
The Cholinergic Selectivity of FDA-Approved and Metabolite Compounds Examined with Molecular-Docking-Based Virtual Screening

Abstract

1. Introduction
2. Results
2.1. Molecular Docking
2.1.1. FDA-Approved Dataset—AChE
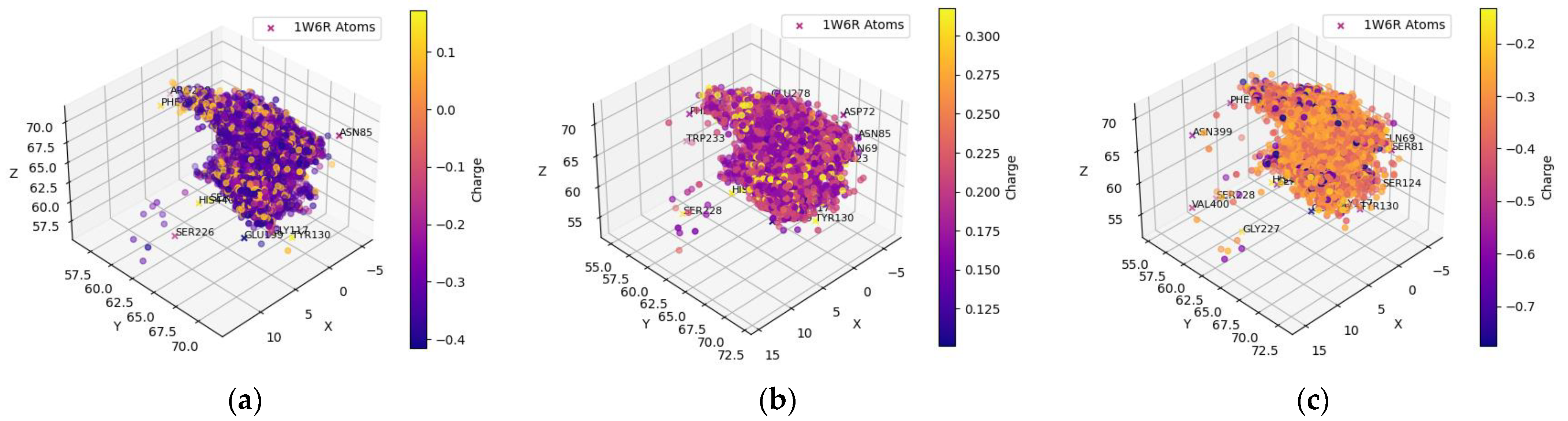
2.1.2. FDA-Approved Dataset—BChE
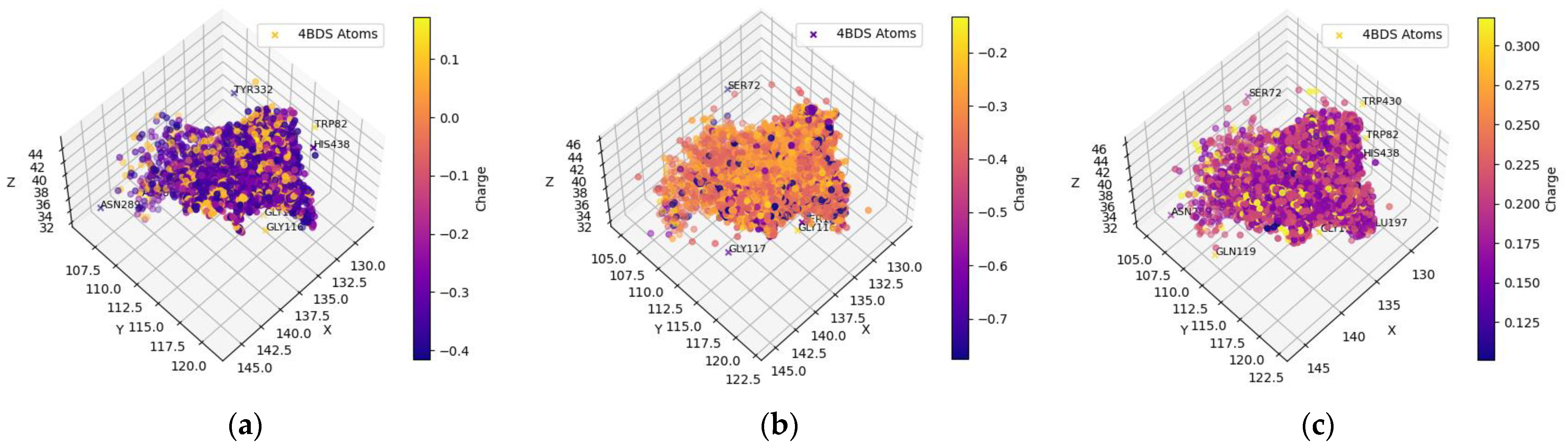

2.1.3. Clean-Metabolites-In-Vivo Dataset—AChE
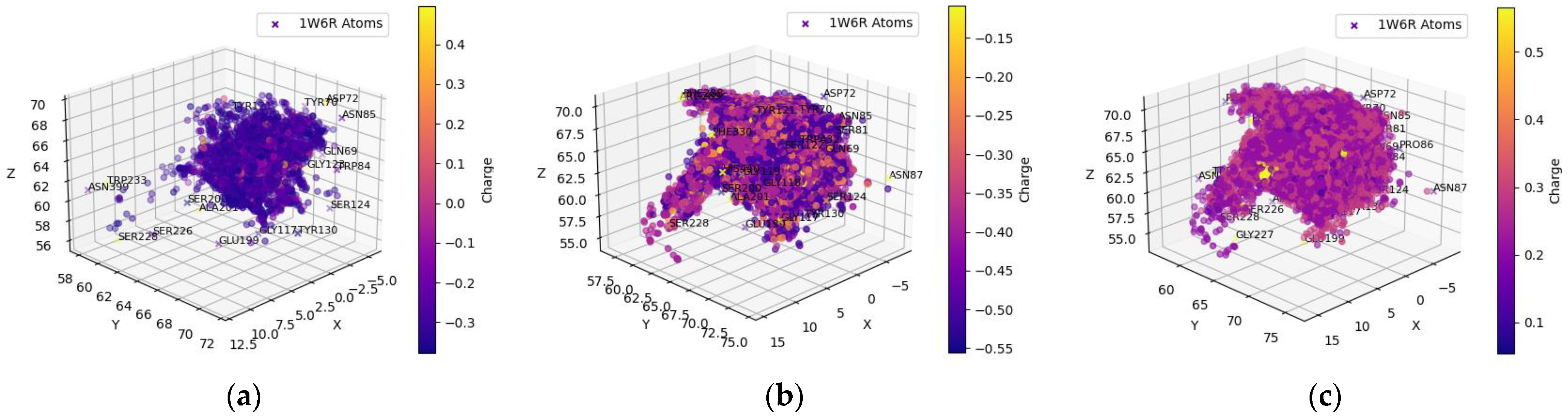
2.1.4. Clean-Metabolites-In-Vivo Dataset—BChE
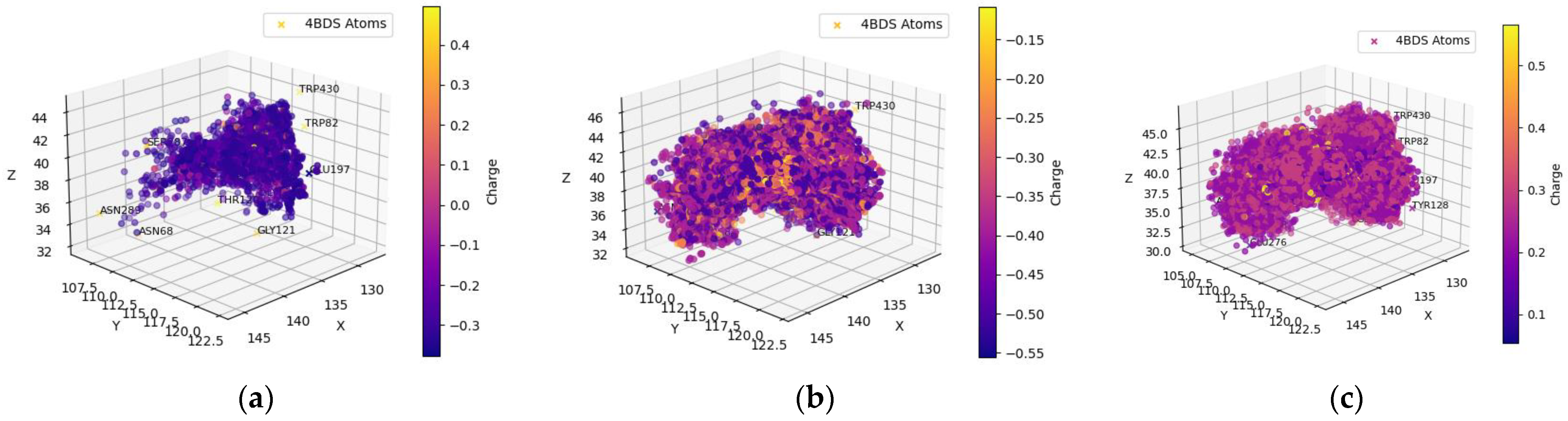

3. Discussion
3.1. Molecular Docking
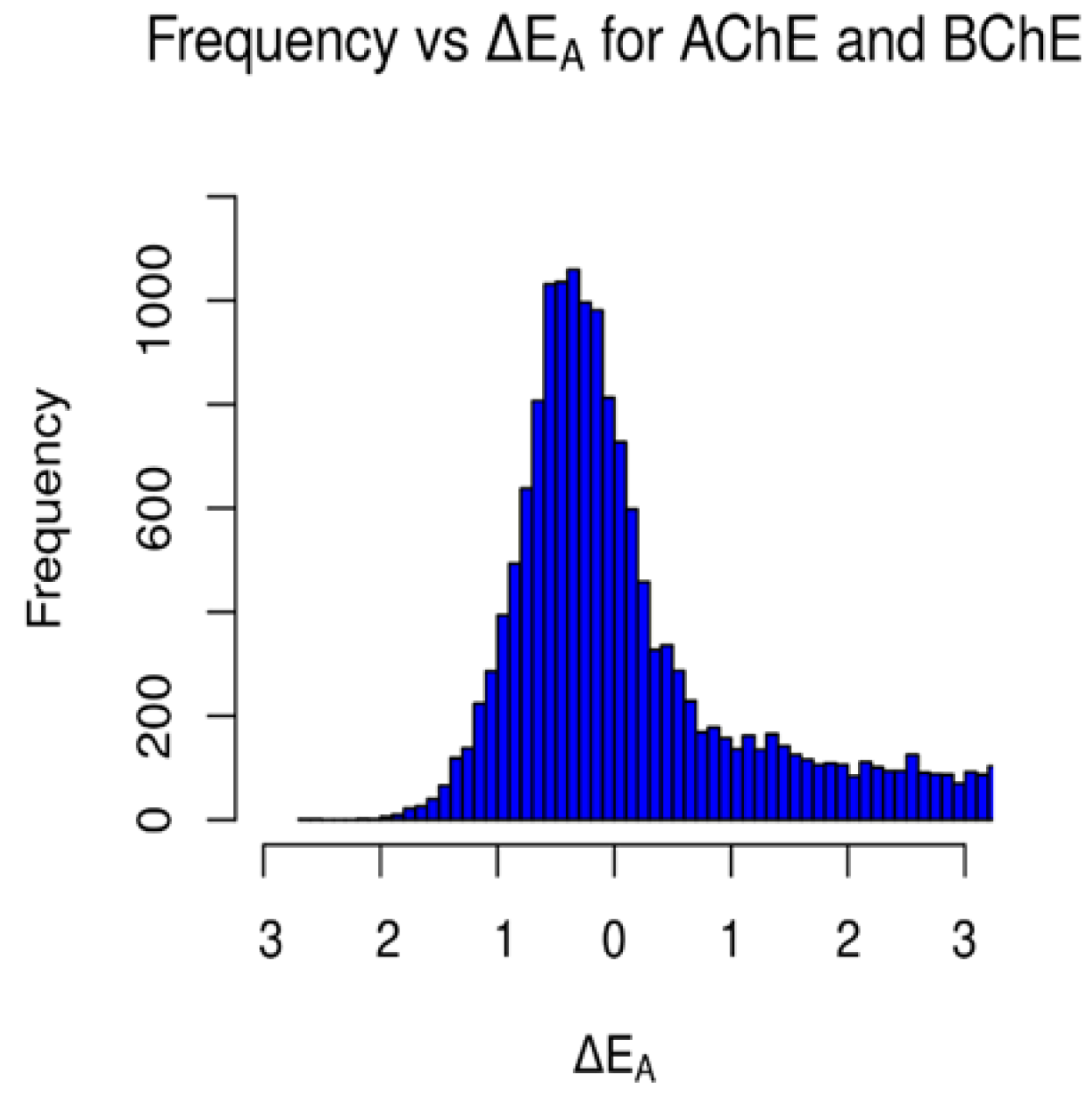
3.2. The Selectivity between AChE and BChE
4. Materials and Methods
4.1. Molecular Docking
4.1.1. File Preparation
4.1.2. Docking Parameters
4.2. Docking Analysis
4.2.1. Binding Affinity
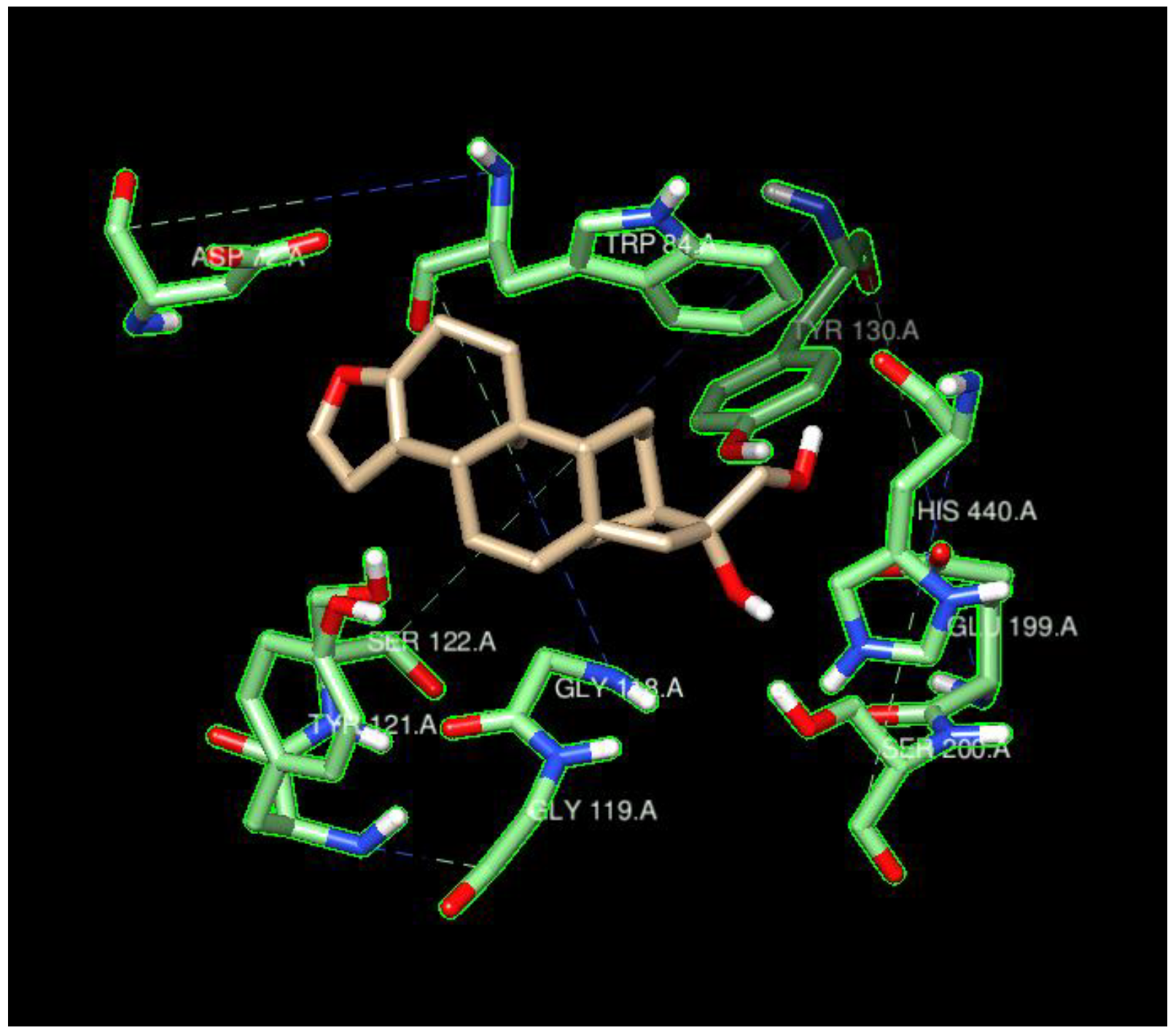
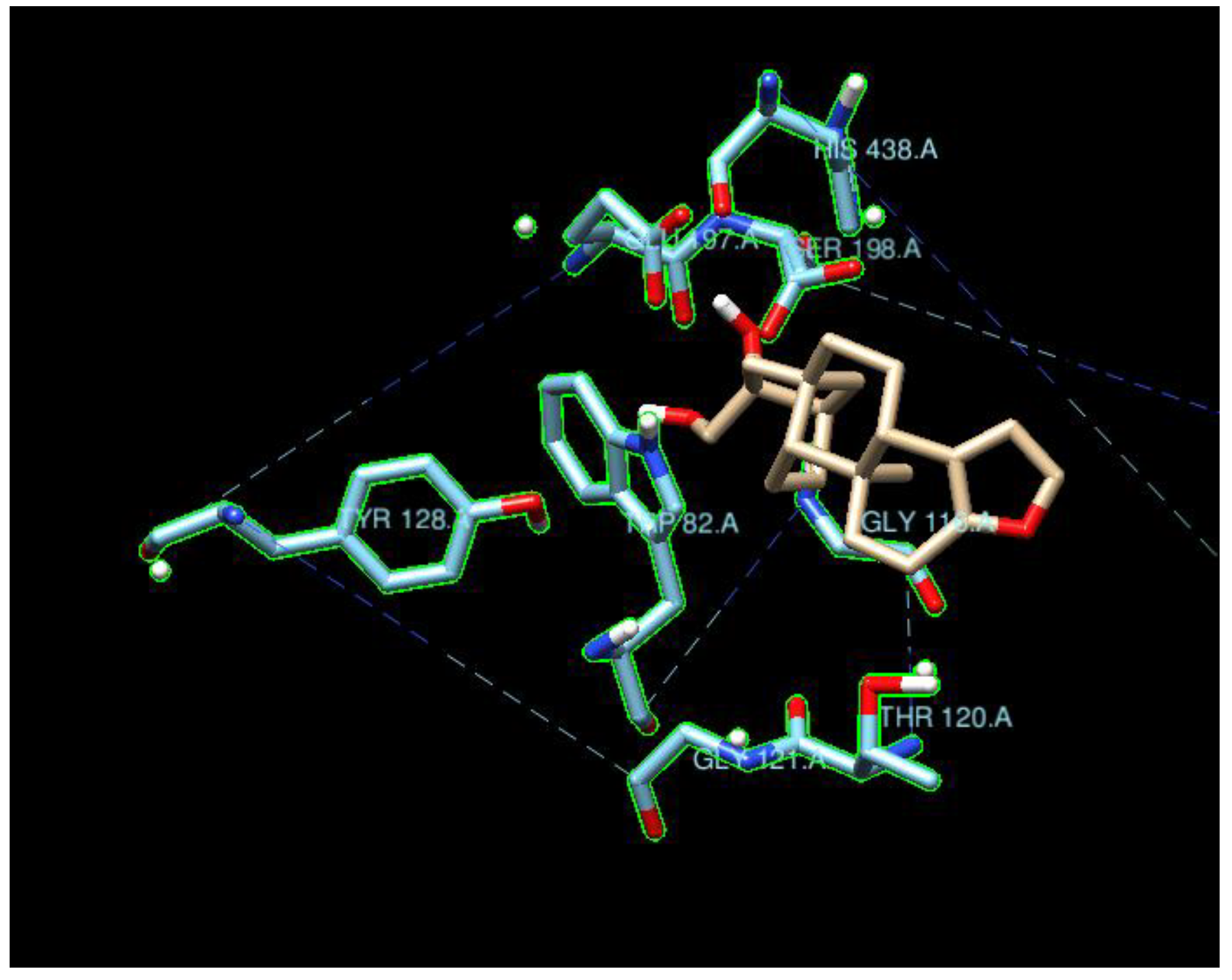
4.2.2. Residues of Interest
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pezzementi, L.; Chatonnet, A. Evolution of cholinesterases in the animal kingdom. Chem.-Biol. Interact. 2010, 187, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Haga, T. Molecular properties of muscarinic acetylcholine receptors. Proc. Jpn. Acad. Ser. B 2013, 89, 226–256. [Google Scholar] [CrossRef]
- Unwin, N. Nicotinic acetylcholine receptor and the structural basis of neuromuscular transmission: Insights from Torpedo postsynaptic membranes. Q. Rev. Biophys. 2013, 46, 283–322. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A Review of Butyrylcholinesterase as a Therapeutic Target in the Treatment of Alzheimer’s Disease. Prim. Care Companion CNS Disord. 2013, 15, 26731. [Google Scholar] [CrossRef]
- Li, Q.; Yang, H.-Y.; Chen, Y.; Sun, H. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 132, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Redman, A.M.G.; Jiang, X.; Lockridge, O.; Docto, B.P. Differences in active-site gorge dimensions of cholinesterases revealed by binding of inhibitors to human butyrylcholinesterase. Chem.-Biol. Interact. 1999, 119–120, 61–69. [Google Scholar] [CrossRef]
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D Structure to Function. Chem.-Biol. Interact. 2010, 187, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M.; Dobes, P. Caffeine Inhibits Acetylcholinesterase but Not Butyrylcholinesterase. Int. J. Mol. Sci. 2013, 14, 9873–9882. [Google Scholar] [CrossRef] [PubMed]
- Radić, Z.; Pickering, N.A.; Vellom, D.C.; Camp, S.; Taylor, P. Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry 1993, 32, 12074–12084. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Greenblatt, H.M.; Guillou, C.; Guénard, D.; Argaman, A.; Botti, S.; Badet, B.; Thal, C.; Silman, I.; Sussman, J.L. The Complex of a Bivalent Derivative of Galanthamine with Torpedo Acetylcholinesterase Displays Drastic Deformation of the Active-Site Gorge: Implications for Structure-Based Drug Design. J. Am. Chem. Soc. 2004, 126, 15405–15411. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.-Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: Elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Boerner, T.J.; Deems, S.; Furlani, T.R.; Knuth, S.L.; Towns, J. ACCESS: Advancing Innovation: NSF’s Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support. In Proceedings of the PEARC ’23: Practice and Experience in Advanced Research Computing, Portland, OR, USA, 23–27 July 2023; ACM: New York, NY, USA, 2023. 4p. Available online: https://dl.acm.org/doi/10.1145/3569951.3597559 (accessed on 24 March 2024).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | ΔEA | 1st EA | <EA> | <rmsd l.b.> | <rmsd u.b.> | |
|---|---|---|---|---|---|---|
| Ambrisentan | 1.9 | −10.8 | −9.0 | 0.97 | 1.855 | 3.895 |
| Methadone | 1.9 | −9.9 | −8.9 | 0.60 | 1.696 | 4.326 |
| Triamterene | 1.8 | −9.6 | −8.4 | 0.45 | 2.176 | 4.693 |
| Prednisone | 1.7 | −11.0 | −8.7 | 1.20 | 1.639 | 4.357 |
| Metaxalone | 1.6 | −8.8 | −8.0 | 0.59 | 2.085 | 3.697 |
| Doxapram | 1.5 | −10.9 | −9.2 | 1.07 | 1.617 | 3.620 |
| Ligand | ΔEA | 1st EA | <EA> | <rmsd l.b.> | <rmsd u.b.> | |
|---|---|---|---|---|---|---|
| Ergotamine | 12.6 | −12.0 | −11.5 | 0.31 | 2.568 | 8.325 |
| Ciclesonide | 9.9 | −11.8 | −11.0 | 0.49 | 2.789 | 5.359 |
| Suvorexant | 7.0 | −10.1 | −9.1 | 0.30 | 4.740 | 8.674 |
| Nintedanib | 6.8 | −10.6 | −10.2 | 0.24 | 2.664 | 6.214 |
| Lurasidone | 6.0 | −11.1 | −11.1 | 0.55 | 2.985 | 5.905 |
| Amcinonide | 5.0 | −10.9 | −10.2 | 0.40 | 3.320 | 6.541 |
| Ligand | ΔEA | 1st EA | <EA> | <rmsd l.b.> | <rmsd u.b.> | |
|---|---|---|---|---|---|---|
| Afzelchin | 2.1 | −10.2 | −8.3 | 0.95 | 1.937 | 6.013 |
| Aminoclonazepam | 1.9 | −10.7 | −9.3 | 0.78 | 2.708 | 4.454 |
| Dihydroisorhamnetin | 1.7 | −10.2 | −9.0 | 0.68 | 1.254 | 5.850 |
| Bisphenol A | 1.5 | −9.3 | −8.7 | 0.42 | 1.989 | 5.145 |
| N-cinnamoyloctopamin | 1.3 | −10.4 | −10.0 | 0.24 | 2.816 | 4.520 |
| Ligand | ΔEA | 1st EA | <EA> | <rmsd l.b.> | <rmsd u.b.> | |
|---|---|---|---|---|---|---|
| Azukisapogenol | 15.0 | −10.5 | −10.1 | 0.23 | 1.926 | 2.790 |
| D-maslinic acid | 11.0 | −10.6 | −10.2 | 0.25 | 1.687 | 4.801 |
| Isoliensinine | 7.4 | −11.3 | −10.9 | 0.26 | 2.491 | 6.986 |
| Dukunolide D | 4.8 | −11.0 | −10.8 | 0.13 | 2.211 | 5.002 |
| Zanthobisquinolone | 3.2 | −11.1 | −10.7 | 0.23 | 1.770 | 5.954 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambardella, M.D.; Wang, Y.; Pang, J. The Cholinergic Selectivity of FDA-Approved and Metabolite Compounds Examined with Molecular-Docking-Based Virtual Screening. Molecules 2024, 29, 2333. https://doi.org/10.3390/molecules29102333
Gambardella MD, Wang Y, Pang J. The Cholinergic Selectivity of FDA-Approved and Metabolite Compounds Examined with Molecular-Docking-Based Virtual Screening. Molecules. 2024; 29(10):2333. https://doi.org/10.3390/molecules29102333
Chicago/Turabian StyleGambardella, Michael D., Yigui Wang, and Jiongdong Pang. 2024. "The Cholinergic Selectivity of FDA-Approved and Metabolite Compounds Examined with Molecular-Docking-Based Virtual Screening" Molecules 29, no. 10: 2333. https://doi.org/10.3390/molecules29102333
APA StyleGambardella, M. D., Wang, Y., & Pang, J. (2024). The Cholinergic Selectivity of FDA-Approved and Metabolite Compounds Examined with Molecular-Docking-Based Virtual Screening. Molecules, 29(10), 2333. https://doi.org/10.3390/molecules29102333