

Pan-EGFR Inhibitor Dacomitinib Resensitizes Paclitaxel and Induces Apoptosis via Elevating Intracellular ROS Levels in Ovarian Cancer SKOV3-TR Cells


Abstract
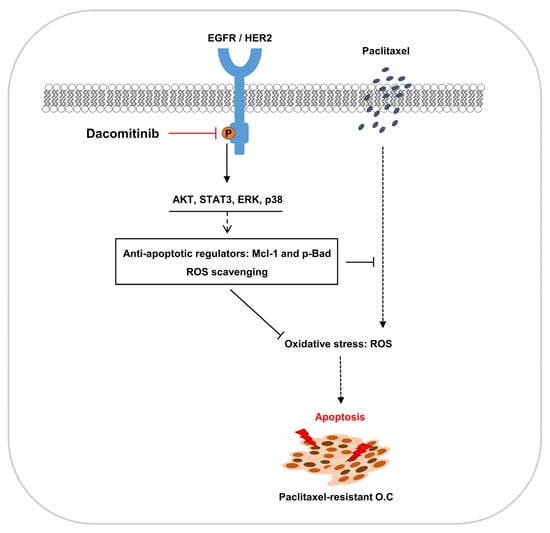
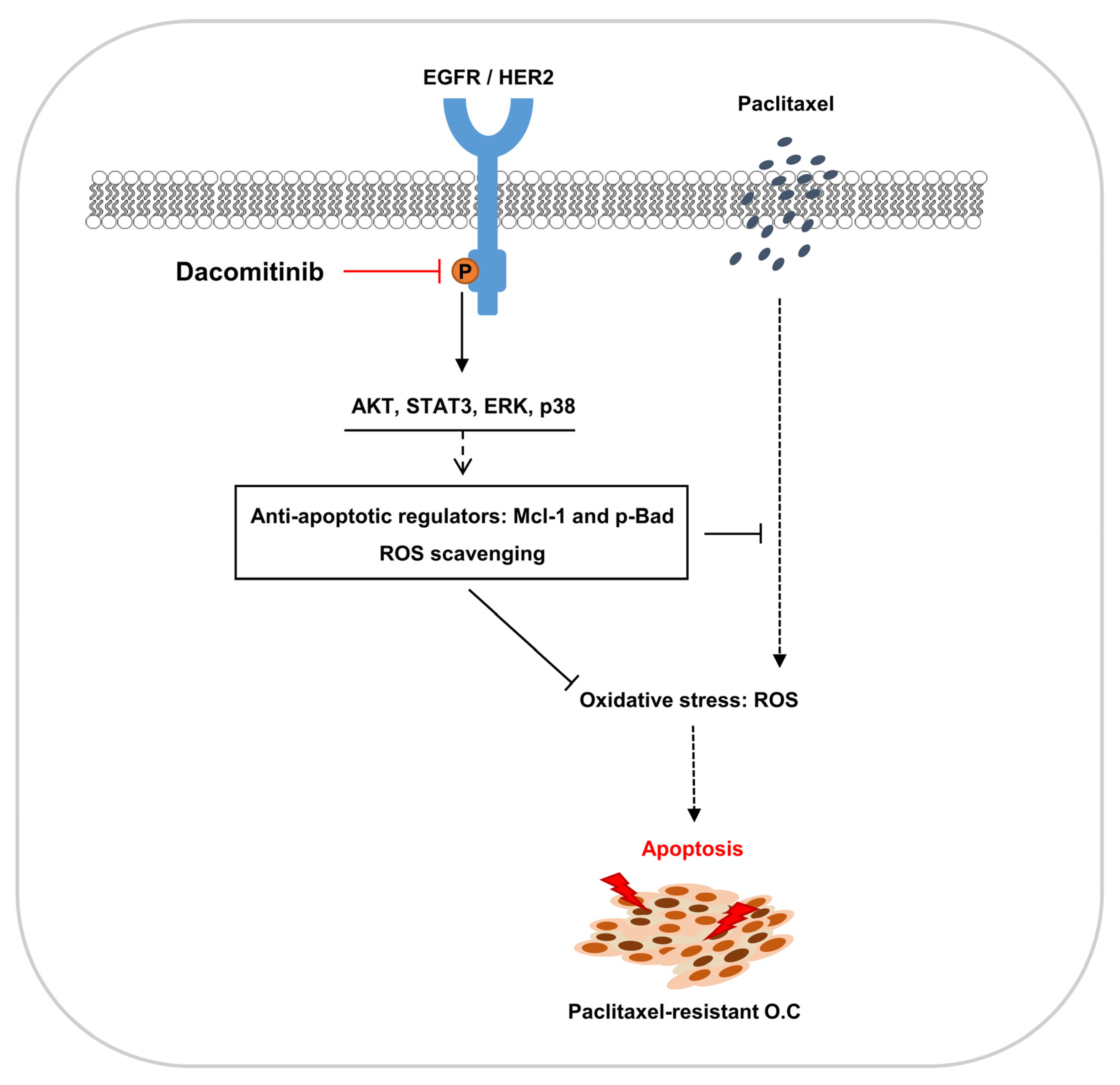
1. Introduction
2. Results
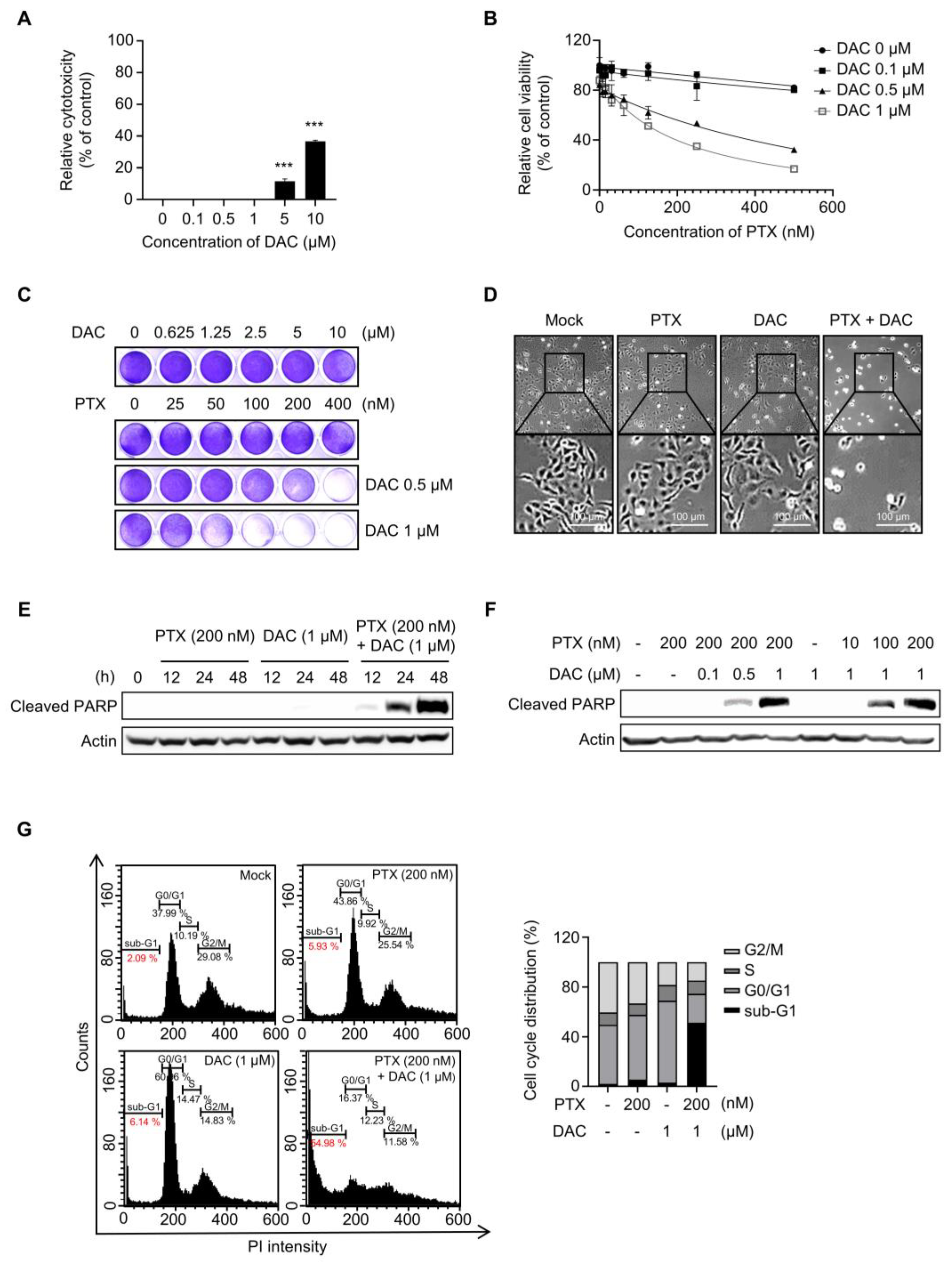
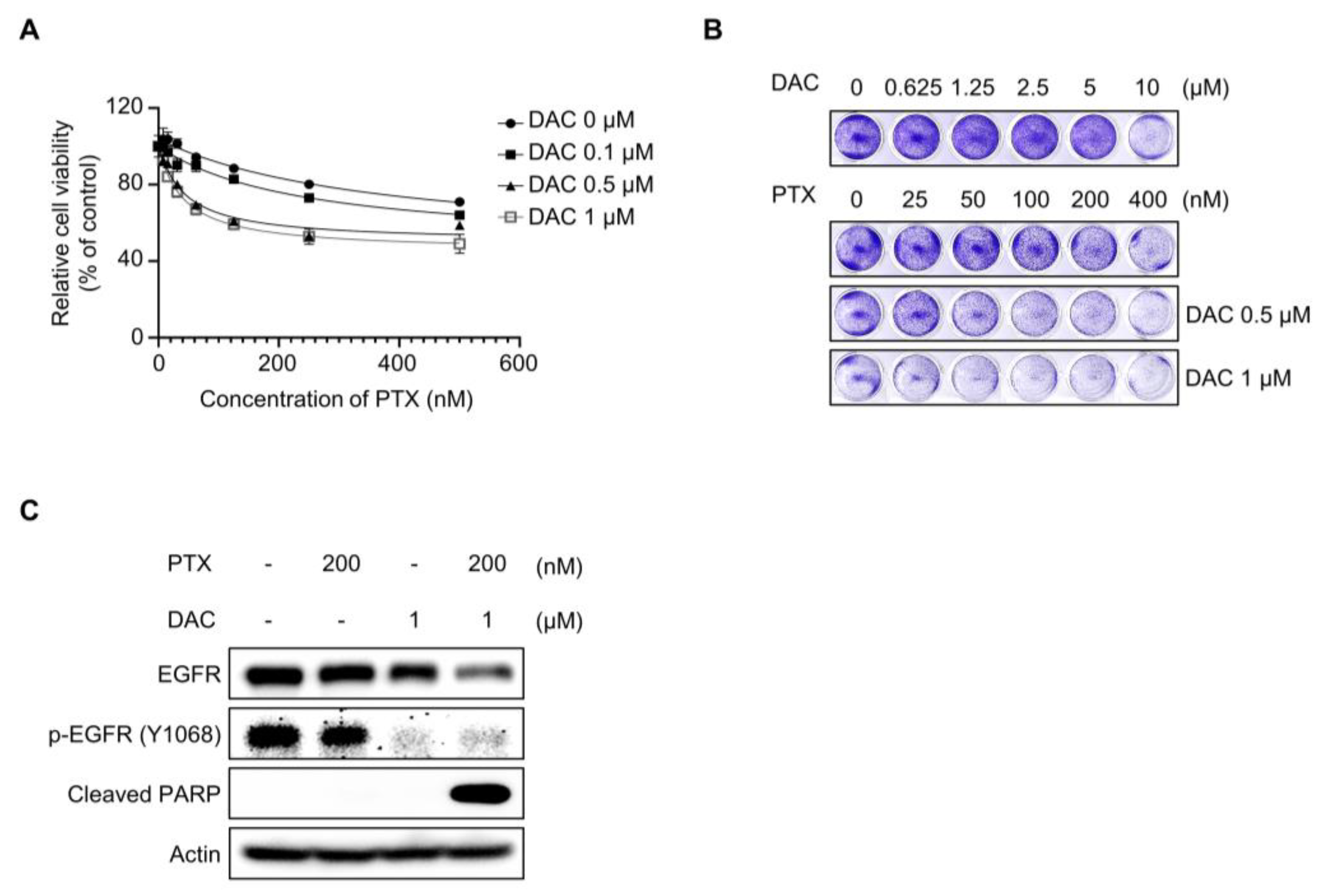
2.1. Dacomitinib Induced Cytotoxicity in Paclitaxel-Resistant Ovarian Cancer SKOV3-TR Cells
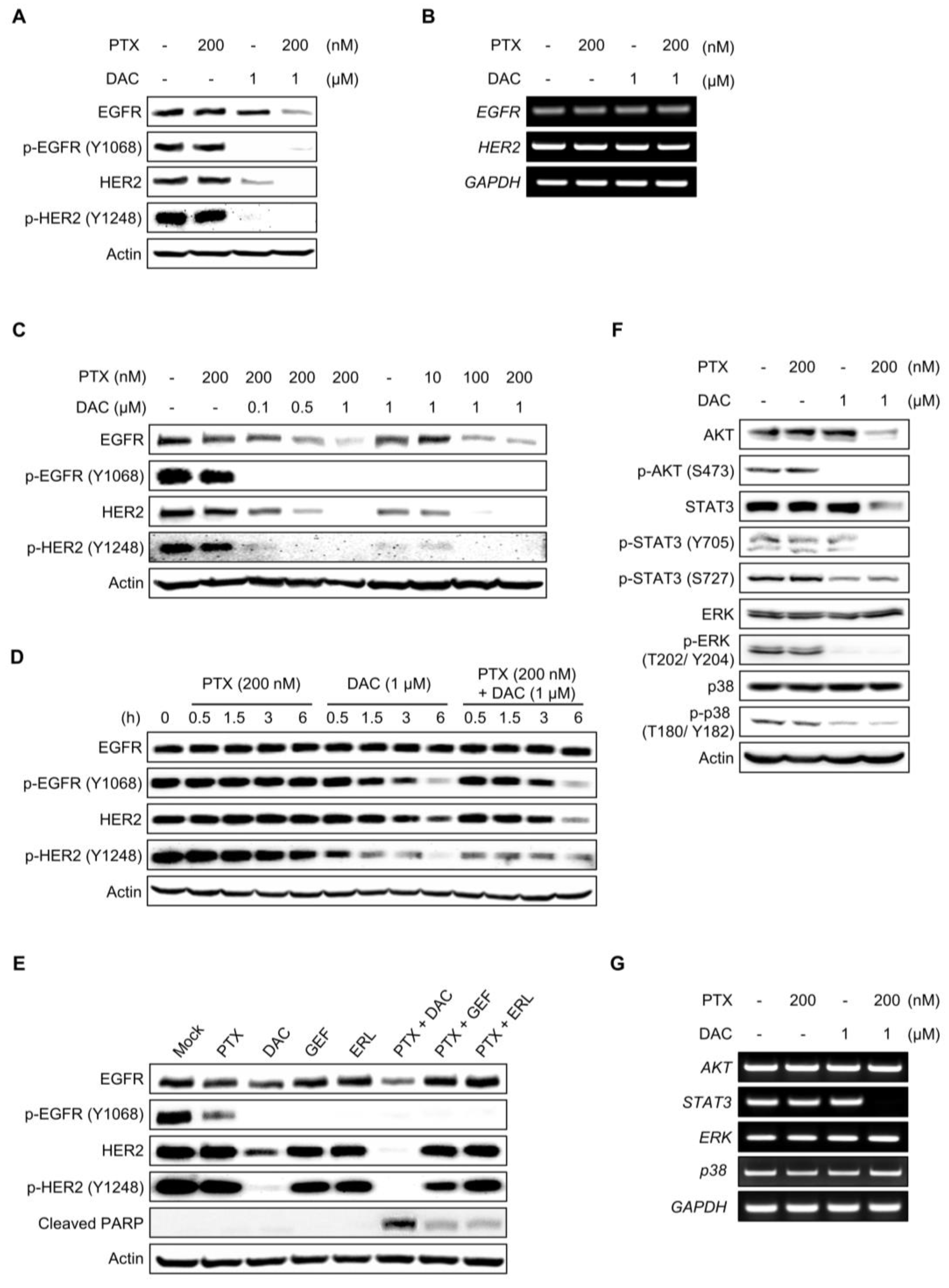
2.2. Dacomitinib Suppressed EGFR Signaling in Ovarian Cancer SKOV3-TR Cells
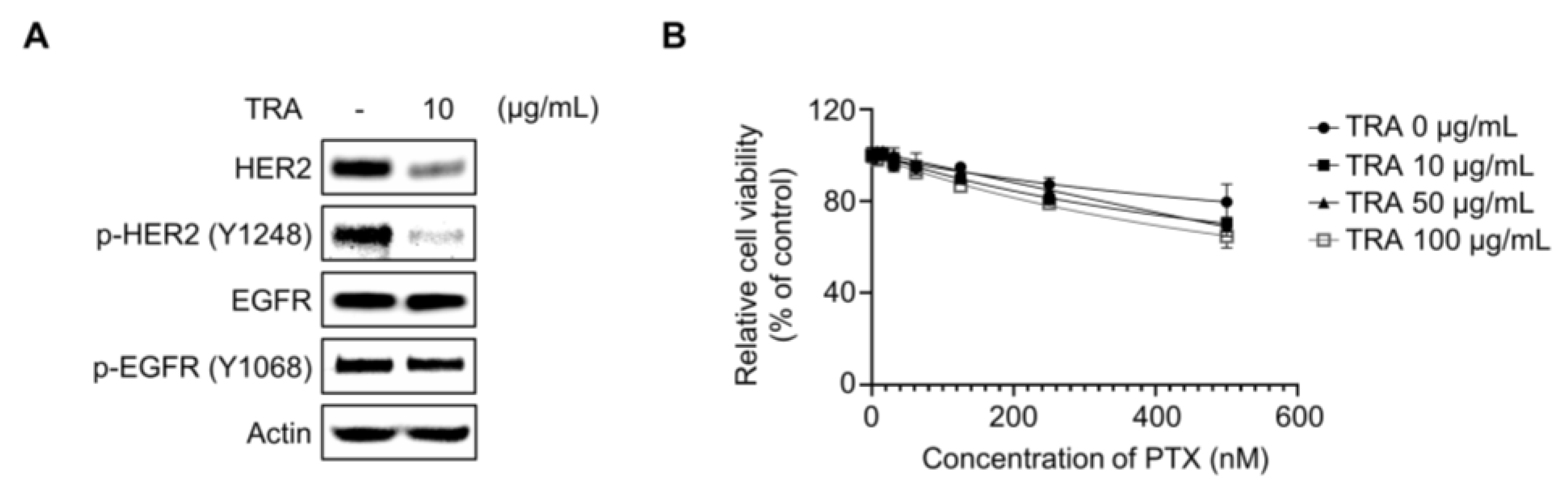
2.3. Inhibition of HER2 Did Not Resensitize Paclitaxel in SKOV3-TR Cells
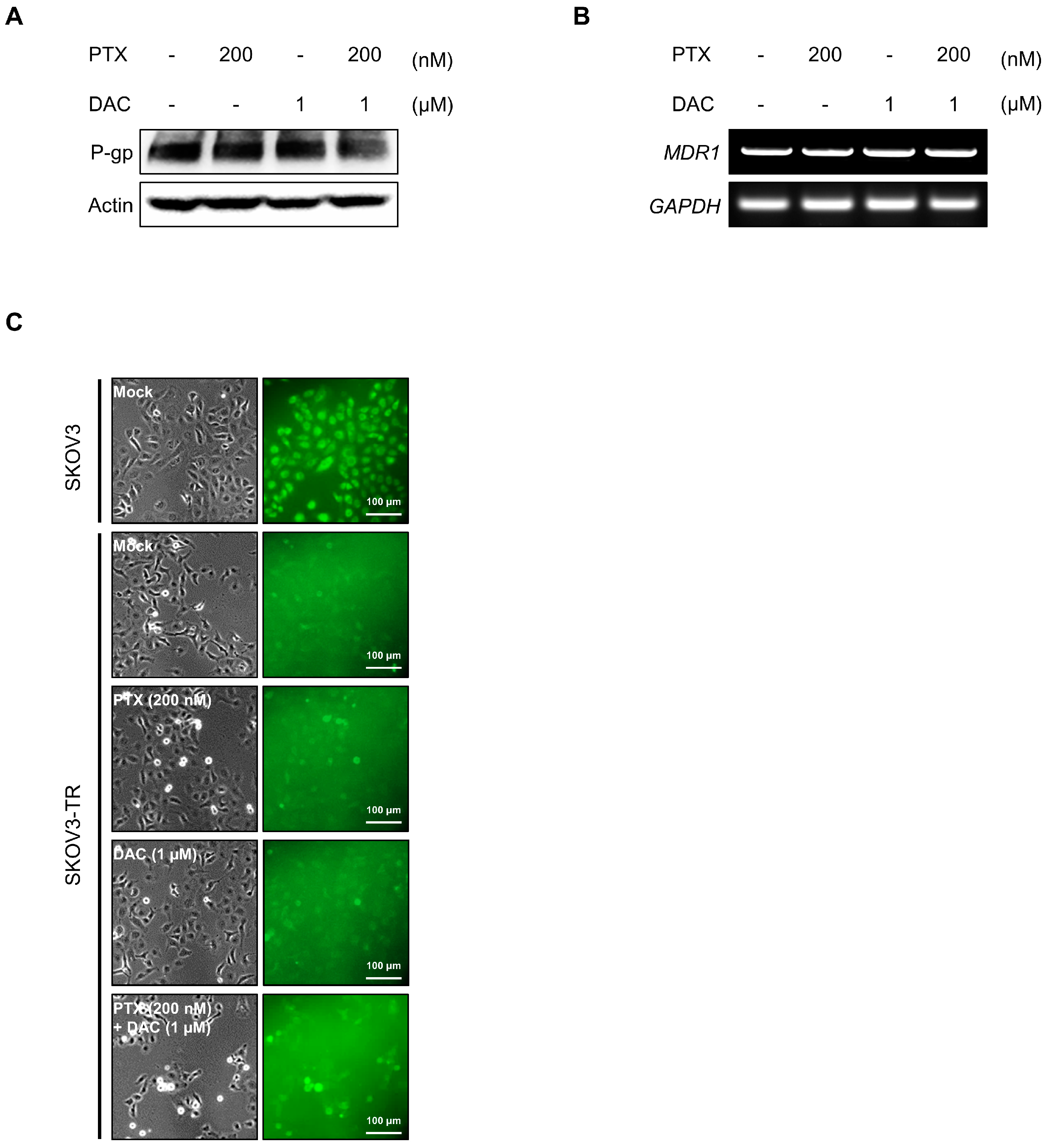
2.4. Dacomitinib Did Not Affect the Expression and Function of P-gp in SKOV3-TR Cells
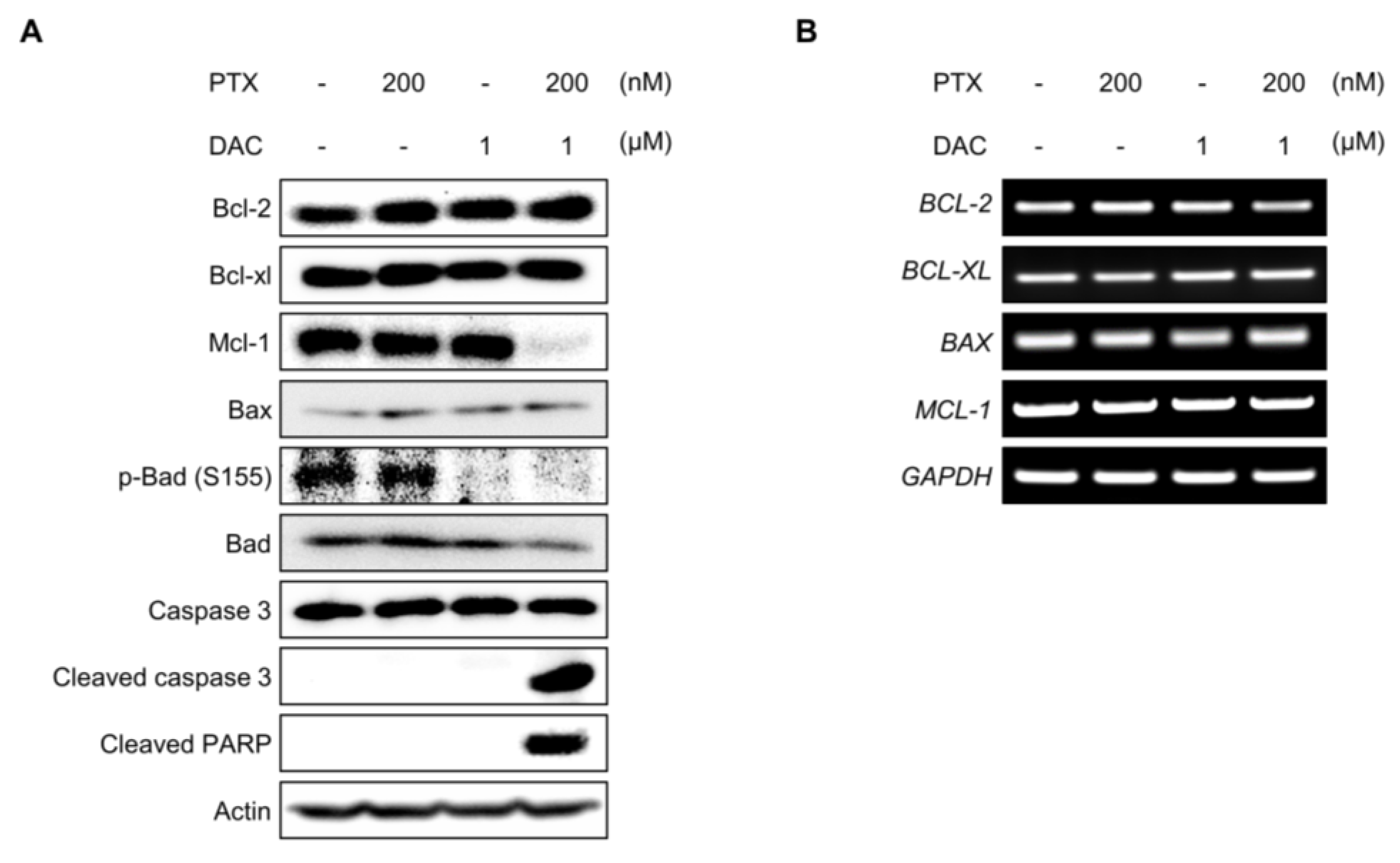
2.5. Dacomitinib Downregulated the Expression of Mcl-1 and the Phosphorylation of Bad in Paclitaxel-Treated SKOV3-TR Cells
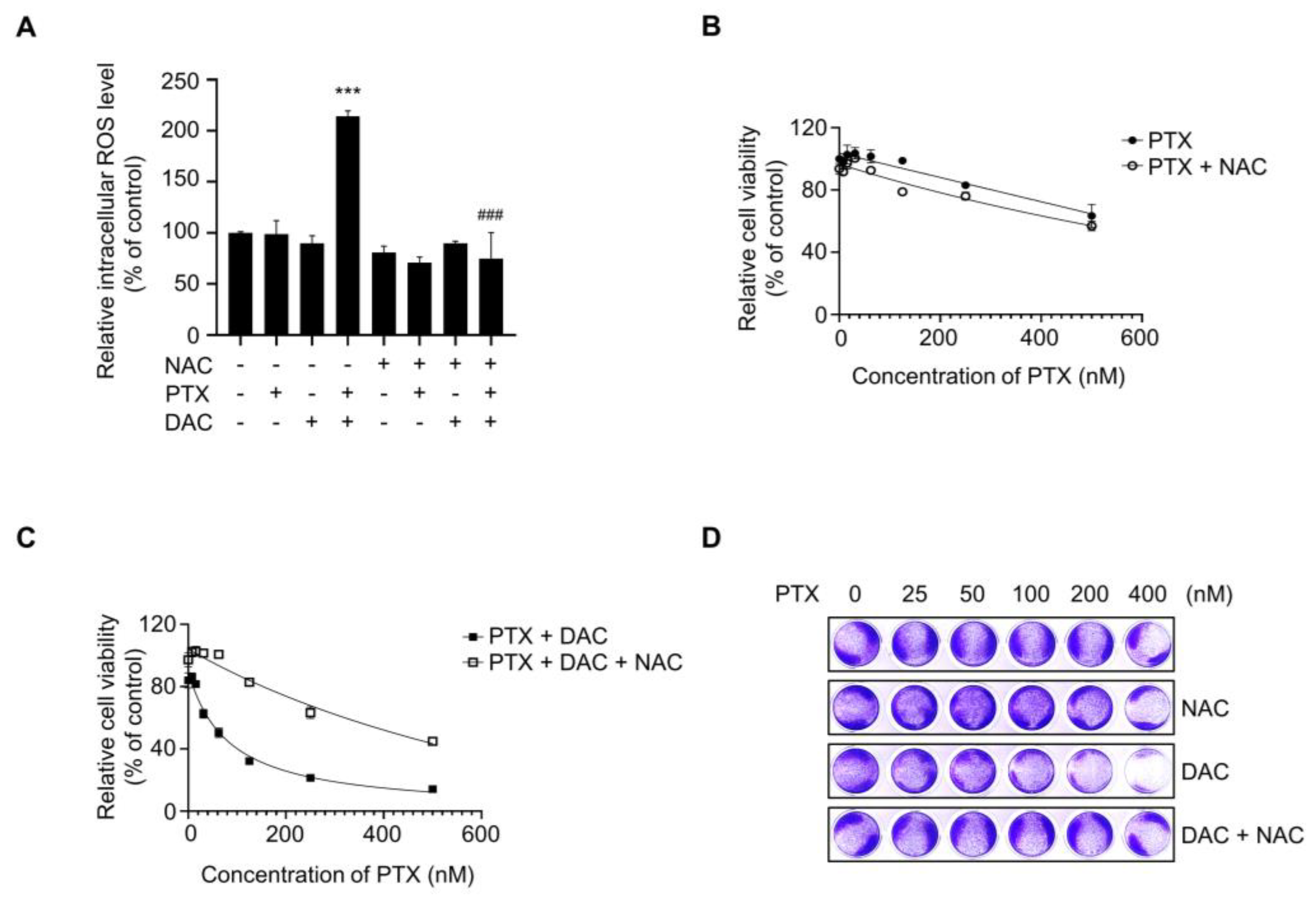
2.6. Dacomitinib Increased Intracellular ROS Levels in Paclitaxel-Treated SKOV3-TR Cells
2.7. Dacomitinib Induced Cytotoxicity in Paclitaxel-Resistant Ovarian Cancer HeyA8-MDR Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Lactate Dehydrogenase (LDH) Assay
4.3. Water-Soluble Tetrazolium Salt-1 (WST-1) Assay
4.4. Crystal Violet Assay
4.5. Immunoblotting and Antibodies
4.6. Flow Cytometry
4.7. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.8. Fluo-3/Acetoxymethyl (AM) Assay
4.9. 2′-7′-Dichlorodihydrofluorescein Diacetate (DCFH-DA) Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Penson, R.T.; Domchek, S.M.; Kaufman, B.; Shapira-Frommer, R.; Audeh, M.W.; Kaye, S.; Molife, L.R.; Gelmon, K.A.; Robertson, J.D.; et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: A multistudy analysis of response rates and safety. Ann. Oncol. 2016, 27, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in ovarian cancer therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38. [Google Scholar] [CrossRef]
- Kim, S.; Han, Y.; Kim, S.I.; Kim, H.S.; Kim, S.J.; Song, Y.S. Tumor evolution and chemoresistance in ovarian cancer. NPJ Precis. Oncol. 2018, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- McMullen, M.; Karakasis, K.; Madariaga, A.; Oza, A.M. Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer. Cancers 2020, 12, 1607. [Google Scholar] [CrossRef] [PubMed]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Kampan, N.C.; Madondo, M.T.; McNally, O.M.; Quinn, M.; Plebanski, M. Paclitaxel and Its Evolving Role in the Management of Ovarian Cancer. Biomed. Res. Int. 2015, 2015, 413076. [Google Scholar] [CrossRef]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef]
- Taghian, A.G.; Abi-Raad, R.; Assaad, S.I.; Casty, A.; Ancukiewicz, M.; Yeh, E.; Molokhia, P.; Attia, K.; Sullivan, T.; Kuter, I.; et al. Paclitaxel decreases the interstitial fluid pressure and improves oxygenation in breast cancers in patients treated with neoadjuvant chemotherapy: Clinical implications. J. Clin. Oncol. 2005, 23, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- McGuire, W.P.; Rowinsky, E.K.; Rosenshein, N.B.; Grumbine, F.C.; Ettinger, D.S.; Armstrong, D.K.; Donehower, R.C. Taxol: A unique antineoplastic agent with significant activity in advanced ovarian epithelial neoplasms. Ann. Intern. Med. 1989, 111, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Singla, A.K.; Garg, A.; Aggarwal, D. Paclitaxel and its formulations. Int. J. Pharm. 2002, 235, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Baust, C.A.; Becker, K.G.; Wood Iii, W.H.; Zhang, Y.; Morin, P.J. Gene expression and pathway analysis of ovarian cancer cells selected for resistance to cisplatin, paclitaxel, or doxorubicin. J. Ovarian Res. 2011, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Takara, K.; Sakaeda, T.; Okumura, K. An update on overcoming MDR1-mediated multidrug resistance in cancer chemotherapy. Curr. Pharm. Des. 2006, 12, 273–286. [Google Scholar] [CrossRef]
- Koh, E.H.; Chung, H.C.; Lee, K.B.; Lim, H.Y.; Kim, J.H.; Roh, J.K.; Min, J.S.; Lee, K.S.; Kim, B.S. The value of immunohistochemical detection of P-glycoprotein in breast cancer before and after induction chemotherapy. Yonsei Med. J. 1992, 33, 137–142. [Google Scholar] [CrossRef]
- Segawa, Y.; Ohnoshi, T.; Hiraki, S.; Ueoka, H.; Kiura, K.; Kamei, H.; Tabata, M.; Shibayama, T.; Miyatake, K.; Genba, K.; et al. Immunohistochemical detection of P-glycoprotein and carcinoembryonic antigen in small cell lung cancer: With reference to predictability of response to chemotherapy. Acta Medica Okayama 1993, 47, 181–189. [Google Scholar] [CrossRef]
- Chan, H.S.; Haddad, G.; Thorner, P.S.; DeBoer, G.; Lin, Y.P.; Ondrusek, N.; Yeger, H.; Ling, V. P-glycoprotein expression as a predictor of the outcome of therapy for neuroblastoma. N. Engl. J. Med. 1991, 325, 1608–1614. [Google Scholar] [CrossRef]
- Thottassery, J.V.; Zambetti, G.P.; Arimori, K.; Schuetz, E.G.; Schuetz, J.D. p53-dependent regulation of MDR1 gene expression causes selective resistance to chemotherapeutic agents. Proc. Natl. Acad. Sci. USA 1997, 94, 11037–11042. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y. The EGFR family and its ligands in human cancer: Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E. SH2 and PTB domain interactions in tyrosine kinase signal transduction. Curr. Opin. Chem. Biol. 1997, 1, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Frey, M.R.; Dise, R.S.; Bernard, J.K.; Polk, D.B. Specific epidermal growth factor receptor autophosphorylation sites promote mouse colon epithelial cell chemotaxis and restitution. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G368–G376. [Google Scholar] [CrossRef] [PubMed]
- Tvorogov, D.; Carpenter, G. EGF-dependent association of phospholipase C-γ1 with c-Cbl. Exp. Cell Res. 2002, 277, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Waterman, H.; Katz, M.; Rubin, C.; Shtiegman, K.; Lavi, S.; Elson, A.; Jovin, T.; Yarden, Y. A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J. 2002, 21, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Batzer, A.; Rotin, D.; Urena, J.; Skolnik, E.; Schlessinger, J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 1994, 14, 5192–5201. [Google Scholar]
- Sakaguchi, K.; Okabayashi, Y.; Kido, Y.; Kimura, S.; Matsumura, Y.; Inushima, K.; Kasuga, M. Shc phosphotyrosine-binding domain dominantly interacts with epidermal growth factor receptors and mediates Ras activation in intact cells. Mol. Endocrinol. 1998, 12, 536–543. [Google Scholar] [CrossRef]
- David, M.; Wong, L.; Flavell, R.; Thompson, S.A.; Wells, A.; Larner, A.C.; Johnson, G.R. STAT Activation by Epidermal Growth Factor (EGF) and Amphiregulin: Requirement for the EGF Receptor Kinase but Not for Tyrosine Phosphorylation Sites or JAK1 (∗). J. Biol. Chem. 1996, 271, 9185–9188. [Google Scholar] [CrossRef]
- Porter, A.C.; Vaillancourt, R.R. Tyrosine kinase receptor-activated signal transduction pathways which lead to oncogenesis. Oncogene 1998, 17, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. EGF Recept. Fam. 2003, 2003, 33–55. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar] [CrossRef]
- Jänne, P.A.; Yang, J.C.-H.; Kim, D.-W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.-J.; Kim, S.-W.; Su, W.-C.; Horn, L. AZD9291 in EGFR inhibitor–resistant non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Zhang, M.; Cong, Q.; Zhang, X.Y.; Zhang, M.X.; Lu, Y.Y.; Xu, C.J. Pyruvate dehydrogenase kinase 1 contributes to cisplatin resistance of ovarian cancer through EGFR activation. J. Cell. Physiol. 2019, 234, 6361–6370. [Google Scholar] [CrossRef]
- Xu, L.; Xu, Y.; Zheng, J.; Zhao, Y.; Wang, H.; Qi, Y. Dacomitinib improves chemosensitivity of cisplatin-resistant human ovarian cancer cells. Oncol. Lett. 2021, 22, 569. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Update 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, M.A.; de la Cruz-Ojeda, P.; Lopez-Grueso, M.J.; Navarro-Villaran, E.; Requejo-Aguilar, R.; Castejon-Vega, B.; Negrete, M.; Gallego, P.; Vega-Ochoa, A.; Victor, V.M.; et al. Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer. Redox Biol. 2020, 36, 101510. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yi, J. Cancer cell killing via ROS: To increase or decrease, that is the question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Raza, M.H.; Siraj, S.; Arshad, A.; Waheed, U.; Aldakheel, F.; Alduraywish, S.; Arshad, M. ROS-modulated therapeutic approaches in cancer treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 1789–1809. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Kim, S.-J.; Kim, B.-J.; Rah, S.-Y.; Chung, S.M.; Im, M.-J.; Kim, U.-H. Doxorubicin-induced reactive oxygen species generation and intracellular Ca2+ increase are reciprocally modulated in rat cardiomyocytes. Exp. Mol. Med. 2006, 38, 535–545. [Google Scholar] [CrossRef]
- Magda, D.; Miller, R.A. Motexafin gadolinium: A novel redox active drug for cancer therapy. In Proceedings of the Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2006; pp. 466–476. [Google Scholar]
- Alexandre, J.; Batteux, F.; Nicco, C.; Chéreau, C.; Laurent, A.; Guillevin, L.; Weill, B.; Goldwasser, F. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. Int. J. Cancer 2006, 119, 41–48. [Google Scholar] [CrossRef]
- Alexandre, J.r.m.; Hu, Y.; Lu, W.; Pelicano, H.; Huang, P. Novel Action of Paclitaxel against Cancer Cells: Bystander Effect Mediated by Reactive Oxygen Species. Cancer Res. 2007, 67, 3512–3517. [Google Scholar] [CrossRef]
- Shirley, M. Dacomitinib: First Global Approval. Drugs 2018, 78, 1947–1953. [Google Scholar] [CrossRef]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Lee, M.; Linke, R.; Rosell, R.; Corral, J.; et al. Improvement in Overall Survival in a Randomized Study That Compared Dacomitinib with Gefitinib in Patients with Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. J. Clin. Oncol. 2018, 36, 2244–2250. [Google Scholar] [CrossRef] [PubMed]
- Abdul Razak, A.R.; Soulieres, D.; Laurie, S.A.; Hotte, S.J.; Singh, S.; Winquist, E.; Chia, S.; Le Tourneau, C.; Nguyen-Tan, P.F.; Chen, E.X.; et al. A phase II trial of dacomitinib, an oral pan-human EGF receptor (HER) inhibitor, as first-line treatment in recurrent and/or metastatic squamous-cell carcinoma of the head and neck. Ann. Oncol. 2013, 24, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Grivas, P.D.; Day, K.C.; Karatsinides, A.; Paul, A.; Shakir, N.; Owainati, I.; Liebert, M.; Kunju, L.P.; Thomas, D.; Hussain, M.; et al. Evaluation of the antitumor activity of dacomitinib in models of human bladder cancer. Mol. Med. 2013, 19, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G.; Camidge, D.R.; Giaccone, G.; Hida, T.; Li, B.T.; O’Connell, J.; Taylor, I.; Zhang, H.; Arcila, M.E.; Goldberg, Z.; et al. Targeting HER2 aberrations as actionable drivers in lung cancers: Phase II trial of the pan-HER tyrosine kinase inhibitor dacomitinib in patients with HER2-mutant or amplified tumors. Ann. Oncol. 2015, 26, 1421–1427. [Google Scholar] [CrossRef]
- Kalous, O.; Conklin, D.; Desai, A.J.; O’Brien, N.A.; Ginther, C.; Anderson, L.; Cohen, D.J.; Britten, C.D.; Taylor, I.; Christensen, J.G.; et al. Dacomitinib (PF-00299804), an irreversible Pan-HER inhibitor, inhibits proliferation of HER2-amplified breast cancer cell lines resistant to trastuzumab and lapatinib. Mol. Cancer Ther. 2012, 11, 1978–1987. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007, 67, 11924–11932. [Google Scholar] [CrossRef]
- Mohiuddin, M.; Kasahara, K. Paclitaxel impedes EGFR-mutated PC9 cell growth via reactive oxygen species-mediated DNA damage and EGFR/PI3K/AKT/mTOR signaling pathway suppression. Cancer Genom. Proteom. 2021, 18, 645–659. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, N.; Wang, R.; Huang, F.; Li, G. Paclitaxel induces apoptosis and reduces proliferation by targeting epidermal growth factor receptor signaling pathway in oral cavity squamous cell carcinoma. Oncol. Lett. 2015, 10, 2378–2384. [Google Scholar] [CrossRef]
- Cohen, M.H.; Johnson, J.R.; Chen, Y.-F.; Sridhara, R.; Pazdur, R. FDA Drug Approval Summary: Erlotinib (Tarceva®) Tablets. Oncologist 2005, 10, 461–466. [Google Scholar] [CrossRef]
- Herbst, R.S.; Fukuoka, M.; Baselga, J. Gefitinib—A novel targeted approach to treating cancer. Nat. Rev. Cancer 2004, 4, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Turkson, J. Targeting STAT3 in cancer: How successful are we? Expert Opin. Investig. Drugs 2009, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Pouyssegur, J. Targeting the ERK signaling pathway in cancer therapy. Ann. Med. 2006, 38, 200–211. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Yusuf, R.; Duan, Z.; Lamendola, D.; Penson, R.; Seiden, M. Paclitaxel resistance: Molecular mechanisms and pharmacologic manipulation. Curr. Cancer Drug Targets 2003, 3, 1–19. [Google Scholar] [CrossRef]
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef]
- Eckford, P.D.; Sharom, F.J. ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009, 109, 2989–3011. [Google Scholar] [CrossRef] [PubMed]
- Fujii, R.i.; Mutoh, M.; Niwa, K.; Yamada, K.; Aikou, T.; Nakagawa, M.; Kuwano, M.; Akiyama, S.I. Active efflux system for cisplatin in cisplatin-resistant human KB cells. Jpn. J. Cancer Res. 1994, 85, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Holló, Z.; Homolya, L.; Davis, C.W.; Sarkadi, B. Calcein accumulation as a fluorometric functional assay of the multidrug transporter. Biochim. Biophys. Acta (BBA)-Biomembr. 1994, 1191, 384–388. [Google Scholar] [CrossRef]
- Homolya, L.; Holló, Z.; Germann, U.A.; Pastan, I.; Gottesman, M.M.; Sarkadi, B. Fluorescent cellular indicators are extruded by the multidrug resistance protein. J. Biol. Chem. 1993, 268, 21493–21496. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Willingham, M.C.; Reed, J.C.; Miyashita, T.; Ray, S.; Ponnathpur, V.; Huang, Y.; Mahoney, M.E.; Bullock, G.; Bhalla, K. High levels of p26BCL-2 oncoprotein retard taxol-induced apoptosis in human pre-B leukemia cells. Leukemia 1994, 8, 1960–1969. [Google Scholar] [PubMed]
- Huang, Y.; Ibrado, A.; Reed, J.; Bullock, G.; Ray, S.; Tang, C.; Bhalla, K. Co-expression of several molecular mechanisms of multidrug resistance and their significance for paclitaxel cytotoxicity in human AML HL-60 cells. Leukemia 1997, 11, 253–257. [Google Scholar] [CrossRef]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.R.; Fletcher, B.; Page, C.; Hu, C.; Nunez, G.; Baker, V. Bcl-xLIs Expressed in Ovarian Carcinoma and Modulates Chemotherapy-Induced Apoptosis. Gynecol. Oncol. 1998, 70, 398–403. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Kelly, P.N.; Strasser, A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011, 18, 1414–1424. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, X.W.; Liao, Q.L.; Wu, W.T.; Liu, Y.L.; Huang, W.H. Electrochemical Monitoring of Paclitaxel-Induced ROS Release from Mitochondria inside Single Cells. Small 2019, 15, e1901787. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Primers 2016, 2, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Jelovac, D.; Armstrong, D.K. Recent progress in the diagnosis and treatment of ovarian cancer. CA Cancer J. Clin. 2011, 61, 183–203. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Coleman, R.L.; Markman, M. Ovarian cancer. Lancet 2009, 374, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Van Gorp, T.; Parma, G.; Amant, F.; Gatta, G.; Sessa, C.; Vergote, I. Ovarian cancer. Crit. Rev. Oncol./Hematol. 2006, 60, 159–179. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Leung, B.; Jahani-Asl, A.; Yan, X.; Thompson, W.E.; Tsang, B.K. Chemoresistance in human ovarian cancer: The role of apoptotic regulators. Reprod. Biol. Endocrinol. 2003, 1, 66. [Google Scholar] [CrossRef]
- Madden, E.C.; Gorman, A.M.; Logue, S.E.; Samali, A. Tumour cell secretome in chemoresistance and tumour recurrence. Trends Cancer 2020, 6, 489–505. [Google Scholar] [CrossRef]
- Chang, A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer 2011, 71, 3–10. [Google Scholar] [CrossRef]
- Lu, D.; Shi, H.-C.; Wang, Z.-X.; Gu, X.-W.; Zeng, Y. Multidrug resistance-associated biomarkers PGP, GST-π, Topo-II and LRP as prognostic factors in primary ovarian carcinoma. Br. J. Biomed. Sci. 2011, 68, 69–74. [Google Scholar] [CrossRef]
- Helena, A.Y.; Riely, G.J. Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in lung cancers. J. Natl. Compr. Cancer Netw. 2013, 11, 161–169. [Google Scholar]
- Duggirala, K.B.; Lee, Y.; Lee, K. Chronicles of EGFR Tyrosine Kinase Inhibitors: Targeting EGFR C797S Containing Triple Mutations. Biomol. Ther. 2022, 30, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Lee, K.W.; Cho, J.Y.; Kang, W.K.; Im, S.A.; Kim, J.W.; Bang, Y.J. Phase II trial of dacomitinib in patients with HER2-positive gastric cancer. Gastric Cancer 2016, 19, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Ather, F.; Hamidi, H.; Fejzo, M.S.; Letrent, S.; Finn, R.S.; Kabbinavar, F.; Head, C.; Wong, S.G. Dacomitinib, an irreversible Pan-ErbB inhibitor significantly abrogates growth in head and neck cancer models that exhibit low response to cetuximab. PLoS ONE 2013, 8, e56112. [Google Scholar] [CrossRef]
- Bijman, M.N.; van Berkel, M.P.; Kok, M.; Janmaat, M.L.; Boven, E. Inhibition of functional HER family members increases the sensitivity to docetaxel in human ovarian cancer cell lines. Anti-Cancer Drugs 2009, 20, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Yu, Y.; Le, X.-F.; Boyer, C.; Mills, G.B.; Bast, R.C., Jr. The outcome of heregulin-induced activation of ovarian cancer cells depends on the relative levels of HER-2 and HER-3 expression. Clin. Cancer Res. 1999, 5, 3653–3660. [Google Scholar] [PubMed]
- Hee Choi, Y.; Yu, A.-M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef]
- Kitazaki, T.; Oka, M.; Nakamura, Y.; Tsurutani, J.; Doi, S.; Yasunaga, M.; Takemura, M.; Yabuuchi, H.; Soda, H.; Kohno, S. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer 2005, 49, 337–343. [Google Scholar] [CrossRef]
- Wang, T.H.; Wang, H.S.; Soong, Y.K. Paclitaxel-induced cell death: Where the cell cycle and apoptosis come together. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2000, 88, 2619–2628. [Google Scholar] [CrossRef]
- Shi, P.; Oh, Y.-T.; Deng, L.; Zhang, G.; Qian, G.; Zhang, S.; Ren, H.; Wu, G.; Legendre Jr, B.; Anderson, E. Overcoming acquired resistance to AZD9291, a third-generation EGFR inhibitor, through modulation of MEK/ERK-dependent Bim and Mcl-1 degradation. Clin. Cancer Res. 2017, 23, 6567–6579. [Google Scholar] [CrossRef]
- Song, L.; Coppola, D.; Livingston, S.; Cress, W.D.; Haura, E.B. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol. Ther. 2005, 4, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Gong, Y.-B.; Kang, X.-H.; Lu, Z.-H.; Wang, Y.; Zhao, K.-L.; Miao, Z.-H.; Liao, M.-J.; Xu, Z.-Y. Degradation of MCL-1 by bufalin reverses acquired resistance to osimertinib in EGFR-mutant lung cancer. Toxicol. Appl. Pharmacol. 2019, 379, 114662. [Google Scholar] [CrossRef] [PubMed]
- Zang, H.; Qian, G.; Arbiser, J.; Owonikoko, T.K.; Ramalingam, S.S.; Fan, S.; Sun, S.Y. Overcoming acquired resistance of EGFR-mutant NSCLC cells to the third generation EGFR inhibitor, osimertinib, with the natural product honokiol. Mol. Oncol. 2020, 14, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Deng, Y.; Qian, L.; Vallega, K.A.; Zhang, G.; Deng, X.; Owonikoko, T.K.; Ramalingam, S.S.; Fang, D.D.; Zhai, Y.; et al. Overcoming acquired resistance to third-generation EGFR inhibitors by targeting activation of intrinsic apoptotic pathway through Mcl-1 inhibition, Bax activation, or both. Oncogene 2022, 41, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Gao, F.; Li, W.; Zhou, L.; Liu, W.; Li, M. Formononetin inhibits tumor growth by suppression of EGFR-Akt-Mcl-1 axis in non-small cell lung cancer. J. Exp. Clin. Cancer Res. CR 2020, 39, 62. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yu, X.; Li, M.; Zhou, L.; Liu, W.; Li, W.; Liu, H. Deguelin suppresses non-small cell lung cancer by inhibiting EGFR signaling and promoting GSK3β/FBW7-mediated Mcl-1 destabilization. Cell Death Dis. 2020, 11, 143. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Griffith, O.W.; Meister, A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (Sn-butyl homocysteine sulfoximine). J. Biol. Chem. 1979, 254, 7558–7560. [Google Scholar] [CrossRef]
- Shan, F.; Shao, Z.; Jiang, S.; Cheng, Z. Erlotinib induces the human non–small-cell lung cancer cells apoptosis via activating ROS-dependent JNK pathways. Cancer Med. 2016, 5, 3166–3175. [Google Scholar] [CrossRef]
- Yan, S.; Zhang, B.; Feng, J.; Wu, H.; Duan, N.; Zhu, Y.; Zhao, Y.; Shen, S.; Zhang, K.; Wu, W. FGFC1 selectively inhibits erlotinib-resistant non-small cell lung cancer via elevation of ROS mediated by the EGFR/PI3K/Akt/mTOR pathway. Front. Pharmacol. 2022, 12, 764699. [Google Scholar] [CrossRef]
- Ge, X.; Zhang, Y.; Huang, F.; Wu, Y.; Pang, J.; Li, X.; Fan, F.; Liu, H.; Li, S. EGFR tyrosine kinase inhibitor Almonertinib induces apoptosis and autophagy mediated by reactive oxygen species in non-small cell lung cancer cells. Hum. Exp. Toxicol. 2021, 40, S49–S62. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zhao, B.; Chang, H.; Xiao, M.; Wu, Y.; Liu, Y. Paclitaxel suppresses proliferation and induces apoptosis through regulation of ROS and the AKT/MAPK signaling pathway in canine mammary gland tumor cells. Mol. Med. Rep. 2018, 17, 8289–8299. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yin, L.; Wu, L.; Zhu, Y.; Wang, X. Paclitaxel inhibits proliferation and promotes apoptosis through regulation ROS and endoplasmic reticulum stress in osteosarcoma cell. Mol. Cell. Toxicol. 2020, 16, 377–384. [Google Scholar] [CrossRef]
- Luo, H.; Yang, Y.; Duan, J.; Wu, P.; Jiang, Q.; Xu, C. PTEN-regulated AKT/FoxO3a/Bim signaling contributes to reactive oxygen species-mediated apoptosis in selenite-treated colorectal cancer cells. Cell Death Dis. 2013, 4, e481. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.; Brunet, A. FOXO transcription factors in ageing and cancer. Acta Physiol. 2008, 192, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Sajadimajd, S.; Khazaei, M. Oxidative stress and cancer: The role of Nrf2. Curr. Cancer Drug Targets 2018, 18, 538–557. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1–Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef]
- Ling, J.; Kumar, R. Crosstalk between NFkB and glucocorticoid signaling: A potential target of breast cancer therapy. Cancer Lett. 2012, 322, 119–126. [Google Scholar] [CrossRef]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef]
- Pan, C.W.; Jin, X.; Zhao, Y.; Pan, Y.; Yang, J.; Karnes, R.J.; Zhang, J.; Wang, L.; Huang, H. AKT-phosphorylated FOXO 1 suppresses ERK activation and chemoresistance by disrupting IQGAP 1-MAPK interaction. EMBO J. 2017, 36, 995–1010. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Chen, N.; Zhao, F.; Wang, X.-J.; Kong, B.; Zheng, W.; Zhang, D.D. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. 2010, 70, 5486–5496. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Liang, C.; Hua, J.; Zhang, B.; Liu, J.; Zhang, Y.; Wei, M.; Yu, X.; Xu, J.; Shi, S. A miR-146a-5p/TRAF6/NF-kB p65 axis regulates pancreatic cancer chemoresistance: Functional validation and clinical significance. Theranostics 2020, 10, 3967. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | Sequence of Primer |
|---|---|
| EGFR | F: 5′-CGCAAGTGTAAGAAGTGCGAA-3′ |
| R: 5′-CGTAGCATTTATGGAGAGTGAGTCT-3′ | |
| HER2 | F: 5′-TAAGGACCCTCCCTTCTGCG-3′ |
| R: 5′-AAAGACCACCCCCAAGACCA-3′ | |
| AKT | F: 5′-ATGAGCGACGTGGCTATTGTG-3′ |
| R: 5′-GAGGCCGTCAGCCACAGTCTG-3′ | |
| STAT3 | F: 5′-GATCCAGTCCGTGGAACCAT-3′ |
| R: 5′-TGGTCTTCAGGTATGGGGCA-3′ | |
| ERK | F: 5′-CCTAAGGAAAAGCTCAAAGA-3′ |
| R: 5′-AAAGTGGATAAGCCAAGAC-3′ | |
| p38 | F: 5′-GATCAGTTGAAGCTCATTTTAA-3′ |
| R: 5′-CACTTGAATAATATTTGGAGAGT-3′ | |
| BCL-2 | F: 5′-ATCGCCCTGTGGATGACTGAGT-3′ |
| R: 5′-GCCAGGAGAAATCAAACAGAGGC-3′ | |
| BCL-XL | F: 5′-GTAAACTGGGGTCGCATTGT-3′ |
| R: 5′-TGGATCCAAGGCTCTAGGTG-3′ | |
| MCL-1 | F: 5′-CTCCCCACCAAGAGTCCACA-3′ |
| R: 5′-CCCAAACCACTTGGGGTGTC-3′ | |
| BAX | F: 5′-CCAGCTGCCTTGGACTGT-3′ |
| R: 5′-ACCCCCTCAAGACCACTCTT-3′ | |
| MDR1 | F: 5′-GCGAGGTCGGAATGGATCTT-3′ |
| R: 5′-AGGGTTAGCTTCCAACCACG-3′ | |
| GAPDH | F: 5′-GTCTCCTCTGACTTCACAGCG-3′ |
| R: 5′-ACCACCCTGTTGCTGTAGCCAA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, Y.J.; Kim, H.S.; Bae, S.; So, K.A.; Kim, T.J.; Lee, J.H. Pan-EGFR Inhibitor Dacomitinib Resensitizes Paclitaxel and Induces Apoptosis via Elevating Intracellular ROS Levels in Ovarian Cancer SKOV3-TR Cells. Molecules 2024, 29, 274. https://doi.org/10.3390/molecules29010274
Lim YJ, Kim HS, Bae S, So KA, Kim TJ, Lee JH. Pan-EGFR Inhibitor Dacomitinib Resensitizes Paclitaxel and Induces Apoptosis via Elevating Intracellular ROS Levels in Ovarian Cancer SKOV3-TR Cells. Molecules. 2024; 29(1):274. https://doi.org/10.3390/molecules29010274
Chicago/Turabian StyleLim, Ye Jin, Hee Su Kim, Seunghee Bae, Kyeong A So, Tae Jin Kim, and Jae Ho Lee. 2024. "Pan-EGFR Inhibitor Dacomitinib Resensitizes Paclitaxel and Induces Apoptosis via Elevating Intracellular ROS Levels in Ovarian Cancer SKOV3-TR Cells" Molecules 29, no. 1: 274. https://doi.org/10.3390/molecules29010274
APA StyleLim, Y. J., Kim, H. S., Bae, S., So, K. A., Kim, T. J., & Lee, J. H. (2024). Pan-EGFR Inhibitor Dacomitinib Resensitizes Paclitaxel and Induces Apoptosis via Elevating Intracellular ROS Levels in Ovarian Cancer SKOV3-TR Cells. Molecules, 29(1), 274. https://doi.org/10.3390/molecules29010274