

New Quinoid Bio-Inspired Materials Using Para-Azaquinodimethane Moiety


Abstract
:
1. Introduction
2. Results and Discussion
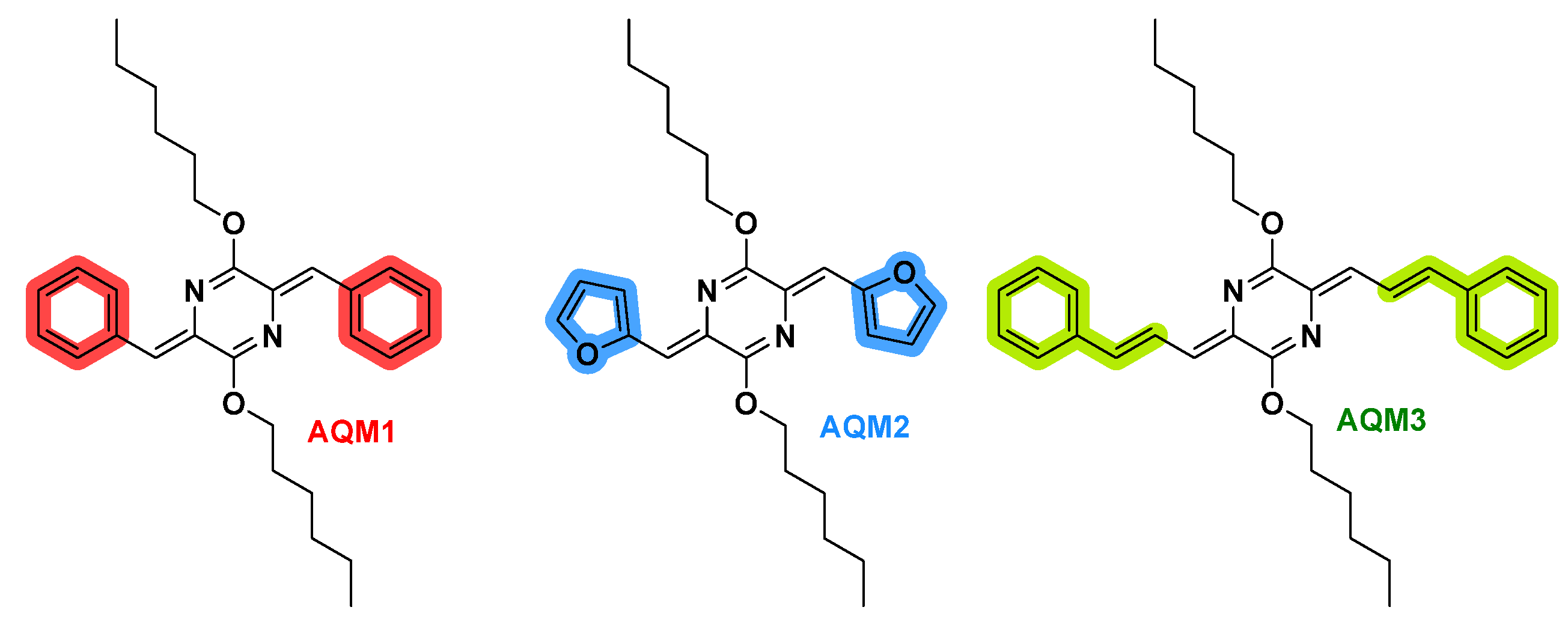
2.1. Synthesis
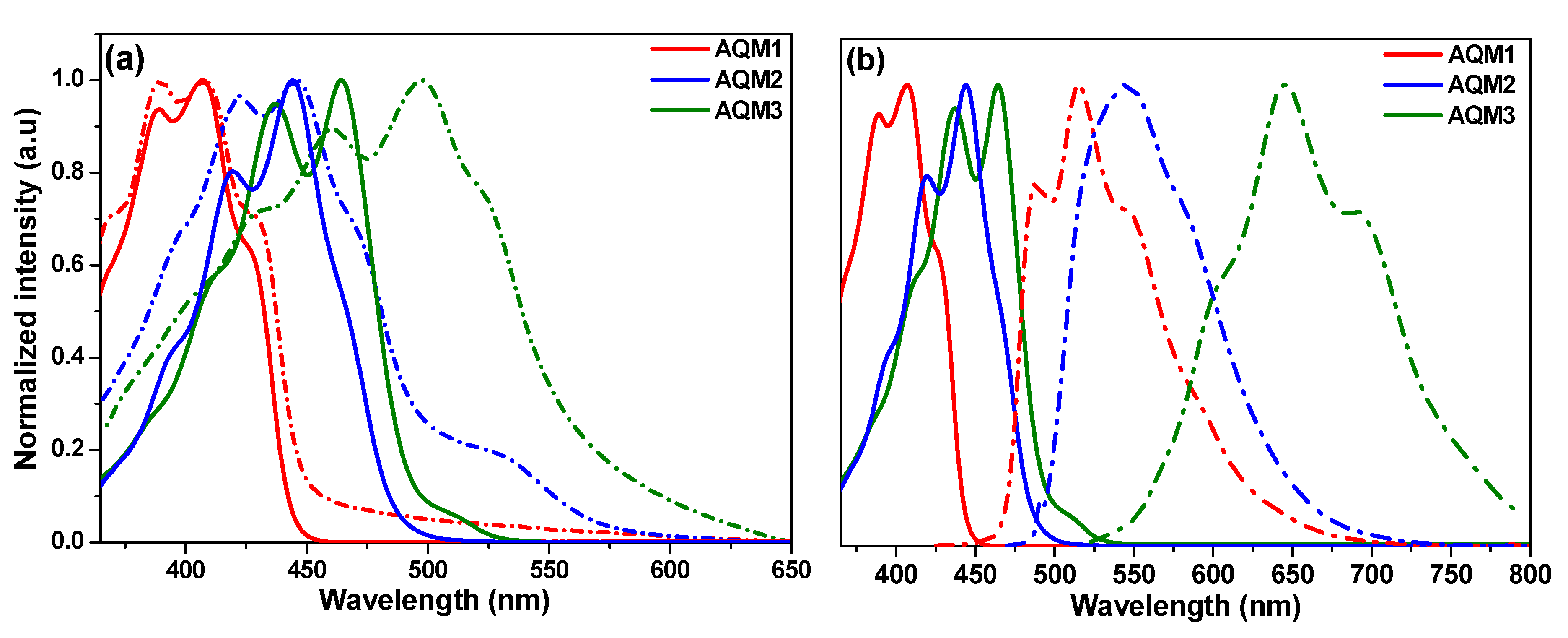
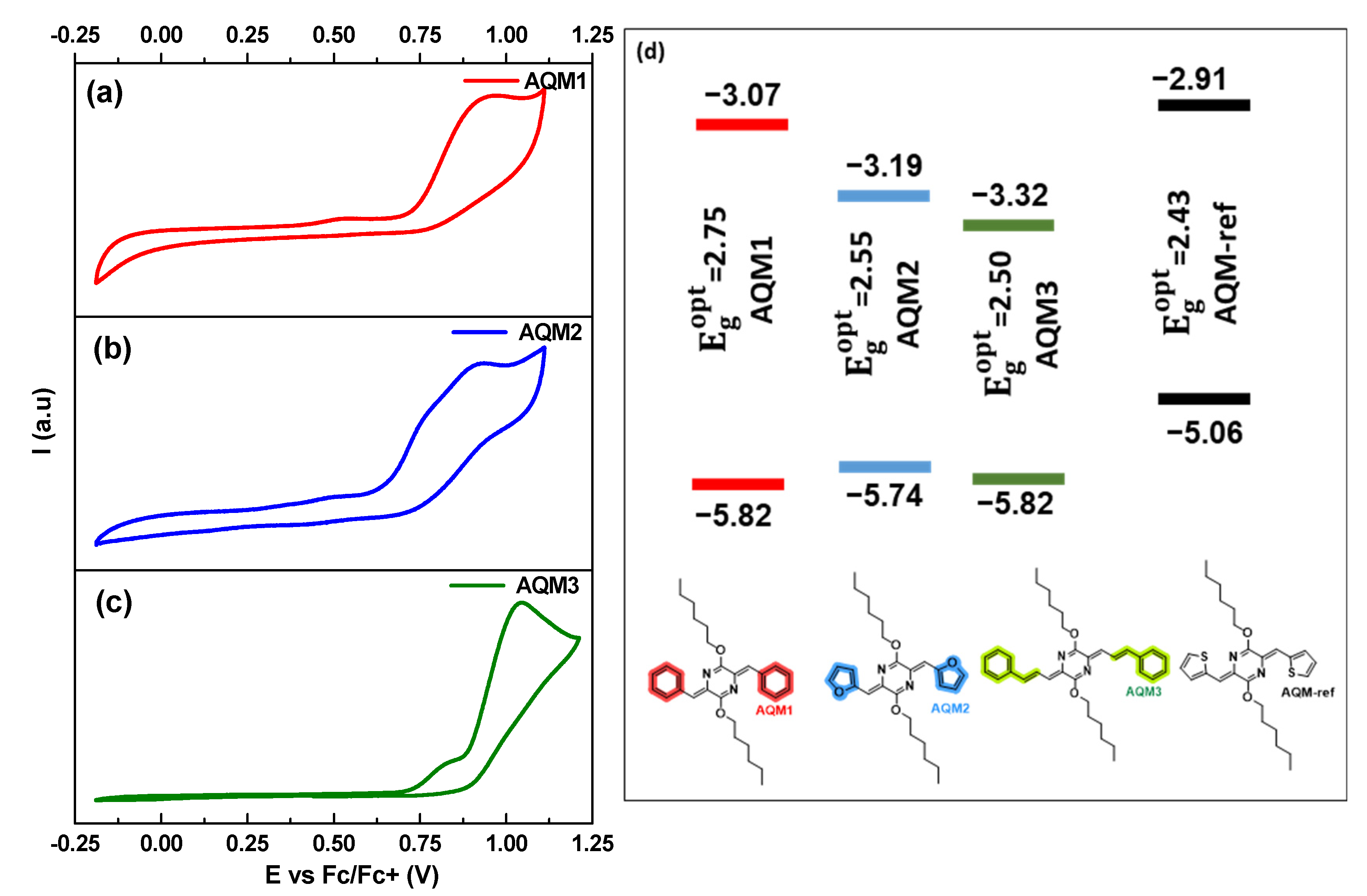
2.2. Optical and Electrochemical Properties

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
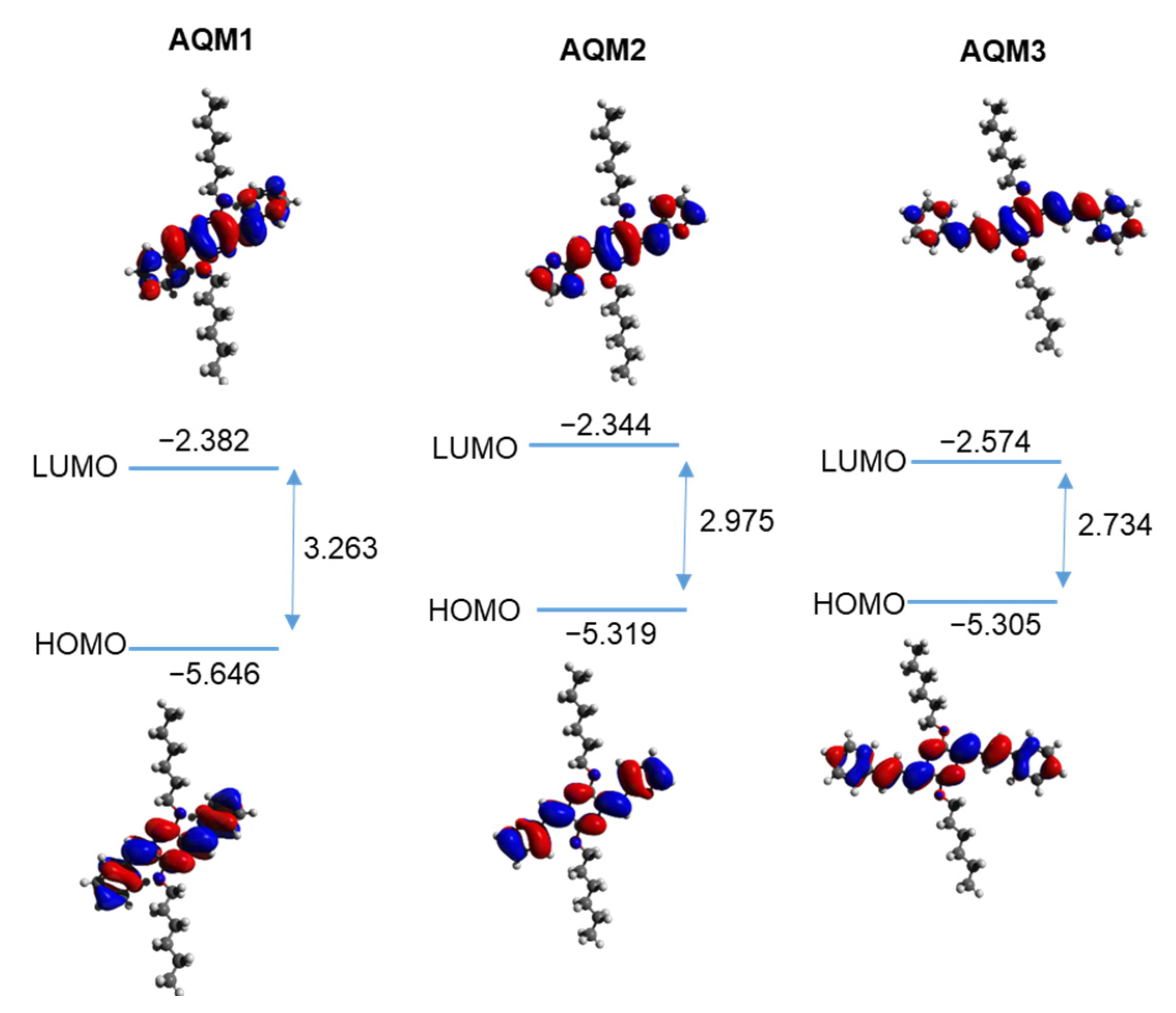
| Absorption | Emission | CV | DFT | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| /nm | /eV | ε b /M−1 cm−1 | /nm | λem c (ϕf) d /nm | Δλ e /nm | EHOMO f/eV | ELUMO g/ eV | λmax /nm | EHOMO/eV | ELUMO /eV | /eV | |
| AQM1 | 407 | 2.75 | 2.2 × 104 | 408 | 515 (0.48) | 108 | −5.82 | −3.07 | 400 | −5.646 | −2.382 | 3.263 |
| AQM2 | 444 | 2.55 | 3.2 × 104 | 445 | 545 (0.2) | 101 | −5.74 | −3.19 | 434 | −5.319 | −2.344 | 2.975 |
| AQM3 | 464 | 2.50 | 6.1 × 104 | 497 | 645 (0.32) | 181 | −5.82 | −3.32 | 465 | −5.305 | −2.574 | 2.734 |
2.3. Thermal Properties
2.4. Theoretical Calculations
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Characterisation
3.3. Experimental Procedures
3.3.1. Synthesis of Intermediates 2a, 2b and 2c by Knoevenagel Condensation
3.3.2. Synthesis of Target p-AQM Molecules by Alkylation
3.4. Computational Details
3.4.1. Ground State Calculation
3.4.2. Absorption and Emission Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, H.; Zhu, S.; Cui, Z.; Li, Z.; Liang, Y.; Zhu, J.; Hu, P.; Zhang, H.-L.; Hu, W. High-Performance Five-Ring-Fused Organic Semiconductors for Field-Effect Transistors. Chem. Soc. Rev. 2022, 51, 3071–3122. [Google Scholar] [CrossRef] [PubMed]
- Holliday, S.; Li, Y.; Luscombe, C.K. Recent Advances in High Performance Donor-Acceptor Polymers for Organic Photovoltaics. Prog. Polym. Sci. 2017, 70, 34–51. [Google Scholar] [CrossRef]
- Liu, C.; Wang, K.; Gong, X.; Heeger, A.J. Low Bandgap Semiconducting Polymers for Polymeric Photovoltaics. Chem. Soc. Rev. 2016, 45, 4825–4846. [Google Scholar] [CrossRef] [PubMed]
- Borges, B.G.A.L.; Gioti, M.; Correa, R.S.; Andreopoulou, A.K.; Veiga, A.G.; Laskarakis, A.; Kallitsis, J.K.; Logothetidis, S.; Rocco, M.L.M. Surface, Interface and Electronic Studies on Anthracene Derived Polymeric Thin Films for OLED Applications. Opt. Mater. 2021, 117, 111145. [Google Scholar] [CrossRef]
- Murawski, C.; Gather, M.C. Emerging Biomedical Applications of Organic Light-Emitting Diodes. Adv. Opt. Mater. 2021, 9, 2100269. [Google Scholar] [CrossRef]
- Chaudhry, M.U.; Muhieddine, K.; Wawrzinek, R.; Li, J.; Lo, S.-C.; Namdas, E.B. Nano-Alignment in Semiconducting Polymer Films: A Path to Achieve High Current Density and Brightness in Organic Light Emitting Transistors. ACS Photonics 2018, 5, 2137–2144. [Google Scholar] [CrossRef]
- Riera-Galindo, S.; Leonardi, F.; Pfattner, R.; Mas-Torrent, M. Organic Semiconductor/Polymer Blend Films for Organic Field-Effect Transistors. Adv. Mater. Technol. 2019, 4, 1900104. [Google Scholar] [CrossRef]
- Chen, H.; Hurhangee, M.; Nikolka, M.; Zhang, W.; Kirkus, M.; Neophytou, M.; Cryer, S.J.; Harkin, D.; Hayoz, P.; Abdi-Jalebi, M.; et al. Dithiopheneindenofluorene (TIF) Semiconducting Polymers with Very High Mobility in Field-Effect Transistors. Adv. Mater. 2017, 29, 1702523. [Google Scholar] [CrossRef]
- Moussalem, C.; Segut, O.; Gohier, F.; Allain, M.; Frère, P. Facile Access via Green Procedures to a Material with the Benzodifuran Moiety for Organic Photovoltaics. ACS Sustain. Chem. Eng. 2014, 2, 1043–1048. [Google Scholar] [CrossRef]
- Cheng, Y.-J.; Yang, S.-H.; Hsu, C.-S. Synthesis of Conjugated Polymers for Organic Solar Cell Applications. Chem. Rev. 2009, 109, 5868–5923. [Google Scholar] [CrossRef]
- Ardila-Fierro, K.J.; Hernández, J.G. Sustainability Assessment of Mechanochemistry by Using the Twelve Principles of Green Chemistry. ChemSusChem 2021, 14, 2145–2162. [Google Scholar] [CrossRef] [PubMed]
- Van Beurden, K.; de Koning, S.; Molendijk, D.; van Schijndel, J. The Knoevenagel Reaction: A Review of the Unfinished Treasure Map to Forming Carbon–Carbon Bonds. Green Chem. Lett. Rev. 2020, 13, 349–364. [Google Scholar] [CrossRef]
- Fu, H.; Yao, J.; Zhang, M.; Xue, L.; Zhou, Q.; Li, S.; Lei, M.; Meng, L.; Zhang, Z.-G.; Li, Y. Low-Cost Synthesis of Small Molecule Acceptors Makes Polymer Solar Cells Commercially Viable. Nat. Commun. 2022, 13, 3687. [Google Scholar] [CrossRef] [PubMed]
- Alves Sobrinho, R.C.M.; de Oliveira, P.M.; Montes D’Oca, C.R.; Russowsky, D.; Montes D’Oca, M.G. Solvent-Free Knoevenagel Reaction Catalysed by Reusable Pyrrolidinium Base Protic Ionic Liquids (PyrrILs): Synthesis of Long-Chain Alkylidenes. RSC Adv. 2017, 7, 3214–3221. [Google Scholar] [CrossRef]
- Burrezo, P.M.; Zafra, J.L.; López Navarrete, J.T.; Casado, J. Quinoidal/Aromatic Transformations in π-Conjugated Oligomers: Vibrational Raman Studies on the Limits of Rupture for π-Bonds. Angew. Chem. Int. Ed. 2017, 56, 2250–2259. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.; Khim, D.; Yun, J.-M.; Jung, E.; Jang, S.-Y.; Jang, Y.H.; Noh, Y.-Y.; Kim, D.-Y. Quinoidal Molecules as a New Class of Ambipolar Semiconductor Originating from Amphoteric Redox Behavior. Adv. Funct. Mater. 2015, 25, 1146–1156. [Google Scholar] [CrossRef]
- Cui, W.; Wudl, F. Dithienylbenzodipyrrolidone: New Acceptor for Donor–Acceptor Low Band Gap Polymers. Macromolecules 2013, 46, 7232–7238. [Google Scholar] [CrossRef]
- Zhang, H.; Neudörfl, J.-M.; Tieke, B. 1,6-Naphthodione-Based Monomers and Polymers. Polym. Chem. 2014, 5, 3754–3757. [Google Scholar] [CrossRef]
- Kawabata, K.; Saito, M.; Osaka, I.; Takimiya, K. Very Small Bandgap π-Conjugated Polymers with Extended Thienoquinoids. J. Am. Chem. Soc. 2016, 138, 7725–7732. [Google Scholar] [CrossRef]
- Gorokh, I.D.; Adonin, S.A.; Novikov, A.S.; Sokolov, M.N.; Samsonenko, D.G.; Fedin, V.P. Polybromides of Pyridinium and Quinolinium-Type Cations: Cation-Induced Structural Diversity and Theoretical Analysis of Br⋯Br Interactions. J. Mol. Struct. 2019, 1179, 725–731. [Google Scholar] [CrossRef]
- Adonin, S.A.; Bondarenko, M.A.; Novikov, A.S.; Abramov, P.A.; Plyusnin, P.E.; Sokolov, M.N.; Fedin, V.P. Halogen Bonding-Assisted Assembly of Bromoantimonate(V) and Polybromide-Bromoantimonate-Based Frameworks. CrystEngComm 2019, 21, 850–856. [Google Scholar] [CrossRef]
- Walzer, K.; Maennig, B.; Pfeiffer, M.; Leo, K. Highly Efficient Organic Devices Based on Electrically Doped Transport Layers. Chem. Rev. 2007, 107, 1233–1271. [Google Scholar] [CrossRef] [PubMed]
- Casado, J. Para-Quinodimethanes: A Unified Review of the Quinoidal-Versus-Aromatic Competition and Its Implications. Top. Curr. Chem. Z 2017, 375, 73. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, B.; Anderson, C.L.; Kang, J.; Chen, T.; Chen, J.; Feng, S.; Zhang, L.; Kolaczkowski, M.A.; Teat, S.J.; et al. Para-Azaquinodimethane: A Compact Quinodimethane Variant as an Ambient Stable Building Block for High-Performance Low Band Gap Polymers. J. Am. Chem. Soc. 2017, 139, 8355–8363. [Google Scholar] [CrossRef]
- Dyaga, B.; Mayarambakam, S.; Ibraikulov, O.A.; Zimmermann, N.; Fall, S.; Boyron, O.; Heiser, T.; Leclerc, N.; Berton, N.; Schmaltz, B. Para-Azaquinodimethane Based Quinoidal Polymers for Opto-Electronic Applications: Impact of Donor Units on the Opto-Electronic Properties. Mater. Adv. 2022, 3, 6853–6861. [Google Scholar] [CrossRef]
- Anderson, C.L.; Li, H.; Jones, C.G.; Teat, S.J.; Settineri, N.S.; Dailing, E.A.; Liang, J.; Mao, H.; Yang, C.; Klivansky, L.M.; et al. Solution-Processable and Functionalizable Ultra-High Molecular Weight Polymers via Topochemical Synthesis. Nat. Commun. 2021, 12, 6818. [Google Scholar] [CrossRef]
- Anderson, C.L.; Dai, N.; Teat, S.J.; He, B.; Wang, S.; Liu, Y. Electronic Tuning of Mixed Quinoidal-Aromatic Conjugated Polyelectrolytes: Direct Ionic Substitution on Polymer Main-Chains. Angew. Chem. Int. Ed. 2019, 58, 17978–17985. [Google Scholar] [CrossRef]
- Liu, X.; Anderson, C.L.; Liu, Y. P-Azaquinodimethane: A Versatile Quinoidal Moiety for Functional Materials Discovery. Acc. Chem. Res. 2023, 56, 1669–1682. [Google Scholar] [CrossRef]
- Wang, L.; Shi, X.; Feng, S.; Liang, W.; Fu, H.; Yao, J. Molecular Design Strategy for Practical Singlet Fission Materials: The Charm of Donor/Acceptor Decorated Quinoidal Structure. CCS Chem. 2022, 4, 2748–2756. [Google Scholar] [CrossRef]
- Aswani Raj, K.; Rajeswara Rao, M. Synthesis of P-Azaquinodimethane-Based Quinoidal Fluorophores. J. Org. Chem. 2023, 88, 14960–14968. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Ai Tran, H.N.; Staderini, M.; Monaco, A.; López-Cobeñas, A.; Bongarzone, S.; Biarnés, X.; López-Alvarado, P.; Cabezas, N.; Caramelli, M.; et al. Discovery of a Class of Diketopiperazines as Antiprion Compounds. ChemMedChem 2010, 5, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Yu, X.; Lu, A.; Cao, H.; Zhou, B.; Zhou, L.; Zhao, Z. Extraction, Chemical Composition, and Antifungal Activity of Essential Oil of Bitter Almond. Int. J. Mol. Sci. 2016, 17, 1421. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, A.; Nishimura, S.; Ebitani, K. Catalytic Transformations of Biomass-Derived Materials into Value-Added Chemicals. Catal. Surv. Asia 2012, 16, 164–182. [Google Scholar] [CrossRef]
- Al-Bayati, F.A.; Mohammed, M. Isolation, Identification, and Purification of Cinnamaldehyde from Cinnamomum Zeylanicum Bark Oil. An Antibacterial Study. Pharm. Biol. 2008, 47, 61–66. [Google Scholar] [CrossRef]
- Gallina, C.; Liberatori, A. Condensation of 1,4-Diacetylpiperazine-2,5-Dione with Aldehydes. Tetrahedron 1974, 30, 667–673. [Google Scholar] [CrossRef]
- Ayers, P.W.; Parr, R.G.; Pearson, R.G. Elucidating the Hard/Soft Acid/Base Principle: A Perspective Based on Half-Reactions. J. Chem. Phys. 2006, 124, 194107. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Meng, X.; Ke, Y.; Ye, H.; Cui, Y. Novel D-π-A Benzocarbazole Dyes with Simple Structures for Efficient Dye-Sensitized Solar Cells. J. Photochem. Photobiol. A Chem. 2019, 376, 127–134. [Google Scholar] [CrossRef]
- Tigreros, A.; Ortiz, A.; Insuasty, B. Effect of π-Conjugated Linkage on Photophysical Properties: Acetylene Linker as the Better Connection Group for Highly Solvatochromic Probes. Dye. Pigment. 2014, 111, 45–51. [Google Scholar] [CrossRef]
- Xiao, Y.; Fu, H.; Li, Z.; Zheng, Y.; Deng, P.; Lei, Y.; Yu, Y. 6 H -[1,2,5]Thiadiazolo [3,4- e ]Thieno[3,2- b ]Indole-Flanked Para -Azaquinodimethane Based Aromatic-Quinoidal Polymer Semiconductors with High Molecular Weights Synthesized via Direct Arylation Polycondensation. Mater. Adv. 2023, 4, 1927–1934. [Google Scholar] [CrossRef]
- Más-Montoya, M.; Janssen, R. The Effect of H- and J-Aggregation on the Photophysical and Photovoltaic Properties of Small Thiophene-Pyridine-DPP Molecules for Bulk-Heterojunction Solar Cells. Adv. Funct. Mater. 2017, 27, 1605779. [Google Scholar] [CrossRef]
- Joule, J.A.; Mills, K. Heterocyclic Chemistry, 5th ed.; John Wiley & Sons: Chichester, UK, 2010. [Google Scholar]
- Shimizu, A.; Ishizaki, Y.; Horiuchi, S.; Hirose, T.; Matsuda, K.; Sato, H.; Yoshida, J. HOMO–LUMO Energy-Gap Tuning of π-Conjugated Zwitterions Composed of Electron-Donating Anion and Electron-Accepting Cation. J. Org. Chem. 2021, 86, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, A.M. Standards for Photoluminescence Quantum Yield Measurements in Solution (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar] [CrossRef]
- Liu, Z.-X.; Chen, Y.-G.; Yang, Z.-Y. Understanding the Thermal-Annealing-Generated Stable Structure of Phthalocyanine Derivative/Polymer Bicomponent Systems through Scanning Tunneling Microscopy and Density Functional Theory Calculations. Polymer 2022, 238, 124375. [Google Scholar] [CrossRef]
- Reiss, H.; Heller, A. The Absolute Potential of the Standard Hydrogen Electrode: A New Estimate. J. Phys. Chem. 1985, 89, 4207–4213. [Google Scholar] [CrossRef]
- Matin, M.A.; Islam, M.M.; Bredow, T.; Aziz, M.A. The Effects of Oxidation States, Spin States and Solvents on Molecular Structure, Stability and Spectroscopic Properties of Fe-Catechol Complexes: A Theoretical Study. Adv. Chem. Eng. Sci. 2017, 7, 137–153. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Becke, A.D. Perspective: Fifty Years of Density-Functional Theory in Chemical Physics. The J. Chem. Phys. 2014, 140, 18A301. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06 Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange–Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Improta, R.; Scalmani, G.; Frisch, M.J.; Barone, V. Toward Effective and Reliable Fluorescence Energies in Solution by a New State Specific Polarizable Continuum Model Time Dependent Density Functional Theory Approach. J. Chem. Phys. 2007, 127, 074504. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zwaihed, W.; Maurel, F.; Kobeissi, M.; Schmaltz, B. New Quinoid Bio-Inspired Materials Using Para-Azaquinodimethane Moiety. Molecules 2024, 29, 186. https://doi.org/10.3390/molecules29010186
Zwaihed W, Maurel F, Kobeissi M, Schmaltz B. New Quinoid Bio-Inspired Materials Using Para-Azaquinodimethane Moiety. Molecules. 2024; 29(1):186. https://doi.org/10.3390/molecules29010186
Chicago/Turabian StyleZwaihed, Walaa, François Maurel, Marwan Kobeissi, and Bruno Schmaltz. 2024. "New Quinoid Bio-Inspired Materials Using Para-Azaquinodimethane Moiety" Molecules 29, no. 1: 186. https://doi.org/10.3390/molecules29010186
APA StyleZwaihed, W., Maurel, F., Kobeissi, M., & Schmaltz, B. (2024). New Quinoid Bio-Inspired Materials Using Para-Azaquinodimethane Moiety. Molecules, 29(1), 186. https://doi.org/10.3390/molecules29010186