Pentafluorophenylfluorophosphines and Pentafluorophenylfluorophosphoranes. J. Chem. Soc. A 1969, 840–843. [Google Scholar] [CrossRef]
- Fey, N.; Garland, M.; Hopewell, J.P.; McMullin, C.L.; Mastroianni, S.; Orpen, A.G.; Pringle, P.G. Stable Fluorophosphines: Predicted and Realized Ligands for Catalysis. Angew. Chem. Int. Ed. 2012, 51, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Szynkiewicz, N.; Ponikiewski, L.; Grubba, R. Symmetrical and unsymmetrical diphosphanes with diversified alkyl, aryl, and amino substituents. Dalton Trans. 2018, 47, 16885–16894. [Google Scholar] [CrossRef] [PubMed]
- Becker, G.; Golla, W.; Grobe, J.; Klinkhammer, K.W. Element-Element Bonds. IX. Structures of Tetrakis(trifluoromethyl)diphosphane and -diarsane: Experimental and Theoretical Investigations. Inorg. Chem. 1999, 38, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Dodds, D.L.; Floure, J.; Garland, M.; Haddow, M.F.; Leonard, T.R.; McMullin, C.L.; Orpen, A.G.; Pringle, P.G. Diphosphanes derived from phobane and phosphatrioxa-adamantane: Similarities, differences and anomalies. Dalton Trans. 2011, 40, 7137–7146. [Google Scholar] [CrossRef] [PubMed]
- Downing, J.H.; Floure, J.; Heslop, K.; Haddow, M.F.; Hopewell, J.; Lusi, M.; Hirahataya Phetmung, H.; Orpen, A.G.; Pringle, P.G.; Pugh, R.I.; et al. General Routes to Alkyl Phosphatrioxaadamantane Ligands. Organometallics 2008, 27, 3216–3224. [Google Scholar] [CrossRef]
- Carreira, M.; Charernsuk, M.; Eberhard, M.; Fey, N.; van Ginkel, R.; Hamilton, A.; Mul, W.P.; Orpen, A.G.; Phetmung, H.; Pringle, P.G. Anatomy of Phobanes. Diastereoselective Synthesis of the Three Isomers of n-Butylphobane and a Comparison of their Donor Properties. J. Am. Chem. Soc. 2009, 131, 3078–30929. [Google Scholar] [CrossRef] [PubMed]
- Lister, J.M.; Carreira, M.; Haddow, M.F.; Hamilton, A.; McMullin, C.L.; Orpen, A.G.; Pringle, P.G.; Stennett, T.E. Unexpectedly High Barriers to M–P Rotation in Tertiary Phobane Complexes: PhobPR Behavior That Is Commensurate with tBu2PR. Organometallics 2014, 33, 702–714. [Google Scholar] [CrossRef]
- Deiters, J.A.; Holmes, R.R.; Holmes, J.M. Fluorine and Chlorine Apicophilicities in Five-Coordinated Phosphorus and Silicon Compounds via Molecular Orbital Calculations, A Model for Nucleophilic Substitution. J. Am. Chem. Soc. 1988, 110, 7672–7681. [Google Scholar] [CrossRef]
- Hófler, M.; Stubenrauch, M.; Rlcharz, E. A New Method for the Preparation of Monofluorophosphane Complexes. Isolation of [(CO)5CrP(NEt2Me)Et2][BF4]. Organometallics 1987, 6, 198–199. [Google Scholar] [CrossRef]
- Yih, K.-H. Syntheses and Characterization of Molybdenum Complexes with the (1,3-Dithioliumyl)diphenylphosphine Containing Ligands. J. Chin. Chem. Soc. 1999, 46, 535–538. [Google Scholar] [CrossRef]
- Yih, K.-H.; Lee, G.-H.; Wang, Y. Syntheses and Crystal Structures of Tungsten Complexes with Various Ligands Containing (1,3-Dithioliumyl)diphenylphosphine. Organometallics 2001, 20, 2604–2610. [Google Scholar] [CrossRef]
- Stelzer, O.; Schmutzler, R. Phosphorus-Fluorine Chemistry. Part XXVIII. Fluorophosphines with Bulky Substituents as Ligands in Transition Metal Carbonyl Complexes. J. Chem. Soc. A 1971, 2867–2873. [Google Scholar] [CrossRef]
- Grobe, J.; Le Van, D.; Meyring, W. Chrom- und Wolframpentacarbonylkomplexe von Bis(Trifluormethyl)Phosphanen des Typs (F3C)2PX’ (X’ = H, F, Cl, Br, I, NEt2). Z. Anorg. Allg. Chem. 1990, 586, 149–158. [Google Scholar] [CrossRef]
- Barlow, C.G.; Nixon, J.F.; Webster, M. The Chemistry of Phosphorus-Fluorine Compounds. Part IX. Preparation and Spectroscopic Studies of Fluorophosphine-Molybdenum Carbonyl Complexes. J. Chem. Soc. A 1968, 2216–2223. [Google Scholar] [CrossRef]
- Clarke, M.L.; Holliday, G.L.; Slawin, A.M.Z.; Woollins, J.D. Highly electron rich alkyl- and dialkyl-N-pyrrolidinyl phosphines: An evaluation of their electronic and structural properties. J. Chem. Soc. Dalton Trans. 2002, 6, 1093–1103. [Google Scholar] [CrossRef]
- Dobbie, R.C. Action of Bistrifluoromethylphosphino-Compounds on Pentacarbonyl-Manganese Hydride. J. Chem. Soc. A 1971, 230–233. [Google Scholar] [CrossRef]
- Hoidn, C.M.; Leitl, J.; Ziegler, C.G.P.; Shenderovich, I.G.; Wolf, R. Halide-Substituted Phosphacyclohexadienyl Iron Complexes: Covalent Structures vs. Ion Pairs. Eur. J. Inorg. Chem. 2019, 2019, 1567–1574. [Google Scholar] [CrossRef]
- Oberhammer, H.; Schmutzler, R.; Stelzer, O. Molecular Structures of Phosphorus Compounds. 6. An Electron Diffraction Study of tert-Butylfluorophosphines ButnPF3-n (n = 1, 2, 3). Inorg. Chem. 1978, 17, 1254–1258. [Google Scholar] [CrossRef]
- Heuer, L.; Schomburg, D. tBu2PF as a Ligand in Tri-osmium Clusters. J. Organomet. Chem. 1995, 495, 53–59. [Google Scholar] [CrossRef]
- Stelzer, O.; Unger, E. Alkyl- and Aryl-Fluorophosphines as Ligands in Transition-Metal Complexes with Metals in Positive Oxidation States. Part I. Nickel(II) and Cobalt(II) Halide Complexes of Di-(t-Butyl)Fluorophosphine. J. Chem. Soc. Dalton Trans. 1973, 17, 1783–1788. [Google Scholar] [CrossRef]
- Dobbie, R.C.; Morton, S. Trifluoromethylphosphine Complexes of Tricarbonylnitrosylcobalt. J. Chem. Soc. Dalton Trans. 1976, 14, 1421–1423. [Google Scholar] [CrossRef]
- Macgregor, S.A.; Roe, D.C.; Marshall, W.J.; Bloch, K.M. The F/Ph Rearrangement Reaction of [(Ph3P)3RhF]. J. Am. Chem. Soc. 2005, 127, 15304–15321. [Google Scholar] [CrossRef] [PubMed]
- Burg, A.B.; Street, G.B. Perfluoromethylphosphine-Nickel Compounds, Including a New Volatile Heterocycle. Inorg. Chem. 1966, 5, 1532–1537. [Google Scholar] [CrossRef]
- Nixon, J.F. The Chemistry of Phosphorus-Fluorine Compounds. Part VIII. Synthesis and Nuclear Magnetic Resonance Spectra of Tetrakisfluorophosphine Derivatives of Zerovalent Nickel. J. Chem. Soc. A 1967, 1136–1139. [Google Scholar] [CrossRef]
- Nixon, J.F.; Sexton, M.D. Phosphorus-Fluorine Compounds. Part XIV. Direct Syntheses of Tetrakis(Fluorophosphine) Complexes of Zerovalent Nickel. J. Chem. Soc. A 1969, 1089–1091. [Google Scholar] [CrossRef]
- Sheldrick, W.S.; Stelzer, O. Preparation, Crystal and Molecular Structure of trans-Dibromobis[di(t-butyl)fluorophosphine]nickel(II). J. Chem. Soc. Dalton 1973, 9, 926–929. [Google Scholar] [CrossRef]
- Pabel, M.; Willis, A.C.; Wild, S.B. First Resolution of a Free Fluorophosphine Chiral at Phosphorus. Resolution and Reactions of Free and Coordinated (±)-Fluorophenylisopropylphosphine. Inorg. Chem. 1996, 35, 1244–1249. [Google Scholar] [CrossRef]
- Nixon, J.F.; Sexton, M.D. Phosphorus-Fluorine Compounds. Part XVII. Fluorophosphine Complexes of Zerovalent Platinum. J. Chem. Soc. A 1970, 321–323. [Google Scholar] [CrossRef]
- Al-Ohaly, A.-R.; Nixon, J.F. 31P Nuclear Magnetic Resonance Spectroscopic Studies on Some Zerovalent Platinum Phosphine Complexes. Inorg. Chim. Acta 1980, 47, 105–109. [Google Scholar] [CrossRef]
- Heuer, L.; Jones, P.G.; Schmutzler, R. Preparation of Bis[2,4-bis(trifluoromethyl)phenyl]fluorophosphine and 2,4-Bis(trifluoromethyl)phenyl-[2,6-bis(trifluoromethyl)phenyl]fluorophosphine—Two Distillable Monofluorophosphines. J. Fluor. Chem. 1990, 46, 243–254. [Google Scholar] [CrossRef]
- Capel, V.L.; Dillon, K.B.; Goeta, A.E.; Howard, J.A.K.; Monks, P.K.; Probert, M.R.; Shepherd, H.J.; Zorina, N.V. Stereochemically Inactive Lone Pairs in Phosphorus(III) Compounds: The Characterisation of Some Derivatives with the 2,5-(CF3)2C6H3 (Ar) Substituent and Their Complexation Behaviour towards Pt(II) Species. Dalton Trans. 2011, 40, 1808–1816. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, P.W.N.M.; Nicolas, D.; Cleément, N.D.; Mathieu, J.-L.; Tschan, M.J.-L. New processes for the selective production of 1-octene. Coord. Chem. Rev. 2011, 255, 1499–1517. [Google Scholar] [CrossRef]
- Mastroianni, S.; Pringle, P.; Garland, M.; Hopewell, J. Method for the Production of Nitrile Compounds from Ethylenically-Unsaturated Compounds. WO 2010/145960 A1, 23 December 2010. [Google Scholar]
- Mastroianni, S.; Pringle, P.; Hopewell, J.; Garland, M. Method for Producing Nitrile Compounds from Ethylenically Unsaturated Compounds. WO 2013/045524 A1, 4 April 2013. [Google Scholar]
- Michalski, J.; Wojciech Dabkowski, W. New chemistry and stereochemistry of tricoordinate phosphorus esters containing phosphorus–fluorine bond. Comptes Rendus. Chim. 2004, 7, 901–907. [Google Scholar] [CrossRef]
- Puckette, T.A. Amido-Fluorophosphite Compounds and Catalysts. US 8492593 B2, 23 July 2013. [Google Scholar]
- Eickhoff, L.; Kramer, P.; Bresien, J.; Michalik, D.; Villinger, A.; Schulz, A. On the Dynamic Behaviour of Pacman Phosphanes—A Case of Cooperativity and Redox Isomerism. Inorg. Chem. 2023, 62, 6768–6778. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miles-Hobbs, A.M.; Pringle, P.G.; Woollins, J.D.; Good, D. Monofluorophos–Metal Complexes: Ripe for Future Discoveries in Homogeneous Catalysis. Molecules 2024, 29, 2368. https://doi.org/10.3390/molecules29102368
Miles-Hobbs AM, Pringle PG, Woollins JD, Good D. Monofluorophos–Metal Complexes: Ripe for Future Discoveries in Homogeneous Catalysis. Molecules. 2024; 29(10):2368. https://doi.org/10.3390/molecules29102368
Chicago/Turabian StyleMiles-Hobbs, Alexandra M., Paul G. Pringle, J. Derek Woollins, and Daniel Good. 2024. "Monofluorophos–Metal Complexes: Ripe for Future Discoveries in Homogeneous Catalysis" Molecules 29, no. 10: 2368. https://doi.org/10.3390/molecules29102368
APA StyleMiles-Hobbs, A. M., Pringle, P. G., Woollins, J. D., & Good, D. (2024). Monofluorophos–Metal Complexes: Ripe for Future Discoveries in Homogeneous Catalysis. Molecules, 29(10), 2368. https://doi.org/10.3390/molecules29102368