
Recent Advances (2015–2020) in Drug Discovery for Attenuation of Pulmonary Fibrosis and COPD


Abstract

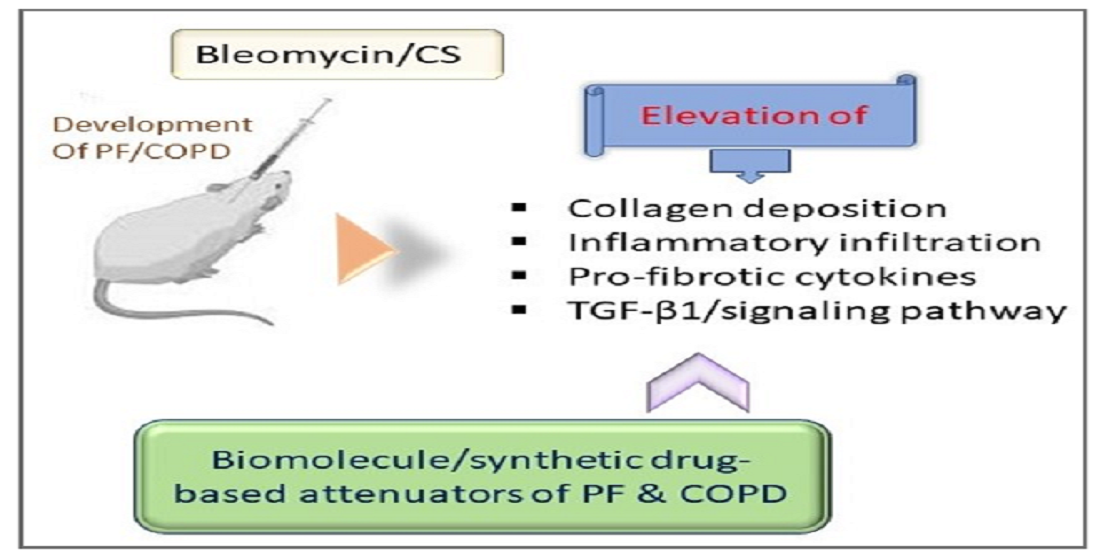
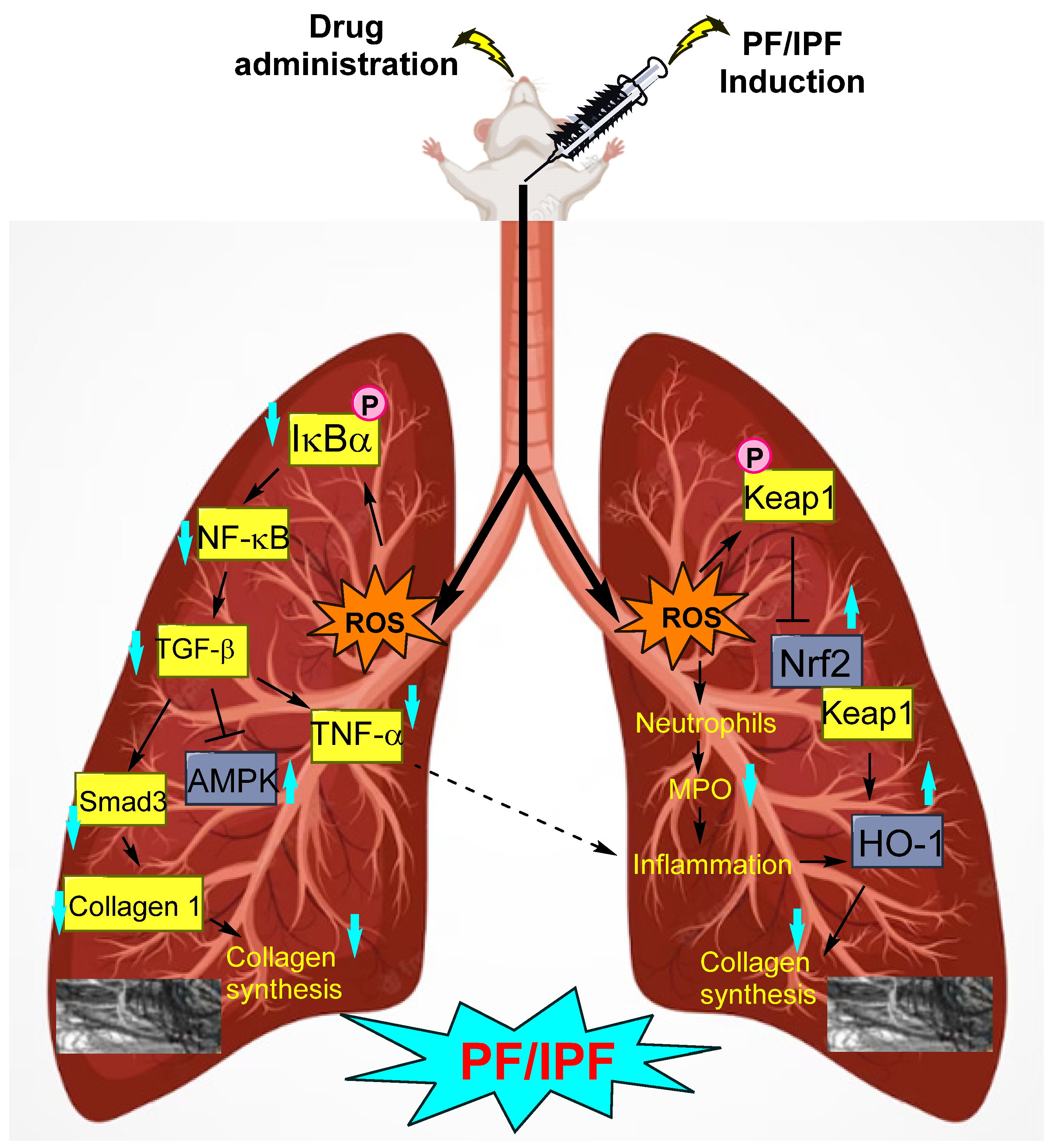
1. Introduction
1.1. Idiopathic Pulmonary Fibrosis (IPF)
1.2. Attenuation of PF by Biomolecule-Based Inhibitors
1.3. Synthetic-Molecule-Based PF Inhibitors
1.4. Miscellaneous
1.5. Chronic Obstructive Pulmonary Disease (COPD)
1.6. Inhibition of Cigarette Smoke (CS)-Induced COPD
2. Discussion
3. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hubbard, R.; Cooper, M.; Antoniak, M.; Venn, A.; Khan, S.; Johnston, I.; Lewis, S.; Britton, J. Risk of cryptogenic fibrosing alveolitis in metal workers. Lancet 2000, 355, 466–467. [Google Scholar] [CrossRef]
- Goemaere, N.N.T.; Grijm, K.; van Hal, P.T.W.; den Bakker, M.A. Nitrofurantoin-induced pulmonary fibrosis: A case report. J. Med. Case Rep. 2008, 2, 169. [Google Scholar] [CrossRef]
- Jin, Y.-K.; Li, X.-H.; Wang, W.; Liu, J.; Zhang, W.; Fang, Y.-S.; Zhang, Z.-F.; Dai, H.-P.; Ning, W.; Wang, C. Follistatin-Like 1 Promotes Bleomycin-Induced Pulmonary Fibrosis through the Transforming Growth Factor Beta 1/Mitogen-Activated Protein Kinase Signaling Pathway. Chin. Med. J. 2018, 131, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Kandhare, A.D.; Kandhare, A.A.; Bodhankar, S.L. Hesperidin ameliorates bleomycin-induced experimental pulmonary fibrosis via inhibition of TGF-beta1/Smad3/AMPK and IkappaBalpha/NF-kappaB pathways. EXCLI J. 2019, 18, 723–745. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy of a Tyrosine Kinase Inhibitor in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2011, 365, 1079–1087. [Google Scholar] [CrossRef]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.S.; Grossfeld, D.; Renna, H.A.; Agarwala, P.; Spiegler, P.; Kasselman, L.J.; Glass, A.D.; DeLeon, J.; Reiss, A.B. Idiopathic pulmonary fibrosis: Molecular mechanisms and potential treatment approaches. Respir. Investig. 2020, 58, 320–335. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-J.; Chen, X.; Yang, W.-N.; Xu, X.-M.; Lu, L.-Y.; Wang, J.; Kong, Y.-X.; Zheng, J.-H. Traditional Chinese medicine for the treatment of pulmonary fibrosis. Medicine 2020, 99, e21310. [Google Scholar] [CrossRef] [PubMed]
- Mahalanobish, S.; Saha, S.; Dutta, S.; Sil, P.C. Matrix metalloproteinase: An upcoming therapeutic approach for idiopathic pulmonary fibrosis. Pharmacol. Res. 2020, 152, 104591. [Google Scholar] [CrossRef]
- Kolilekas, L.; Papiris, S.; Bouros, D. Existing and emerging treatments for idiopathic pulmonary fibrosis. Expert Rev. Respir. Med. 2019, 13, 229–239. [Google Scholar] [CrossRef]
- Homma, S.; Bando, M.; Azuma, A.; Sakamoto, S.; Sugino, K.; Ishii, Y.; Izumi, S.; Inase, N.; Inoue, Y.; Ebina, M.; et al. Japanese guideline for the treatment of idiopathic pulmonary fibrosis. Respir. Investig. 2018, 56, 268–291. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Wilson, Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 755–772. [Google Scholar] [CrossRef]
- Song, S.; Ji, Y.; Zhang, G.; Zhang, X.; Li, B.; Li, D.; Jiang, W. Protective Effect of Atazanavir Sulphate against Pulmonary Fibrosis In Vivo and In Vitro. Basic Clin. Pharmacol. Toxicol. 2018, 122, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Yang, H.-M.; Sun, C.-Y.; Liu, Y.-L.; Zhuo, J.-Y.; Zhang, Z.-B.; Lai, X.-P.; Su, Z.-R.; Li, Y.-C. Scutellarin Enhances Antitumor Effects and Attenuates the Toxicity of Bleomycin in H22 Ascites Tumor-Bearing Mice. Front. Pharmacol. 2018, 9, 615. [Google Scholar] [CrossRef]
- Ding, D.; Cai, X.; Zheng, H.; Guo, S.-W.; Liu, X. Scutellarin Suppresses Platelet Aggregation and Stalls Lesional Progression in Mouse with Induced Endometriosis. Reprod. Sci. 2019, 26, 1417–1428. [Google Scholar] [CrossRef]
- Miao, K.; Pan, T.; Mou, Y.; Zhang, L.; Xiong, W.; Xu, Y.; Yu, J.; Wang, Y. Scutellarein inhibits BLM-mediated pulmonary fibrosis by affecting fibroblast differentiation, proliferation, and apoptosis. Ther. Adv. Chronic Dis. 2020, 11, 204062232094018. [Google Scholar] [CrossRef]
- Desaki, M.; Okazaki, H.; Sunazuka, T.; Omura, S.; Yamamoto, K.; Takizawa, H. Molecular Mechanisms of Anti-Inflammatory Action of Erythromycin in Human Bronchial Epithelial Cells: Possible Role in the Signaling Pathway That Regulates Nuclear Factor-κB Activation. Antimicrob. Agents Chemother. 2004, 48, 1581–1585. [Google Scholar] [CrossRef]
- Li, Y.J.; Azuma, A.; Usuki, J.; Abe, S.; Matsuda, K.; Sunazuka, T.; Shimizu, T.; Hirata, Y.; Inagaki, H.; Kawada, T.; et al. EM703 improves bleomycin-induced pulmonary fibrosis in mice by the inhibition of TGF-β signaling in lung fibroblasts. Respir. Res. 2006, 7, 16. [Google Scholar] [CrossRef]
- Huang, X.; Zou, L.; Yu, X.; Chen, M.; Guo, R.; Cai, H.; Yao, D.; Xu, X.; Chen, Y.; Ding, C.; et al. Salidroside attenuates chronic hypoxia-induced pulmonary hypertension via adenosine A2a receptor related mitochondria-dependent apoptosis pathway. J. Mol. Cell. Cardiol. 2015, 82, 153–166. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.-Z.; Lu, A.-X.; Zhang, K.-F.; Li, B.-J. Anticancer effect of salidroside on A549 lung cancer cells through inhibition of oxidative stress and phospho-p38 expression. Oncol. Lett. 2014, 7, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Gao, L.; Mao, J.; He, H.; Liu, J.; Cai, X.; Lin, H.; Wu, T. Salidroside protects against bleomycin-induced pulmonary fibrosis: Activation of Nrf2-antioxidant signaling, and inhibition of NF-κB and TGF-β1/Smad-2/-3 pathways. Cell Stress Chaperones 2016, 21, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, E.; Garcia-Gonzalez, E.; Selvi, E.; Akhmetshina, A.; Palumbo, K.; Lorenzini, S.; Maggio, R.; Lucattelli, M.; Galeazzi, M.; Distler, J.W.H. The cannabinoid WIN55, 212–2 abrogates dermal fibrosis in scleroderma bleomycin model. Ann. Rheum. Dis. 2011, 70, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.G.; Selvi, E.; Balistreri, E.; Akhmetshina, A.; Palumbo, K.; Lorenzini, S.; Lazzerini, P.E.; Montilli, C.; Capecchi, P.L.; Lucattelli, M.; et al. Synthetic cannabinoid ajulemic acid exerts potent antifibrotic effects in experimental models of systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Lucattelli, M.; Fineschi, S.; Selvi, E.; Gonzalez, E.G.; Bartalesi, B.; de Cunto, G.; Lorenzini, S.; Galeazzi, M.; Lungarella, G. Ajulemic acid exerts potent anti-fibrotic effect during the fibrogenic phase of bleomycin lung. Respir. Res. 2016, 17, 49. [Google Scholar] [CrossRef]
- Sax, P.E.; Zolopa, A.; Brar, I.; Elion, R.; Ortiz, R.; Post, F.; Wang, H.; Callebaut, C.; Martin, H.; Fordyce, M.W.; et al. Tenofovir Alafenamide vs. Tenofovir Disoproxil Fumarate in Single Tablet Regimens for Initial HIV-1 Therapy. Am. J. Ther. 2014, 67, 52–58. [Google Scholar] [CrossRef]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Schall, R.A.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef]
- Li, L.; Zhao, J.; Zhou, L.; Chen, J.; Ma, Y.; Yu, Y.; Cheng, J. Tenofovir alafenamide fumarate attenuates bleomycin-induced pulmonary fibrosis by upregulating the NS5ATP9 and TGF-β1/Smad3 signaling pathway. Respir. Res. 2019, 20, 163. [Google Scholar] [CrossRef]
- Chung, K.-T.; Wong, T.Y.; Wei, C.-I.; Huang, Y.-W.; Lin, Y. Tannins and Human Health: A Review. Crit. Rev. Food Sci. Nutr. 1998, 38, 421–464. [Google Scholar] [CrossRef]
- Chu, L.; Li, P.; Song, T.; Han, X.; Zhang, X.; Song, Q.; Liu, T.; Zhang, Y.; Zhang, J. Protective effects of tannic acid on pressure overload-induced cardiac hypertrophy and underlying mechanisms in rats. J. Pharm. Pharmacol. 2017, 69, 1191–1207. [Google Scholar] [CrossRef]
- Reed, E.B.; Ard, S.; La, J.; Park, C.Y.; Culligan, L.; Fredberg, J.J.; Smolyaninova, L.V.; Orlov, S.N.; Chen, B.; Guzy, R.; et al. Anti-fibrotic effects of tannic acid through regulation of a sustained TGF-beta receptor signaling. Respir. Res. 2019, 20, 168. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-H.; Sun, R.-S.; Hu, J.-M.; Mo, Z.-Y.; Yang, Z.-F.; Jin, G.-Y.; Guan, W.-D.; Zhong, N.-S. Inhibitory Effect OF Emodin on Bleomycin-Induced Pulmonary Fibrosis in Mice. Clin. Exp. Pharmacol. Physiol. 2009, 36, 146–153. [Google Scholar] [CrossRef]
- Tian, S.-L.; Yang, Y.; Liu, X.-L.; Xu, Q.-B. Emodin Attenuates Bleomycin-Induced Pulmonary Fibrosis via Anti-Inflammatory and Anti-Oxidative Activities in Rats. Med. Sci. Monit. 2018, 24, 1–10. [Google Scholar] [CrossRef]
- Bohanon, F.J.; Wang, X.; Ding, C.; Ding, Y.; Radhakrishnan, G.L.; Rastellini, C.; Zhou, J.; Radhakrishnan, R.S. Oridonin inhibits hepatic stellate cell proliferation and fibrogenesis. J. Surg. Res. 2014, 190, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Zhang, T.; Ma, X.; Jiang, K.; Wu, H.; Qiu, C.; Guo, M.; Deng, G. Oridonin attenuates the release of pro-inflammatory cytokines in lipopolysaccharide-induced RAW264.7 cells and acute lung injury. Oncotarget 2017, 8, 68153–68164. [Google Scholar] [CrossRef]
- Li, L.-C.; Kan, L.-D. Traditional Chinese medicine for pulmonary fibrosis therapy: Progress and future prospects. J. Ethnopharmacol. 2017, 198, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhao, P.; Xie, Z.; Wang, L.; Chen, S. Oridonin Inhibits Myofibroblast Differentiation and Bleomycin-induced Pulmonary Fibrosis by Regulating Transforming Growth Factor β (TGFβ)/Smad Pathway. Med. Sci. Monit. 2018, 24, 7548–7555. [Google Scholar] [CrossRef]
- Guan, E.; Wang, Y.; Wang, C.; Zhang, R.; Zhao, Y.; Hong, J. Necrostatin-1 attenuates lipopolysaccharide-induced acute lung injury in mice. Exp. Lung Res. 2017, 43, 378–387. [Google Scholar] [CrossRef]
- Pan, L.; Yao, D.-C.; Yu, Y.-Z.; Li, S.-J.; Chen, B.-J.; Hu, G.-H.; Xi, C.; Wang, Z.-H.; Wang, H.-Y.; Li, J.-H.; et al. Necrostatin-1 protects against oleic acid-induced acute respiratory distress syndrome in rats. Biochem. Biophys. Res. Commun. 2016, 478, 1602–1608. [Google Scholar] [CrossRef]
- Mou, F.; Mou, C. Necrostatin-1 Alleviates Bleomycin-Induced Pulmonary Fibrosis and Extracellular Matrix Expression in Interstitial Pulmonary Fibrosis. Med. Sci. Monit. 2020, 26, e919739-1–e919739-9. [Google Scholar] [CrossRef]
- Ahn, J.-Y.; Kim, M.-H.; Lim, M.-J.; Park, S.; Lee, S.-L.; Yun, Y.-S.; Song, J.-Y. The inhibitory effect of ginsan on TGF-β mediated fibrotic process. J. Cell. Physiol. 2011, 226, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Huang, X.; Liu, Z.; Xu, X.; Xiao, H.; Zhang, X.; Dai, H.; Wang, C. Ouabain ameliorates bleomycin induced pulmonary fibrosis by inhibiting proliferation and promoting apoptosis of lung fibroblasts. Am. J. Transl. Res. 2018, 10, 2967–2974. [Google Scholar] [PubMed]
- La, J.; Reed, E.; Chan, L.; Smolyaninova, L.V.; Akomova, O.A.; Mutlu, G.M.; Orlov, S.N.; Dulin, N.O. Downregulation of TGF-β Receptor-2 Expression and Signaling through Inhibition of Na/K-ATPase. PLoS ONE 2016, 11, e0168363. [Google Scholar] [CrossRef] [PubMed]
- La, J.; Reed, E.B.; Koltsova, S.; Akimova, O.; Hamanaka, R.B.; Mutlu, G.M.; Orlov, S.N.; Dulin, N.O. Regulation of myofibroblast differentiation by cardiac glycosides. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L815–L823. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Geng, J.; Xu, X.; Huang, X.; Leng, D.; Jiang, D.; Liang, J.; Wang, C.; Jiang, D.; Dai, H. miR-130b-3p Modulates Epithelial-Mesenchymal Crosstalk in Lung Fibrosis by Targeting IGF-1. PLoS ONE 2016, 11, e0150418. [Google Scholar] [CrossRef]
- Choi, J.-E.; Lee, S.-S.; Sunde, D.A.; Huizar, I.; Haugk, K.L.; Thannickal, V.J.; Vittal, R.; Plymate, S.R.; Schnapp, L.M. Insulin-like Growth Factor-I Receptor Blockade Improves Outcome in Mouse Model of Lung Injury. Am. J. Respir. Crit. Care Med. 2009, 179, 212–219. [Google Scholar] [CrossRef]
- Aljada, A.; Mousa, S.A. Metformin and neoplasia: Implications and indications. Pharmacol. Ther. 2012, 133, 108–115. [Google Scholar] [CrossRef]
- Xiao, H.; Huang, X.; Wang, S.; Liu, Z.; Dong, R.; Song, D.; Dai, H. Metformin ameliorates bleomycin-induced pulmonary fibrosis in mice by suppressing IGF-1. Am. J. Transl. Research 2020, 12, 940–949. [Google Scholar]
- Saghir, S.A.M.; Al-Gabri, N.A.; Khafaga, A.F.; El-Shaer, N.H.; Alhumaidh, K.A.; Elsadek, M.F.; Ahmed, B.M.; Alkhawtani, D.M.; El-Hack, M.E.A. Thymoquinone-PLGA-PVA Nanoparticles Ameliorate Bleomycin-Induced Pulmonary Fibrosis in Rats via Regulation of Inflammatory Cytokines and iNOS Signaling. Animals 2019, 9, 951. [Google Scholar] [CrossRef]
- Chen, H.; Yang, R.; Tang, Y.; Fu, X. Effects of curcumin on artery blood gas index of rats with pulmonary fibrosis caused by paraquat poisoning and the expression of Smad 4, Smurf 2, interleukin-4 and interferon-γ. Exp. Ther. Med. 2019, 17, 3664–3670. [Google Scholar] [CrossRef]
- Qiu, Y.; Pan, X.; Hu, Y. Polydatin ameliorates pulmonary fibrosis by suppressing inflammation and the epithelial mesenchymal transition via inhibiting the TGF-β/Smad signaling pathway. RSC Adv. 2019, 9, 8104–8112. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-L.; Chen, B.-Y.; Nie, J.; Zhao, G.-H.; Zhuo, J.-Y.; Yuan, J.; Li, Y.-C.; Wang, L.-L.; Chen, Z.-W. Polydatin prevents bleomycin-induced pulmonary fibrosis by inhibiting the TGF-β/Smad/ERK signaling pathway. Exp. Ther. Med. 2020, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Guo, X.-Y.; Gao, F.; Jin, M.-L.; Song, X.-N. Identification of the Perturbed Metabolic Pathways Associating with Renal Fibrosis and Evaluating Metabolome Changes of Pretreatment with Astragalus polysaccharide through Liquid Chromatography Quadrupole Time-of-Flight Mass Spectrometry. Front. Pharmacol. 2020, 10, 1623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xu, L.; An, X.; Sui, X.; Lin, S. Astragalus polysaccharides attenuate pulmonary fibrosis by inhibiting the epithelial-mesenchymal transition and NF-κB pathway activation. Int. J. Mol. Med. 2020, 46, 331–339. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, T.; Wei, Z.; Lu, G.; Jiang, S.; Xia, Y.; Dai, Y. Paeoniflorin, the main active constituent of Paeonia lactiflora roots, attenuates bleomycin-induced pulmonary fibrosis in mice by suppressing the synthesis of type I collagen. J. Ethnopharmacol. 2013, 149, 825–832. [Google Scholar] [CrossRef]
- Jia, L.; Sun, P.; Gao, H.; Shen, J.; Gao, Y.; Meng, C.; Fu, S.; Yao, H.; Zhang, G. Mangiferin attenuates bleomycin-induced pulmonary fibrosis in mice through inhibiting TLR 4/p65 and TGF-β1/Smad2/3 pathway. J. Pharm. Pharmacol. 2019, 71, 1017–1028. [Google Scholar] [CrossRef]
- Jin, M.; Wu, Y.; Wang, L.; Zang, B.; Tan, L. Hydroxysafflor Yellow a Attenuates Bleomycin-induced Pulmonary Fibrosis in Mice. Phytotherapy Res. 2016, 30, 577–587. [Google Scholar] [CrossRef]
- Jin, M.; Wang, L.; Wu, Y.; Zang, B.; Tan, L. Protective effect of hydroxysafflor yellow a on bleomycin-induced pulmonary inflammation and fibrosis in rats. Chin. J. Integr. Med. 2018, 24, 32–39. [Google Scholar] [CrossRef]
- Song, X.; Liu, W.; Xie, S.; Wang, M.; Cao, G.; Mao, C.; Lv, C. All-transretinoic acid ameliorates bleomycin-induced lung fibrosis by downregulating the TGF-β1/Smad3 signaling pathway in rats. Lab. Investig. 2013, 93, 1219–1231. [Google Scholar] [CrossRef]
- Cho, H.-S.; Lee, M.-H.; Lee, J.W.; No, K.-O.; Park, S.-K.; Lee, H.-S.; Kang, S.; Cho, W.-G.; Park, H.J.; Oh, K.W.; et al. Anti-wrinkling effects of the mixture of vitamin C, vitamin E, pycnogenol and evening primrose oil, and molecular mechanisms on hairless mouse skin caused by chronic ultraviolet B irradiation. Photodermatol. Photoimmunol. Photomed. 2007, 23, 155–162. [Google Scholar] [CrossRef]
- Ko, J.-W.; Shin, N.-R.; Park, S.-H.; Kim, J.-S.; Cho, Y.-K.; Kim, J.-C.; Shin, I.-S.; Shin, D.-H. Pine bark extract (Pycnogenol®) suppresses cigarette smoke-induced fibrotic response via transforming growth factor-β1/Smad family member 2/3 signaling. Lab. Anim. Res. 2017, 33, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Feng, M.; Sun, R.; Li, Z.; Hu, L.; Peng, G.; Xu, X.; Wang, W.; Cui, F.; Yue, W.; et al. Andrographolide ameliorates bleomycin-induced pulmonary fibrosis by suppressing cell proliferation and myofibroblast differentiation of fibroblasts via the TGF-β1-mediated Smad-dependent and -independent pathways. Toxicol. Lett. 2020, 321, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, W.A.; Willems, S.; Vos, R.; Vanaudenaerde, B.M.; De Vleeschauwer, S.I.; Rinaldi, M.; Vanhooren, H.M.; Geudens, N.; Verleden, S.E.; Demedts, M.G.; et al. Azithromycin reduces pulmonary fibrosis in a bleomycin mouse model. Exp. Lung Res. 2010, 36, 602–614. [Google Scholar] [CrossRef]
- Zhang, L.-L.; Jiang, X.-M.; Huang, M.-Y.; Feng, Z.-L.; Chen, X.; Wang, Y.; Li, H.; Li, A.; Lin, L.-G.; Lu, J.-J. Nagilactone E suppresses TGF-β1-induced epithelial–mesenchymal transition, migration and invasion in non-small cell lung cancer cells. Phytomedicine 2019, 52, 32–39. [Google Scholar] [CrossRef]
- Li, A.; Xiao, X.; Feng, Z.-L.; Chen, X.; Liu, L.-J.; Lin, L.-G.; Lu, J.-J.; Zhang, L.-L. Nagilactone D ameliorates experimental pulmonary fibrosis in vitro and in vivo via modulating TGF-β/Smad signaling pathway. Toxicol. Appl. Pharmacol. 2020, 389, 114882. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Cao, Y.; Yu, J.; Liu, R.; Bai, B.; Qi, H.; Zhang, Q.; Guo, W.; Zhu, H.; Qu, L. Baicalin alleviates ischemia-induced memory impairment by inhibiting the phosphorylation of CaMKII in hippocampus. Brain Res. 2016, 1642, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, C.; Li, L.; Liu, J.; Gao, Y.; Mu, K.; Chen, D.; Lu, A.; Ren, Y.; Li, Z. Baicalin alleviates bleomycin-induced pulmonary fibrosis and fibroblast proliferation in rats via the PI3K/AKT signaling pathway. Mol. Med. Rep. 2020, 21, 2321–2334. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Zhang, D.; Yang, R.; Wang, S. Paclitaxel: New uses for an old drug, Drug Design. Drug Des. Dev. Ther. 2014, 8, 279–284. [Google Scholar] [CrossRef]
- Nie, Y.; Zhang, D.; Qian, F.; Wu, Y. Baccatin III ameliorates bleomycin-induced pulmonary fibrosis via suppression of TGF-β1 production and TGF-β1-induced fibroblast differentiation. Int. Immunopharmacol. 2019, 74, 105696. [Google Scholar] [CrossRef]
- Zhao, L.; Zhen, C.; Wu, Z.; Hu, R.; Zhou, C.; Guo, Q. General pharmacological properties, developmental toxicity, and analgesic activity of gambogic acid, a novel natural anticancer agent. Drug Chem. Toxicol. 2010, 33, 88–96. [Google Scholar] [CrossRef]
- Cascão, R.; Vidal, B.; Raquel, H.; Neves-Costa, A.; Figueiredo, N.; Gupta, V.; Fonseca, J.; Moita, L.F. Potent Anti-Inflammatory and Antiproliferative Effects of Gambogic Acid in a Rat Model of Antigen-Induced Arthritis. Mediat. Inflamm. 2014, 2014, 195327. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, G.; Ji, Y.; Zhua, H.; Lv, C.; Jiang, W. Protective role of gambogic acid in experimental pulmonary fibrosis in vitro and in vivo. Phytomedicine 2016, 23, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Ma, Z.; Shi, S.; Hu, Y.; Ma, T.; Rong, G.; Yang, J. Sulforaphane prevents bleomycin-induced pulmonary fibrosis in mice by inhibiting oxidative stress via nuclear factor erythroid 2-related factor-2 activation. Mol. Med. Rep. 2017, 15, 4005–4014. [Google Scholar] [CrossRef] [PubMed]
- Kyung, S.Y.; Kim, D.Y.; Yoon, J.Y.; Son, E.S.; Kim, Y.J.; Park, J.W.; Jeong, S.H. Sulforaphane attenuates pulmonary fibrosis by inhibiting the epithelial-mesenchymal transition. BMC Pharmacol. Toxicol. 2018, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; DU, F.; Chen, W.; Yao, M.; Lv, K.; Liu, Y. Tanshinone IIA blocks epithelial-mesenchymal transition through HIF-1α downregulation, reversing hypoxia-induced chemotherapy resistance in breast cancer cell lines. Oncol. Rep. 2014, 31, 2561–2568. [Google Scholar] [CrossRef]
- Li, J.-H.; Xu, M.; Xie, X.-Y.; Fan, Q.-X.; Mu, D.-G.; Zhang, Y.; Cao, F.-L.; Wang, Y.-X.; Zhao, P.-T.; Zhang, B.; et al. Tanshinone IIA suppresses lung injury and apoptosis, and modulates protein kinase B and extracellular signal-regulated protein kinase pathways in rats challenged with seawater exposure. Clin. Exp. Pharmacol. Physiol. 2011, 38, 269–277. [Google Scholar] [CrossRef]
- Tang, H.; He, H.; Ji, H.; Gao, L.; Mao, J.; Liu, J.; Lin, H.; Wu, T. Tanshinone IIA ameliorates bleomycin-induced pulmonary fibrosis and inhibits transforming growth factor-beta-β–dependent epithelial to mesenchymal transition. J. Surg. Res. 2015, 197, 167–175. [Google Scholar] [CrossRef]
- Xiong, A.; Liu, Y. Targeting Hypoxia Inducible Factors-1α As a Novel Therapy in Fibrosis. Front. Pharmacol. 2017, 8, 326. [Google Scholar] [CrossRef]
- Huang, H.; Wang, X.; Zhang, X.; Wang, H.; Jiang, W. Roxadustat attenuates experimental pulmonary fibrosis in vitro and in vivo. Toxicol. Lett. 2020, 331, 112–121. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, G.-Y.; Liu, T.; Duan, J.-X.; Zhou, H.-F.; Lee, K.S.S.; Hammock, B.D.; Fang, X.; Jiang, J.-X.; Guan, C.-X. Soluble epoxide hydrolase inhibitor 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea attenuates bleomycin-induced pulmonary fibrosis in mice. Cell Tissue Res. 2016, 363, 399–409. [Google Scholar] [CrossRef]
- Huang, W.-S.; Metcalf, C.A.; Sundaramoorthi, R.; Wang, Y.; Zou, D.; Thomas, R.M.; Zhu, X.; Cai, L.; Wen, D.; Liu, S.; et al. Discovery of 3-[2-(Imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a Potent, Orally Active Pan-Inhibitor of Breakpoint Cluster Region-Abelson (BCR-ABL) Kinase Including the T315I Gatekeeper Mutant. J. Med. Chem. 2010, 53, 4701–4719. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, L.; Kang, Z.; Jiang, W.; Lv, C. Ponatinib ameliorates pulmonary fibrosis by suppressing TGF-β1/Smad3 pathway. Pulm. Pharmacol. Ther. 2015, 34, 1–7. [Google Scholar] [CrossRef] [PubMed]
- John, A.E.; Graves, R.H.; Pun, K.T.; Vitulli, G.; Forty, E.J.; Mercer, P.F.; Morrell, J.L.; Barrett, J.W.; Rogers, R.F.; Hafeji, M.; et al. Translational pharmacology of an inhaled small molecule αvβ6 integrin inhibitor for idiopathic pulmonary fibrosis. Nat. Commun. 2020, 11, 4659. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Hu, J.; Yu, P.; Zhu, Z.; Yu, R.; Ge, C.; Li, C.; Wu, G.; Lin, B.; Liu, G.; et al. Peptide PD29 treats bleomycin-induced pulmonary fibrosis by inhibiting the TGF-β/smad signaling pathway. Exp. Lung Res. 2019, 45, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, T.; Uzun, O.; Macit, A.; Comunoglu, C.; Yavuz, O.; Silan, C.; Yuksel, H.; Yildirim, H.A. Pyrrolidine dithiocarbamate attenuates the development of monocrotaline-induced pulmonary arterial hypertension. Pathol.-Res. Pr. 2013, 209, 302–308. [Google Scholar] [CrossRef]
- Zaafan, M.A.; Zaki, H.F.; El-Brairy, A.I.; Kenawy, S.A. Pyrrolidinedithiocarbamate attenuates bleomycin-induced pulmonary fibrosis in rats: Modulation of oxidative stress, fibrosis, and inflammatory parameters. Exp. Lung Res. 2016, 42, 408–416. [Google Scholar] [CrossRef]
- Qiu, W.; Wu, M.; Liu, S.; Chen, B.; Pan, C.; Yang, M.; Wang, K.-J. Suppressive immunoregulatory effects of three antidepressants via inhibition of the nuclear factor-κB activation assessed using primary macrophages of carp (Cyprinus carpio). Toxicol. Appl. Pharmacol. 2017, 322, 1–8. [Google Scholar] [CrossRef]
- Achar, E.; Maciel, T.T.; Collares, C.F.; Teixeira, V.P.; Schor, N. Amitriptyline attenuates interstitial inflammation and ameliorates the progression of renal fibrosis. Kidney Int. 2009, 75, 596–604. [Google Scholar] [CrossRef]
- Zaafan, M.A.; Haridy, A.R.; Abdelhamid, A.M. Amitriptyline attenuates bleomycin-induced pulmonary fibrosis: Modulation of the expression of NF-κβ, iNOS, and Nrf2. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 279–286. [Google Scholar] [CrossRef]
- Tao, W.; Li, P.-S.; Yang, L.-Q.; Ma, Y.-B. Effects of a Soluble Epoxide Hydrolase Inhibitor on Lipopolysaccharide-Induced Acute Lung Injury in Mice. PLoS ONE 2016, 11, e0160359. [Google Scholar] [CrossRef]
- Macías, J.; Mancebo, M.; Merino, D.; Téllez, F.; Montes-Ramírez, M.L.; Pulido, F.; Rivero-Juárez, A.; Raffo, M.; Pérez-Pérez, M.; Merchante, N.; et al. Changes in Liver Steatosis After Switching from Efavirenz to Raltegravir Among Human Immunodeficiency Virus–Infected Patients With Nonalcoholic Fatty Liver Disease. Clin. Infect. Dis. 2017, 65, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, H.; Zhang, G.; Li, D.; Wang, H.; Jiang, W. Raltegravir Attenuates Experimental Pulmonary Fibrosis In Vitro and In Vivo. Front. Pharmacol. 2019, 10, 903. [Google Scholar] [CrossRef] [PubMed]
- Roversi, S.; Corbetta, L.; Clini, E. GOLD 2017 recommendations for COPD patients: Toward a more personalized approach. COPD Res. Pr. 2017, 3, 5. [Google Scholar] [CrossRef]
- Vogelmeier, C.F.; Criner, G.J.; Martinez, F.J.; Anzueto, A.; Barnes, P.J.; Bourbeau, J.; Celli, B.R.; Chen, R.; Decramer, M.; Fabbri, L.M.; et al. Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Lung Disease 2017 Report. Respirology 2017, 22, 575–601. [Google Scholar] [CrossRef] [PubMed]
- Vestbo, J.; Hurd, S.S.; Agustí, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M.; et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2013, 187, 347–365. [Google Scholar] [CrossRef]
- Krempaska, K.; Barnowski, S.; Gavini, J.; Hobi, N.; Ebener, S.; Simillion, C.; Stokes, A.; Schliep, R.; Knudsen, L.; Geiser, T.K.; et al. Azithromycin has enhanced effects on lung fibroblasts from idiopathic pulmonary fibrosis (IPF) patients compared to controls. Respir. Res. 2020, 21, 25. [Google Scholar] [CrossRef]
- Jiao, H.; Song, J.; Sun, X.; Sun, D.; Zhong, M. Sodium Arsenite Inhibits Lung Fibroblast Differentiation and Pulmonary Fibrosis. Pharmacology 2019, 104, 368–376. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhu, D. Molecular Hydrogen Therapy Ameliorates Organ Damage Induced by Sepsis. Oxidative Med. Cell. Longev. 2016, 2016, 1–6. [Google Scholar] [CrossRef]
- Gao, L.; Jiang, D.; Geng, J.; Dong, R.; Dai, H. Hydrogen inhalation attenuated bleomycin-induced pulmonary fibrosis by inhibiting transforming growth factor-β1 and relevant oxidative stress and epithelial-to-mesenchymal transition. Exp. Physiol. 2019, 104, 1942–1951. [Google Scholar] [CrossRef]
- Decramer, M.; Janssens, W.; Miravitlles, M. Chronic obstructive pulmonary disease. Lancet 2012, 379, 1341–1351. [Google Scholar] [CrossRef]
- Chronic Obstructive Pulmonary Disease (COPD). (n.d.) Available online: https://www.who.int/en/news-room/fact-sheets/detail/chronic-obstructive-pulmonary-disease-(copd) (accessed on 12 November 2020).
- Gartlehner, G.; Hansen, R.A.; Carson, S.S.; Lohr, K.N. Efficacy and Safety of Inhaled Corticosteroids in Patients With COPD: A Systematic Review and Meta-Analysis of Health Outcomes. Ann. Fam. Med. 2006, 4, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Kew, K.M.; Seniukovich, A. Inhaled steroids and risk of pneumonia for chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2014, 2014, CD010115. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.-S.; Yang, M.; Liu, H.-C.; Zuo, C.; Li, H.-J.; Fan, J.-M. Ginsenoside Rg1, a major active component isolated from Panax notoginseng, restrains tubular epithelial to myofibroblast transition in vitro. J. Ethnopharmacol. 2009, 122, 35–41. [Google Scholar] [CrossRef]
- Yu, M.; Yu, X.; Guo, D.; Yu, B.; Li, L.; Liao, Q.; Xing, R. Ginsenoside Rg1 attenuates invasion and migration by inhibiting transforming growth factor-β1-induced epithelial to mesenchymal transition in HepG2 cells. Mol. Med. Rep. 2015, 11, 3167–3173. [Google Scholar] [CrossRef]
- Guan, S.; Xu, W.; Han, F.; Gu, W.; Song, L.; Ye, W.; Liu, Q.; Guo, X. Ginsenoside Rg1 Attenuates Cigarette Smoke-Induced Pulmonary Epithelial-Mesenchymal Transition via Inhibition of the TGF- β 1/Smad Pathway. BioMed Res. Int. 2017, 2017, 7171404. [Google Scholar] [CrossRef]
- Park, C.; Hong, S.H.; Kim, G.-Y.; Choi, Y.H. So-Cheong-Ryong-Tang induces apoptosis through activation of the intrinsic and extrinsic apoptosis pathways, and inhibition of the PI3K/Akt signaling pathway in non-small-cell lung cancer A549 cells. BMC Complement. Altern. Med. 2015, 15, 113. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.-R.; Kim, C.; Seo, C.-S.; Ko, J.-W.; Cho, Y.-K.; Kim, J.-C.; Kim, J.-S.; Shin, I.-S. So-Cheong-Ryoung-Tang Attenuates Pulmonary Inflammation Induced by Cigarette Smoke in Bronchial Epithelial Cells and Experimental Mice. Front. Pharmacol. 2018, 9, 1064. [Google Scholar] [CrossRef]
- Trappoliere, M.; Caligiuri, A.; Schmid, M.; Bertolani, C.; Failli, P.; Vizzutti, F.; Novo, E.; di Manzano, C.; Marra, F.; Loguercio, C.; et al. Silybin, a component of sylimarin, exerts anti-inflammatory and anti-fibrogenic effects on human hepatic stellate cells. J. Hepatol. 2009, 50, 1102–1111. [Google Scholar] [CrossRef]
- Ko, J.-W.; Shin, N.-R.; Park, S.-H.; Lee, I.-C.; Ryu, J.-M.; Kim, H.-J.; Cho, Y.-K.; Kim, J.-C.; Shin, I.-S. Silibinin inhibits the fibrotic responses induced by cigarette smoke via suppression of TGF-β1/Smad 2/3 signaling. Food Chem. Toxicol. 2017, 106, 424–429. [Google Scholar] [CrossRef]
- Tang, F.; Tang, Q.; Tian, Y.; Fan, Q.; Huang, Y.; Tan, X. Network pharmacology-based prediction of the active ingredients and potential targets of Mahuang Fuzi Xixin decoction for application to allergic rhinitis. J. Ethnopharmacol. 2015, 176, 402–412. [Google Scholar] [CrossRef]
- Ko, J.-W.; Seo, C.-S.; Shin, N.-R.; Kim, J.-S.; Lee, S.-I.; Kim, J.-C.; Kim, S.-H.; Shin, I.-S. Modificated Mahuang-Tang, a traditional herbal medicine suppresses inflammatory responses induced by cigarette smoke in human airway epithelial cell and mice. Phytomedicine 2019, 59, 152777. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Chunhua, M.; Shumin, W. Effects of acteoside on lipopolysaccharide-induced inflammation in acute lung injury via regulation of NF-κB pathway in vivo and in vitro. Toxicol. Appl. Pharmacol. 2015, 285, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-W.; Shin, N.-R.; Park, J.-W.; Park, S.-Y.; Kwon, O.-K.; Lee, H.-S.; Kim, J.H.; Lee, H.J.; Lee, J.; Zhang, Z.-Y.; et al. Callicarpa japonica Thunb. attenuates cigarette smoke-induced neutrophil inflammation and mucus secretion. J. Ethnopharmacol. 2015, 175, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.-R.; Kim, C.; Seo, C.-S.; Ko, J.-W.; Cho, Y.-K.; Shin, I.-S.; Kim, J.-S. Galgeun-tang Attenuates Cigarette Smoke and Lipopolysaccharide Induced Pulmonary Inflammation via IκBα/NF-κB Signaling. Molecules 2018, 23, 2489. [Google Scholar] [CrossRef]
- Kanaji, N.; Basma, H.; Nelson, A.; Farid, M.; Sato, T.; Nakanishi, M.; Wang, X.; Michalski, J.; Li, Y.; Gunji, Y.; et al. Fibroblasts that resist cigarette smoke-induced senescence acquire profibrotic phenotypes. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L364–L373. [Google Scholar] [CrossRef]
- Shin, N.-R.; Park, J.-W.; Lee, I.-C.; Ko, J.-W.; Park, S.-H.; Kim, J.-S.; Kim, J.-C.; Ahn, K.-S.; Shin, I.-S. Melatonin suppresses fibrotic responses induced by cigarette smoke via downregulation of TGF-β1. Oncotarget 2017, 8, 95692–95703. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Zhang, Y.-N.; Zhou, H.; Guan, L.; Li, Y.; Sun, M.-J. IL-17A Promotes Initiation and Development of Intestinal Fibrosis through EMT. Dig. Dis. Sci. 2018, 63, 2898–2909. [Google Scholar] [CrossRef]
- Thakur, V.; Pritchard, M.T.; McMullen, M.R.; Nagy, L.E. Adiponectin normalizes LPS-stimulated TNF-α production by rat Kupffer cells after chronic ethanol feeding. Am. J. Physiol. Liver Physiol. 2006, 290, G998–G1007. [Google Scholar] [CrossRef]
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary fibrosis: Patterns and perpetrators. J. Clin. Investig. 2012, 122, 2756–2762. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.-J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1028–1040. [Google Scholar] [CrossRef]
- Khafaga, A.F.; Abu-Ahmed, H.M.; El-Khamary, A.N.; Elmehasseb, I.M.; Shaheen, H.M. Enhancement of Equid Distal Limb Wounds Healing by Topical Application of Silver Nanoparticles. J. Equine Veter. Sci. 2018, 61, 76–87. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Konzack, A.; Pihlajaniemi, T.; Heljasvaara, R.; Kietzmann, T. Redox-fibrosis: Impact of TGFβ1 on ROS generators, mediators and functional consequences. Redox Biol. 2015, 6, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Campo, G.; Pavasini, R.; Malagù, M.; Mascetti, S.; Biscaglia, S.; Ceconi, C.; Papi, A.; Contoli, M. Chronic Obstructive Pulmonary Disease and Ischemic Heart Disease Comorbidity: Overview of Mechanisms and Clinical Management. Cardiovasc Drugs Ther. 2015, 29, 147–157. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Entry | Structure | Number in the Text | Common Name | IUPAC Name | CAS Number |
|---|---|---|---|---|---|
| 1 |  | 1 | Scutellarin | 4′,5,6-Trihydroxyflavon-7-yl β-D-glucopyranosiduronic acid | 27740-01-8 |
| 2 |  | 2 | Erythromycin 703 | (2R,3R,6R,7S,8S,9R,10R)-3-((2R,3R)-2,3-Dihydroxypentan-2-yl)-7-(((4R,5S,6S)-5-hydroxy-4-methoxy-4,6-dimethyltetrahydro-2H-pyran-2-yl)oxy)-9-(((2S,4S,6R)-3-hydroxy-6-methyl-4-(methylamino)tetrahydro-2H-pyran-2-yl)oxy)-2,6,8,10,12-pentamethyl-4,13-dioxabicyclo [8.2.1]tridec-1(12)-en-5-one | |
| 3 |  | 3 | Salidroside | 2-(4-Hydroxyphenyl)ethyl β-D-glucopyranoside | 10338-51-9 |
| 4 |  | 4 | Ajulemic acid | (6aR,10aR)-3-(1,1-Dimethylheptyl)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6-dimethyl-6H-dibenzo[b,d]pyran-9-carboxylic acid | 137945-48-3 |
| 5 |  | 5 | Tenofovir alafenamide fumarate | Isopropyl (2S)-2-[[[(1R)-2-(6-aminopurin-9-yl)-1-methyl-ethoxy]methyl-phenoxy-phosphoryl]amino]propanoate | 379270-37-8 |
| 6 |  | 6 | Tannic acid | 1,2,3,4,6-Penta-O-{3,4-dihydroxy-5-[(3,4,5-trihydroxybenzoyl)oxy]benzoyl}-D-glucopyranose | 1401-55-4 |
| 7 |  | 7 | Emodin | 1,3,8-Trihydroxy-6-methylanthracene-9,10-dione | 518-82-1 |
| 8 |  | 8 | Oridonin | 7a,20-Epoxy-1a,6b,7,14-tetrahydroxy-Kaur-16-en-15-one | 28957-04-2 |
| 9 |  | 9 | Necrostatin-1 | 5-(1H-Indol-3-ylmethyl)-3-methyl-2-thioxo-4-Imidazolidinone | 4311-88-0 |
| 10 |  | 10 | Ouabain | 1β,3β,5β,11α,14,19-Hexahydroxycard-20(22)-enolide 3-(6-deoxy-α-L-mannopyranoside) | 630-60-4 |
| 11 |  | 11 | Metformin | N,N-Dimethylimidodicarbonimidic diamide | 657-24-9 |
| 12 |  | 12 | Thymoquinone | 2-Methyl-5-(propan-2-yl)cyclohexa-2,5-diene-1,4-dione | 490-91-5 |
| 13 |  | 13 | Curcumin | (1E,6E)-1,7-Bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione | 458-37-7 |
| 14 |  | 14 | Polydatin | (2S,3R,4S,5S,6R)-2-{3-Hydroxy-5-[(E)-2-(4-hydroxyphenyl)ethen-1-yl]phenoxy}-6-(hydroxymethyl)oxane-3,4,5-triol | 27208-80-6 |
| 15 |  | 15 | Paeoniflorin | ((1aR,1a1S,2S,3aR,5R,5aR)-5-Hydroxy-2-methyl-1a-(((3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)tetrahydro-1H-3,4-dioxa-2,5-methanocyclobuta[cd]pentalen-1a1(3aH)-yl)methyl benzoate | 23180-57-6 |
| 16 |  | 16 | Mangiferin | 1,3,6,7-Tetrahydroxy-2-[(2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]-9H-xanthen-9-one | 4773-96-0 |
| 17 |  | 17 | Hydroxysafflor yellow A | (2S,E)-2,5-Dihydroxy-6-((E)-1-hydroxy-3-(4-hydroxyphenyl)allylidene)-2,4-bis((3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)cyclohex-4-ene-1,3-dione | 78281-02-4 |
| 18 |  | 18 | All-trans retinoic acid | (2E,4E,6E,8E)-3,7-Dimethyl-9-(2,6,6-trimethylcyclohex-1-en-1-yl)nona-2,4,6,8-tetraenoic acid | 302-79-4 |
| 19 |  | 19 | Andrographolide | 3-[2-[Decahydro-6-hydroxy-5-(hydroxymethyl)-5,8a-dimethyl-2-methylene-1-napthalenyl]ethylidene]dihydro-4-hydroxy-2(3H)-furanone | 5508-58-7 |
| 20 |  | 20 | Nagilactone D | (1aS,2S,2aS,2a1S,4aS,9bR,9cR)-6-Ethyl-2-hydroxy-2a,9b-dimethyl-1a,2,2a,2a1,4a,5,9b,9c-octahydro-3H,8H-oxireno [2’,3’:5,6]isobenzofuro [7,1-fg]isochromene-3,8-dione | 19891-53-3 |
| 21 |  | 21 | Baicalin | (2S,3S,4S,5R,6S)-6-[(5,6-Dihydroxy-4-oxo-2-phenyl-4H-1-benzopyran-7-yl)oxy]-3,4,5-trihydroxyoxane-2-carboxylic acid | 21967-41-9 |
| 22 |  | 22 | Gambogic acid | (2Z)-4-[(1R,3aS,5S,11R,14aS)-8-Hydroxy-3,3,11-trimethyl-13-(3-methylbut-2-en-1-yl)-11-(4-methylpent-3-en-1-yl)-7,15-dioxo-3a,4,5,7-tetrahydro-1H,3H,11H-1,5-methanofuro [3,4-g]pyrano [3,2-b]xanthen-1-yl]-2-methylbut-2-enoic acid | 2752-65-0 |
| 23 |  | 23 | Sulforaphane | 1-Isothiocyanato-4-(methanesulfinyl)butane | 4478-93-7 |
| 24 |  | 24 | Tanshinone IIA | 1,6,6-Trimethyl-6,7,8,9-tetrahydrophenanthro [1,2-b]furan-10,11-dione | 568-72-9 |
| 25 |  | 25 | Roxadustat | 2-[(4-Hydroxy-1-methyl-7-phenoxyisoquinoline-3-carbonyl)amino]acetic acid | 808118-40-3 |
| 26 |  | 26 | 1-(1-Propanoylpiperidin-4-yl)-3-[4-(trifluoromethoxy)phenyl]urea | 1-(1-Propionylpiperidin-4-yl)-3-(4-(trifluoromethoxy)phenyl)urea | 1222780-33-7 |
| 27 |  | 27 | Ponatinib | 3-(2-Imidazo [1,2-b]pyridazin-3-ylethynyl)-4-methyl-N-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]benzamide | 943319-70-8 |
| 28 |  | 28 | GSK3008348 | (S)-3-(3-(3,5-Dimethyl-1H-pyrazol-1-yl)phenyl)-4-((S)-3-(2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethyl)pyrrolidin-1-yl)butanoic acid | |
| 29 |  | 29 | Pyrrolidine dithiocarbamate | Pyrrolidine-1-carbodithioic acid | 25769-03-3 |
| 30 |  | 30 | Amitriptyline | 3-(10,11-Dihydro-5H-dibenzo[a,d]cycloheptene-5-ylidene)-N,N-dimethylpropan-1-amine | 50-48-6 |
| 31 |  | 31 | 12-(3-(Adamantan-1-yl)ureido)dodecanoic acid | 12-(3-(Adamantan-1-yl)ureido)dodecanoic acid | 479413-70-2 |
| 32 |  | 32 | Silymarin | (2R,3R)-3,5,7-Trihydroxy-2-[(2R*,3R*)-3-(4-hydroxy-3-methoxyphenyl)-2-(hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxin-6-yl]chroman-4-one | 1265089-69-7 |
| 33 |  | 33 | Melatonin | N-[2-(5-Methoxy-1H-indol-3-yl)ethyl]acetamide | 73-31-4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorababu, A.; Maraswami, M. Recent Advances (2015–2020) in Drug Discovery for Attenuation of Pulmonary Fibrosis and COPD. Molecules 2023, 28, 3674. https://doi.org/10.3390/molecules28093674
Dorababu A, Maraswami M. Recent Advances (2015–2020) in Drug Discovery for Attenuation of Pulmonary Fibrosis and COPD. Molecules. 2023; 28(9):3674. https://doi.org/10.3390/molecules28093674
Chicago/Turabian StyleDorababu, Atukuri, and Manikantha Maraswami. 2023. "Recent Advances (2015–2020) in Drug Discovery for Attenuation of Pulmonary Fibrosis and COPD" Molecules 28, no. 9: 3674. https://doi.org/10.3390/molecules28093674
APA StyleDorababu, A., & Maraswami, M. (2023). Recent Advances (2015–2020) in Drug Discovery for Attenuation of Pulmonary Fibrosis and COPD. Molecules, 28(9), 3674. https://doi.org/10.3390/molecules28093674