
Influence of the Solvent on the Stability of Aminopurine Tautomers and Properties of the Amino Group

Abstract
1. Introduction
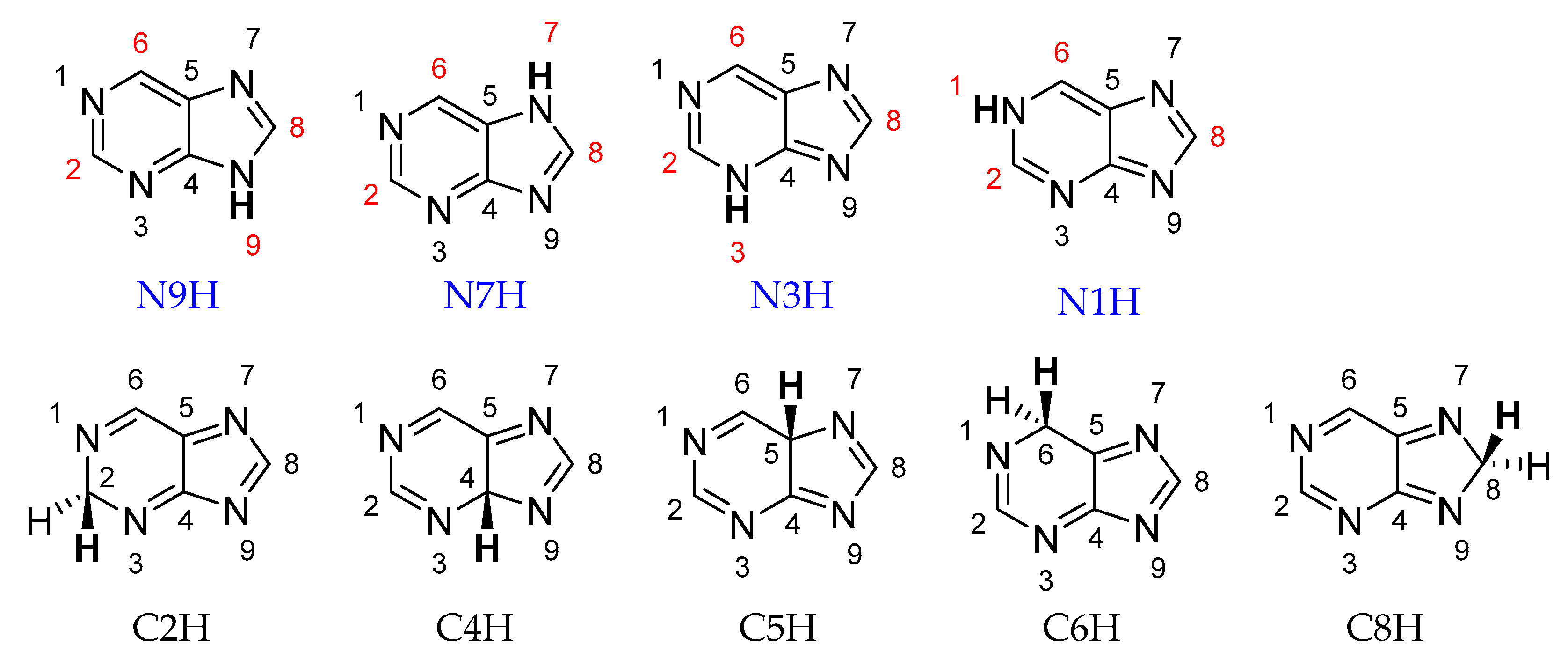
2. Results
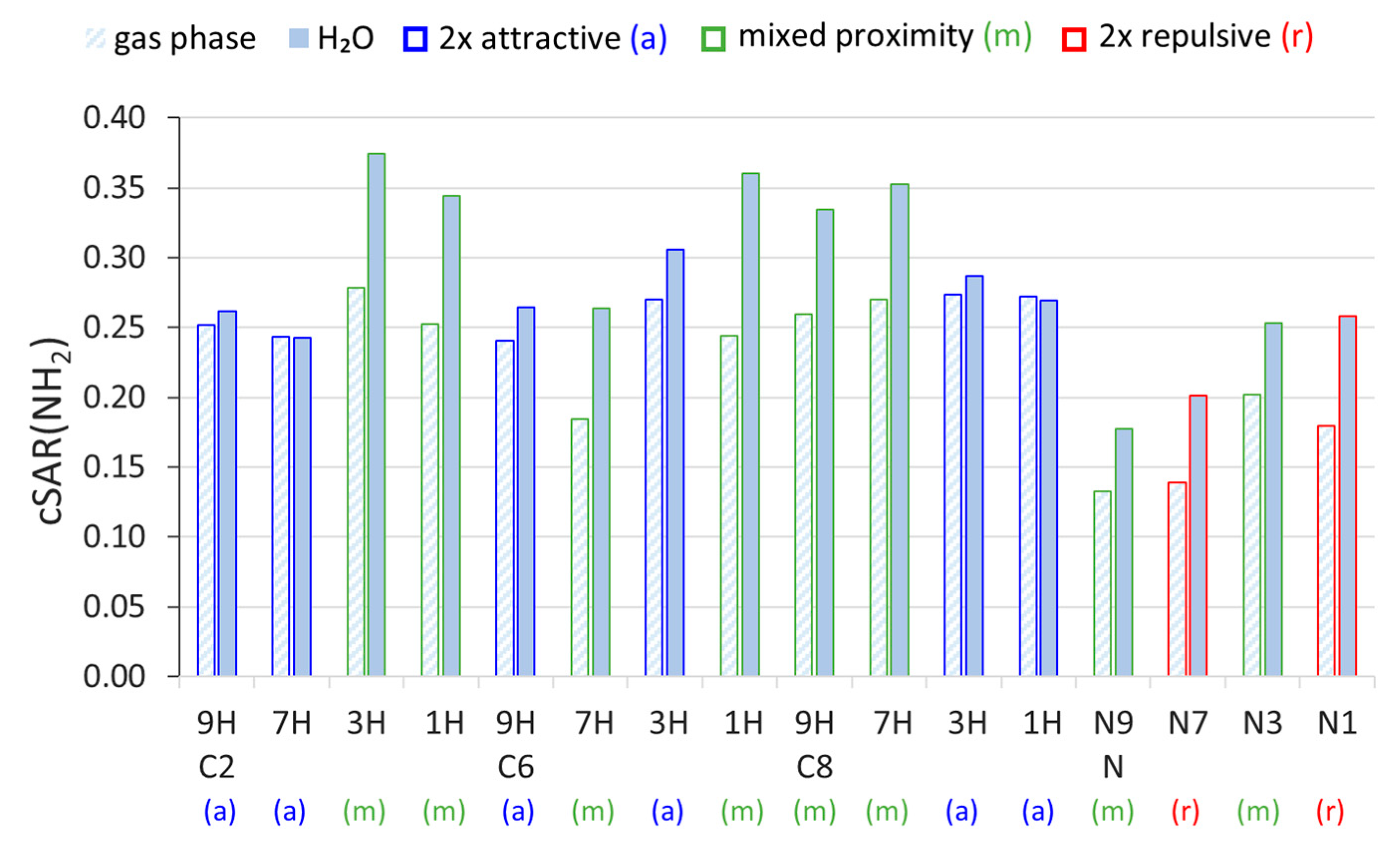
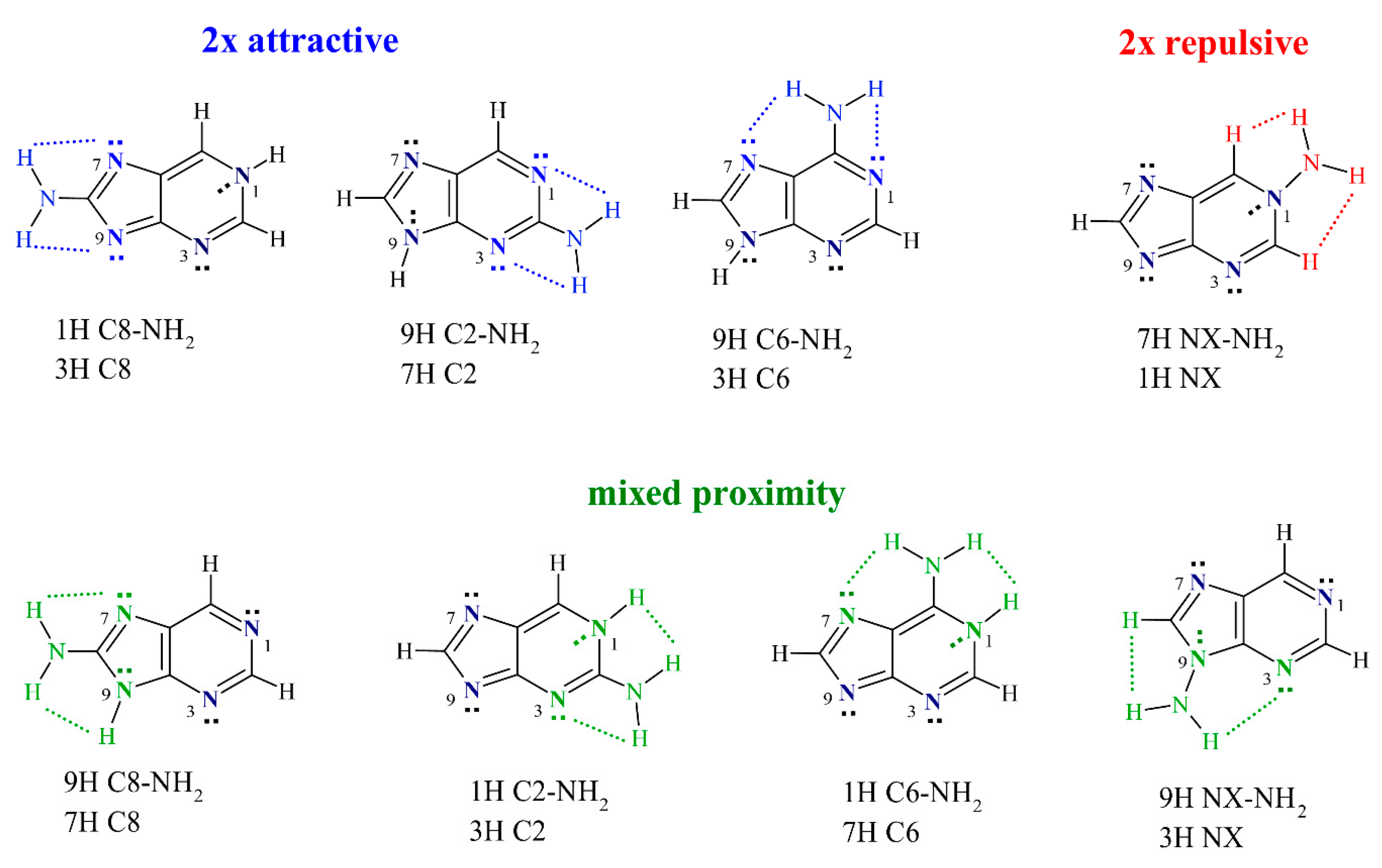
2.1. Electronic Properties of the Amino Group
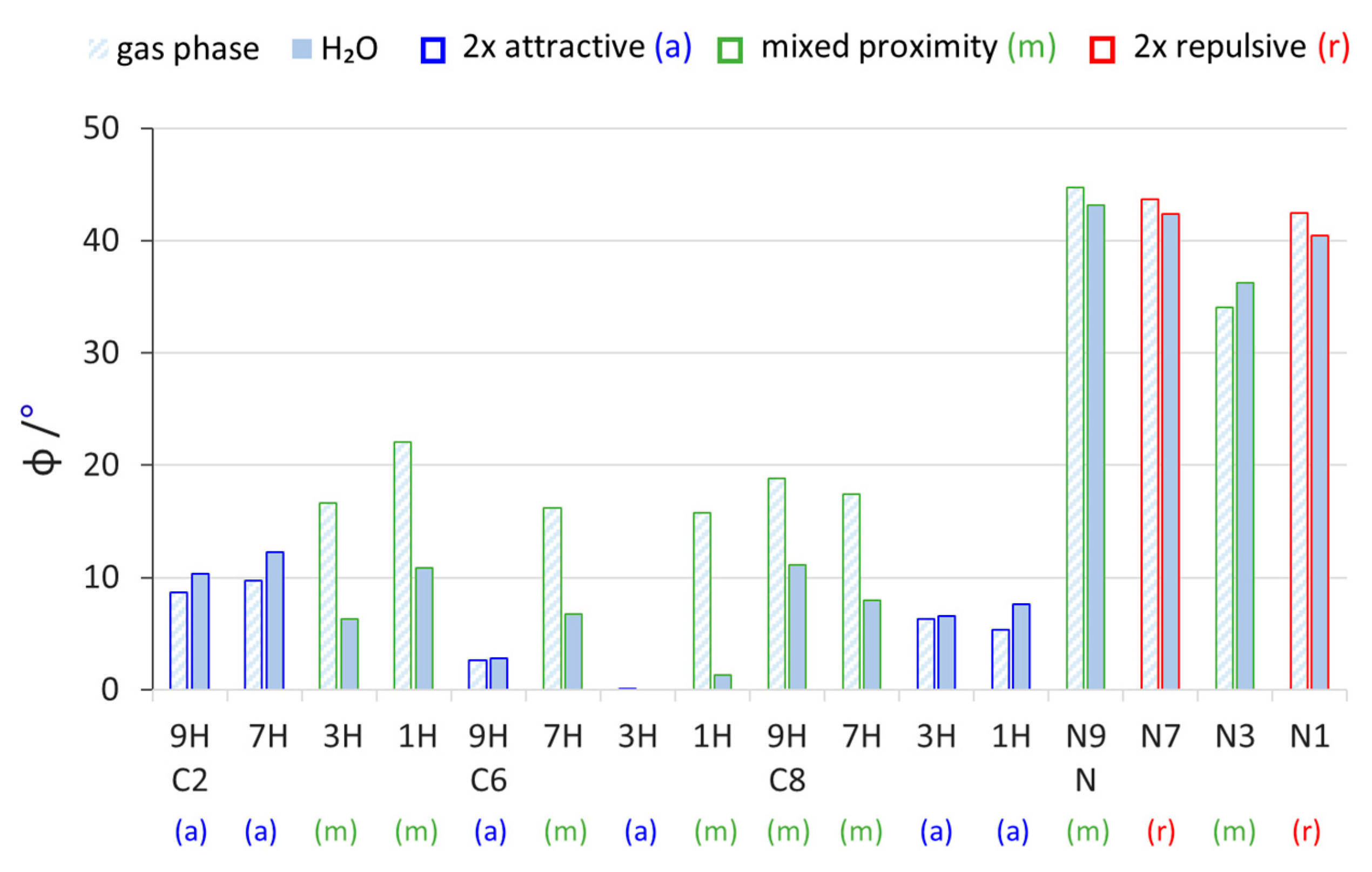
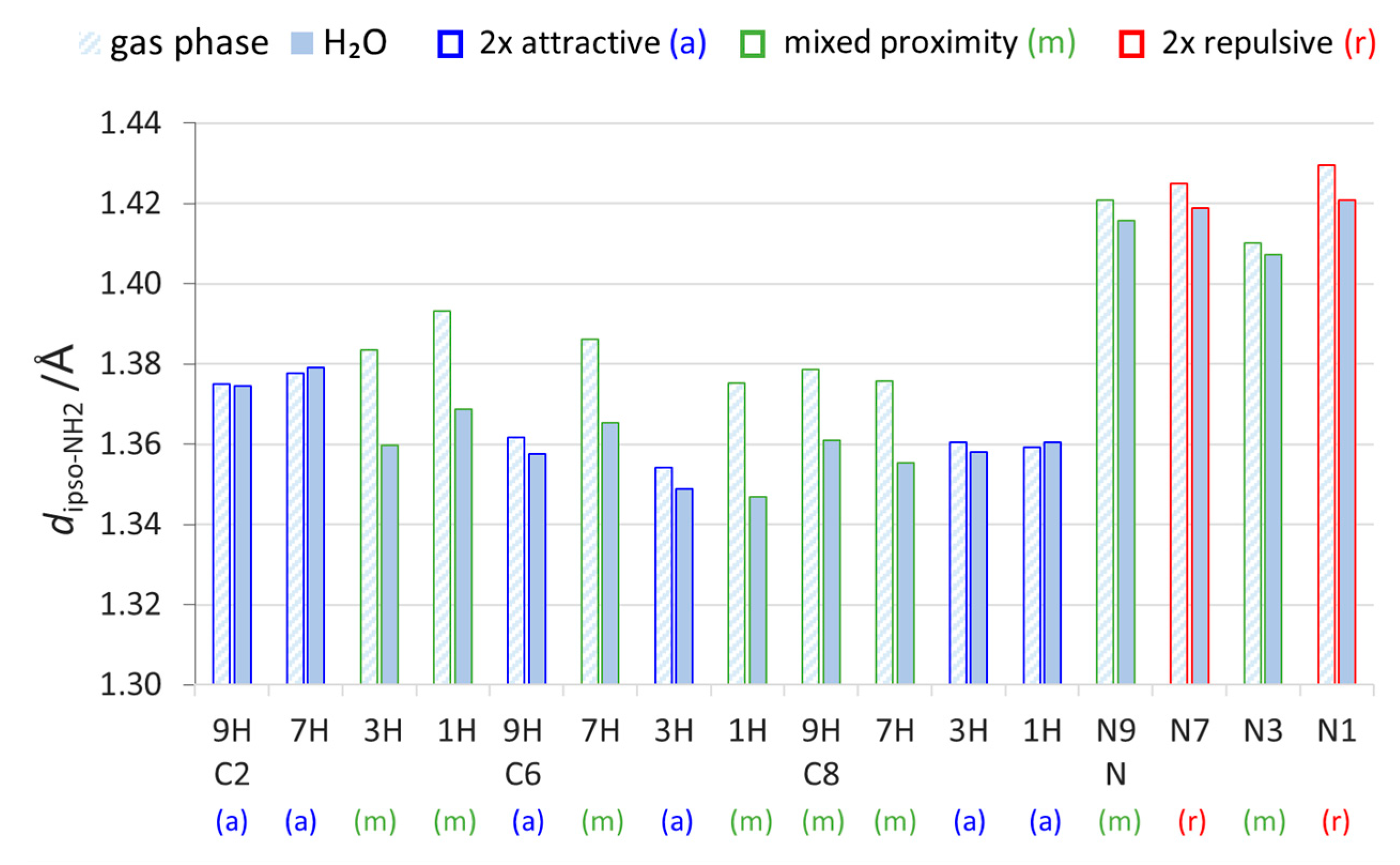
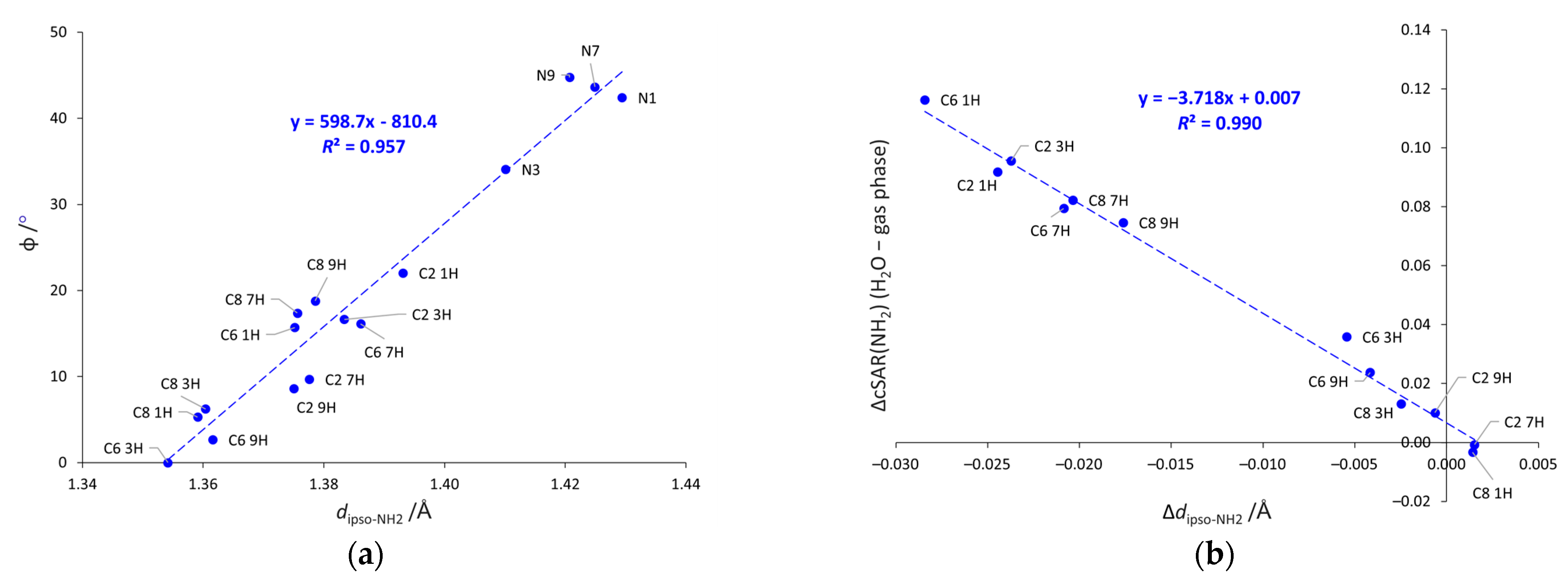
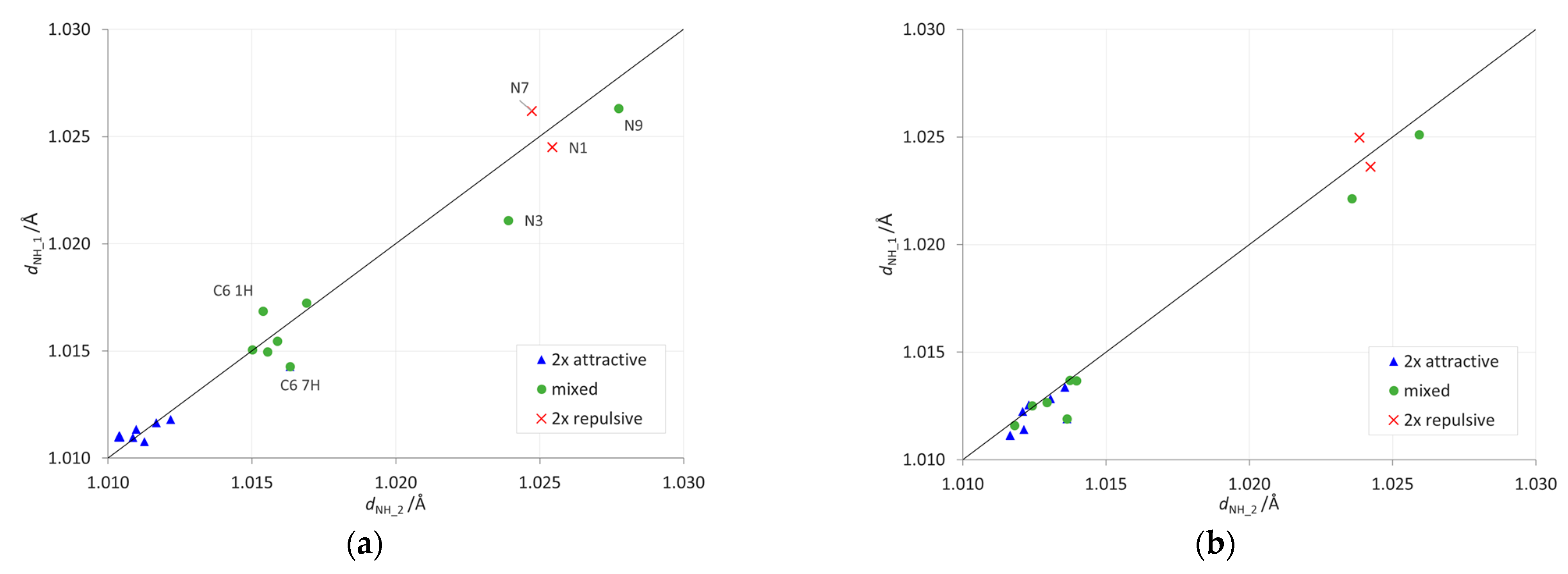
2.2. Geometric Parameters of the Amino Group
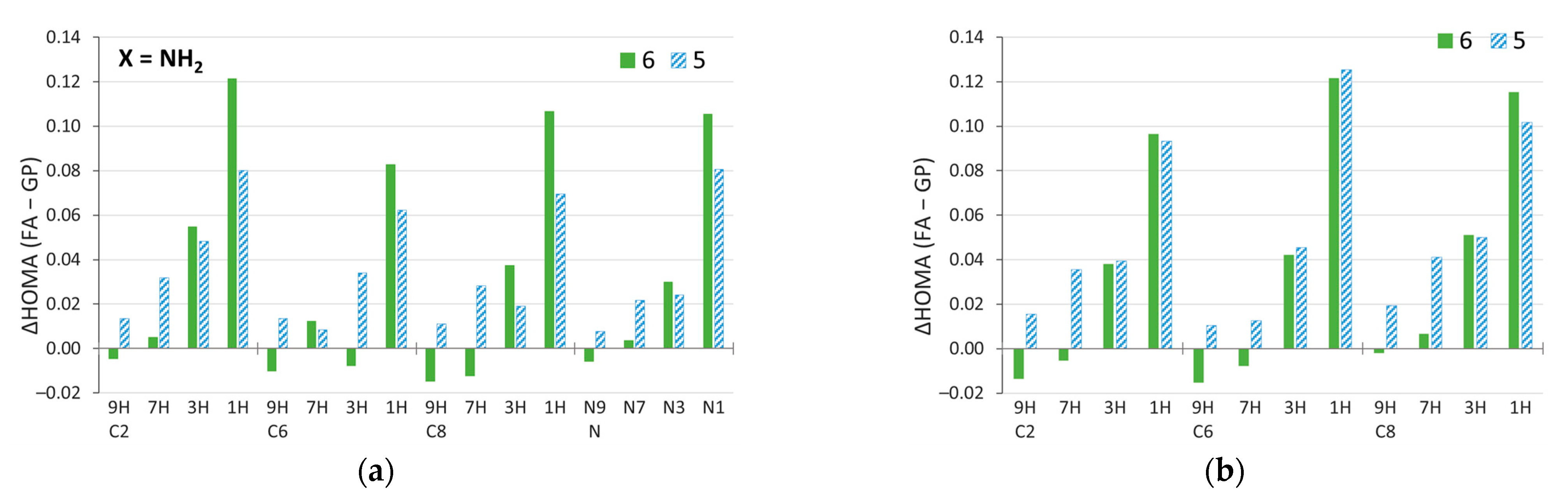
2.3. π-Electron Delocalization in Aminopurine Tautomers
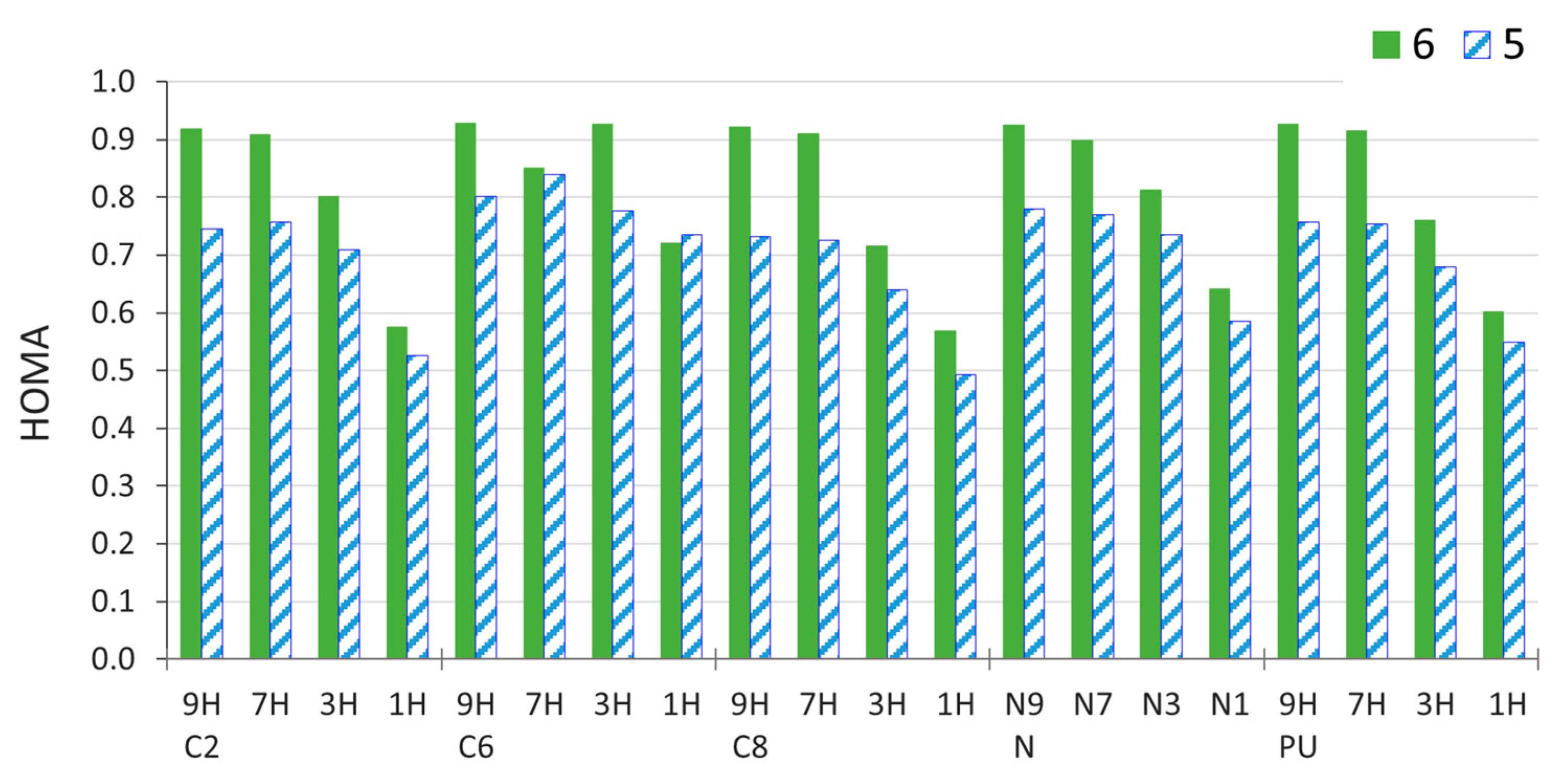
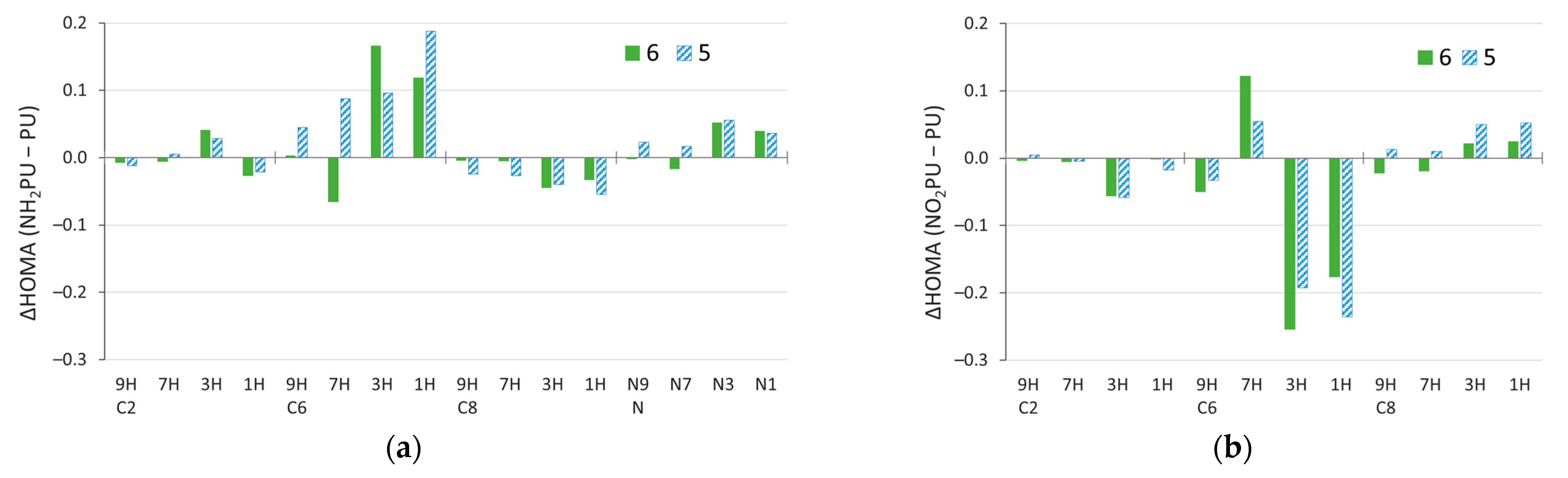
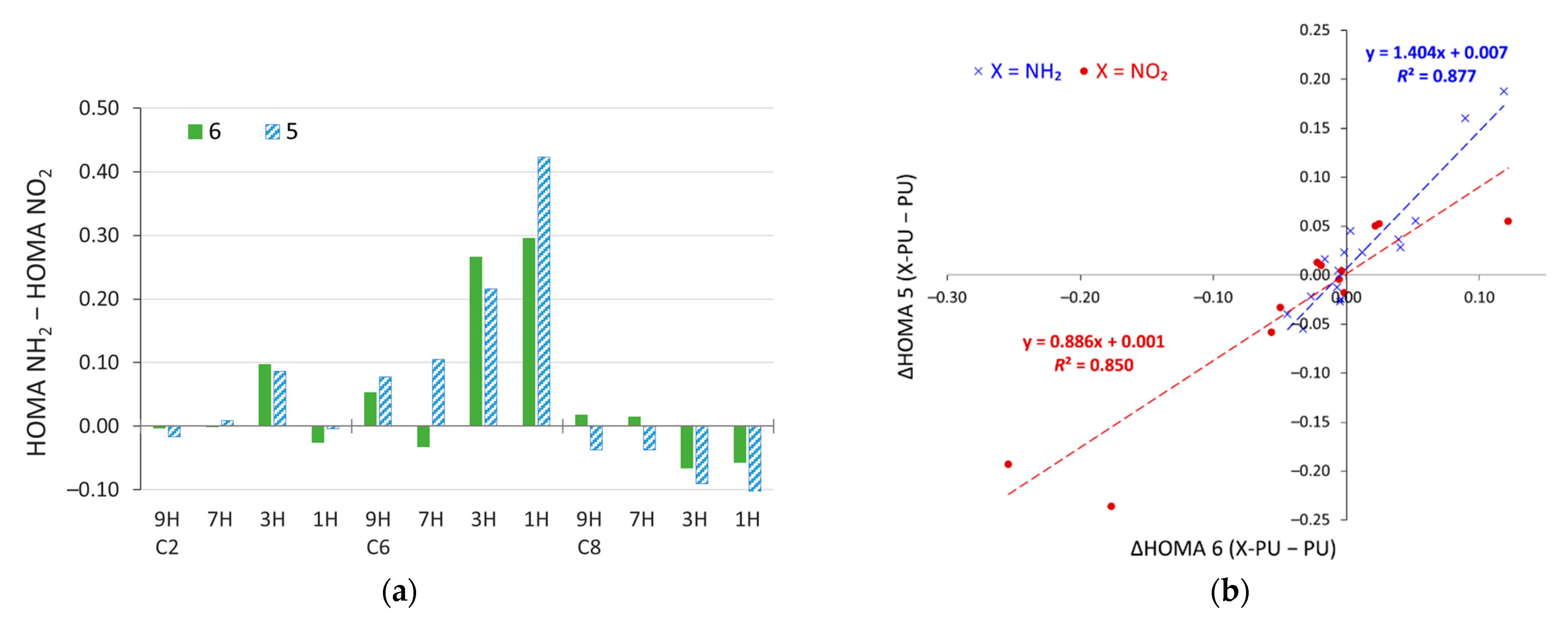
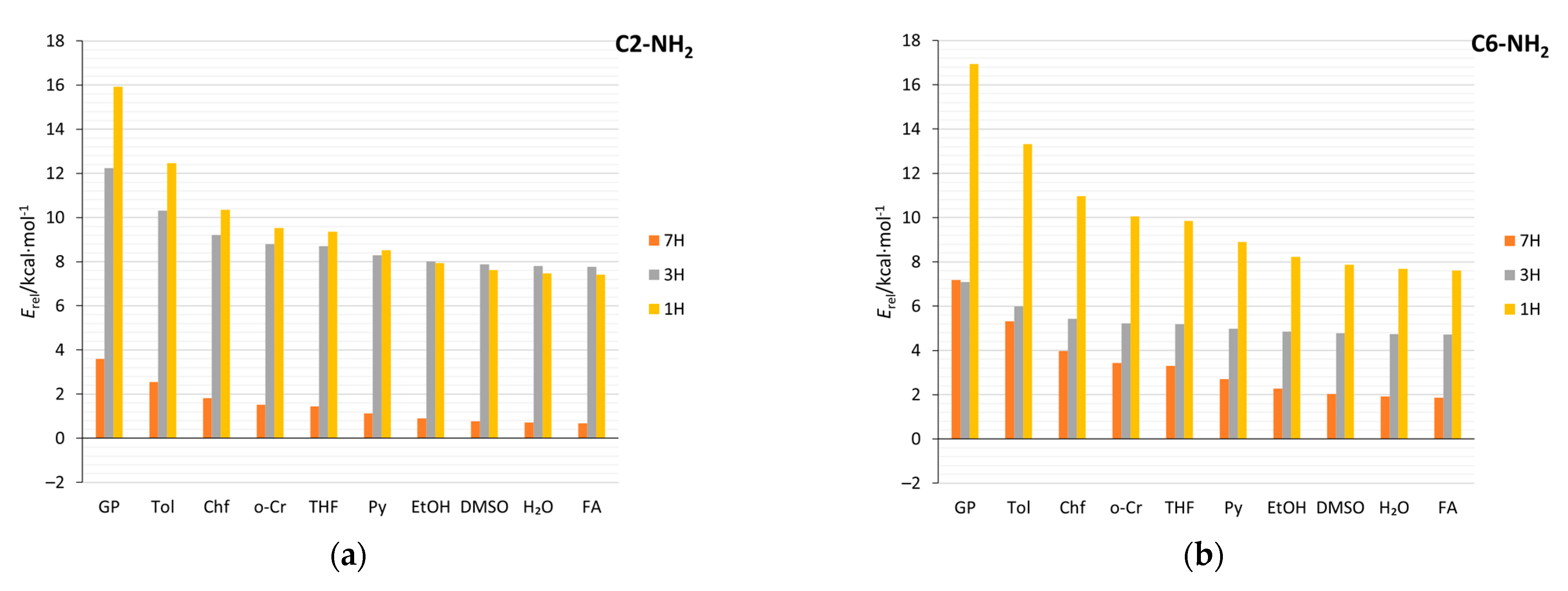
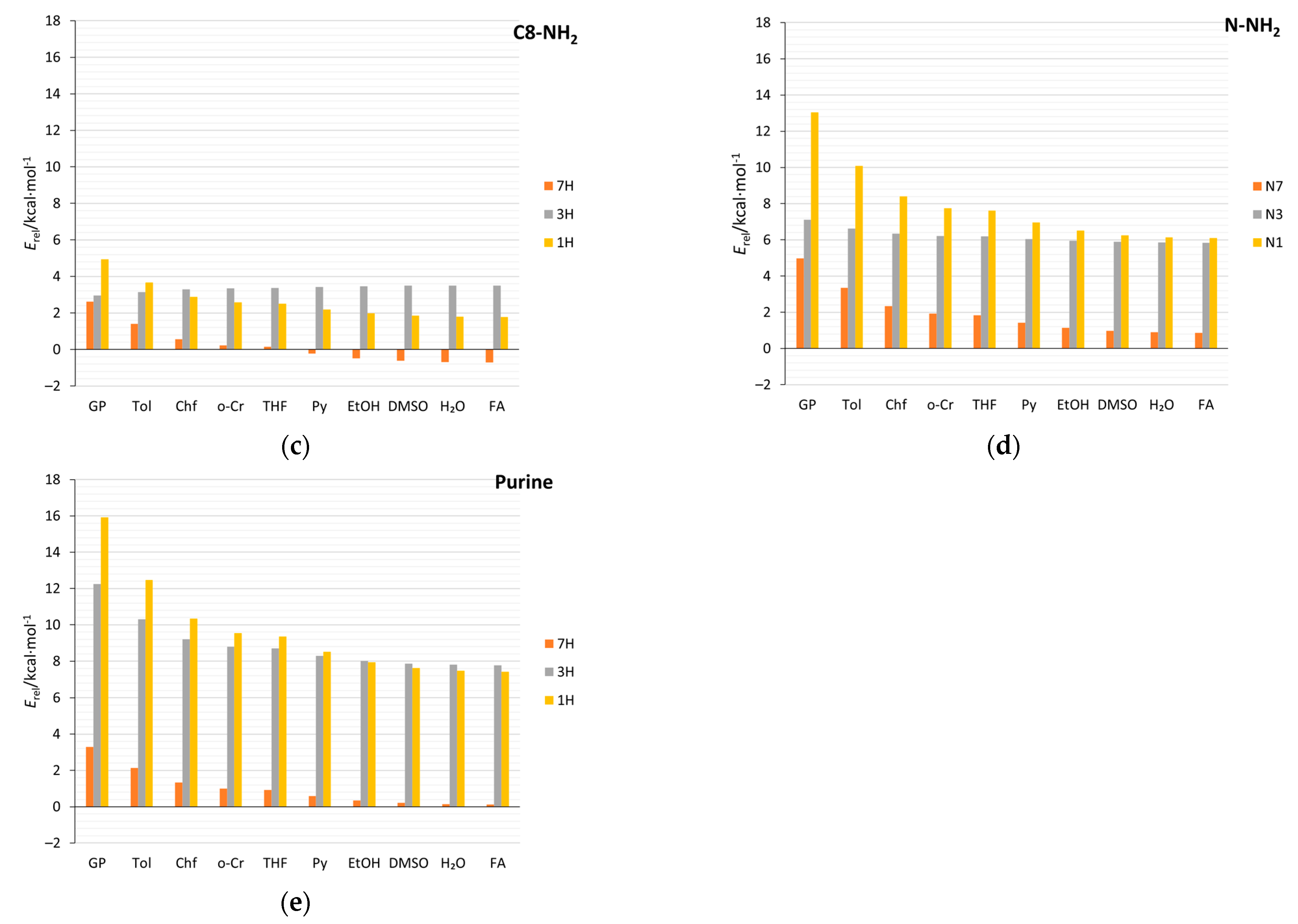
2.4. Stability of Aminopurine Tautomers in Various Solvents
3. Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rosemeyer, H. The Chemodiversity of Purine as a Constituent of Natural Products. Chem. Biodivers. 2004, 1, 361–401. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Legraverend, M.; Grierson, D.S. The purines: Potent and versatile small molecule inhibitors and modulators of key biological targets. Bioorganic Med. Chem. 2006, 14, 3987–4006. [Google Scholar] [CrossRef]
- Pozharskiĭ, A.F.; Soldatenkov, A.T.; Katritzky, A.R. Heterocycles in Life and Society: An Introduction to Heterocyclic Chemistry, Biochemistry, and Applications, 2nd ed.; Wiley: Chichester, UK, 2011. [Google Scholar]
- Raczyńska, E.D.; Kamińska, B. Variations of the tautomeric preferences and π-electron delocalization for the neutral and redox forms of purine when proceeding from the gas phase (DFT) to water (PCM). J. Mol. Model. 2013, 19, 3947–3960. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Kamińska, B. Prototropy and π-electron delocalization for purine and its radical ions—DFT studies. J. Phys. Org. Chem. 2010, 23, 828–835. [Google Scholar] [CrossRef]
- Peral, F.; Gallego, E. A study by ultraviolet spectroscopy on self-association of purine, 6-methylpurine, benzimidazole, and imidazo [1,2-a]pyridine in aqueous solution. Spectrochim. Acta 2000, 56, 747–759. [Google Scholar] [CrossRef]
- Gonnella, N.C.; Roberts, J.D. Studies of the tautomerism of purine and the protonation of purine, and its 7- and 9-methyl derivatives, by nitrogen-15 nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 1982, 104, 3162–3164. [Google Scholar] [CrossRef]
- Burova, T.G.; Ten, G.N.; Kucherova, V.V. Investigations of Tautomeric Purine Forms by the Methods of Vibrational Spectroscopy and Resonance Raman Scattering. II. Quantum-Mechanical Calculations of the Resonance Raman Scattering Spectra of Purine Tautomers. Russ. Phys. J. 2004, 47, 721–725. [Google Scholar] [CrossRef]
- Nowak, M.J.; Lapinski, L.; Kwiatkowski, J.S. An infrared matrix isolation study of tautomerism in purine and adenine. Chem. Phys. Lett. 1989, 157, 14–18. [Google Scholar] [CrossRef]
- Nowak, M.J.; Rostkowska, H.; Lapinski, L.; Kwiatkowski, J.S.; Leszczynski, J. Tautomerism N(9)H .dblharw. N(7)H of Purine, Adenine, and 2-Chloroadenine: Combined Experimental IR Matrix Isolation and Ab Initio Quantum Mechanical Studies. J. Phys. Chem. 1994, 98, 2813–2816. [Google Scholar] [CrossRef]
- Chenon, M.T.; Pugmire, R.J.; Grant, D.M.; Panzica, R.P.; Townsend, L.B. Carbon-13 magnetic resonance. XXVI. Quantitative determination of the tautomeric populations of certain purines. J. Am. Chem. Soc. 1975, 97, 4636–4642. [Google Scholar] [CrossRef] [PubMed]
- Sečkářová, P.; Marek, R.; Maliňáková, K.; Kolehmainen, E.; Hocková, D.; Hocek, M.; Sklenář, V. Direct determination of tautomerism in purine derivatives by low-temperature NMR spectroscopy. Tetrahedron Lett. 2004, 45, 6259–6263. [Google Scholar] [CrossRef]
- Lippert, B.; Gupta, D. Promotion of rare nucleobase tautomers by metal binding. Dalton Trans. 2009, 4619–4634. [Google Scholar] [CrossRef] [PubMed]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C.; Kamińska, B.; Igielska, M.; Kurpiewski, J.; Juras, W. Purine tautomeric preferences and bond-length alternation in relation with protonation-deprotonation and alkali metal cationization. J. Mol. Model. 2020, 26, 93. [Google Scholar] [CrossRef]
- Ouellette, R.J.; Rawn, J.D. Nucleosides, Nucleotides, and Nucleic Acids. In Organic Chemistry: Structure, Mechanism, Syn-Thesis; Elsevier: San Diego, CA, USA, 2018; pp. 973–1000. ISBN 978-0-12-812838-1. [Google Scholar]
- Yang, B.; Srinivasan, B. Recent Developments in the Synthesis of Substituted Purine Nucleosides and Nucleotides. Curr. Org. Chem. 2014, 18, 2072–2107. [Google Scholar] [CrossRef]
- Ratsep, P.C.; Robins, R.K.; Vaghefi, M.M. Introduction of Fluorine Into the C8 Position of Purine Nucleosides. Nucleosides Nucleotides 1990, 9, 197–204. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 2002, 3, 415–424. [Google Scholar] [CrossRef]
- Walters, W.P.; Murcko, M.A. Library Filtering Systems and Prediction of Drug-Like Properties. In Methods and Principles in Medicinal Chemistry; Böhm, H., Schneider, G., Eds.; Wiley: New York, NY, USA, 2000; pp. 15–32. ISBN 978-3-527-30153-9. [Google Scholar]
- Naumann, T.; Matter, H. Structural Classification of Protein Kinases Using 3D Molecular Interaction Field Analysis of Their Ligand Binding Sites: Target Family Landscapes. J. Med. Chem. 2002, 45, 2366–2378. [Google Scholar] [CrossRef]
- Schulze-Gahmen, U.; Brandsen, J.; Jones, H.D.; Morgan, D.; Meijer, L.; Vesely, J.; Kim, S.-H. Multiple modes of ligand recognition: Crystal structures of cyclin-dependent protein kinase 2 in complex with ATP and two inhibitors, olomoucine and isopentenyladenine. Proteins: Struct. Funct. Genet. 1995, 22, 378–391. [Google Scholar] [CrossRef]
- Donohue, J.; Trueblood, K.N. Base pairing in deoxyribonucleic acid. J. Mol. Biol. 1960, 2, 363–371. [Google Scholar] [CrossRef]
- Freese, E. The specific mutagenic effect of base analogues on Phage T4. J. Mol. Biol. 1959, 1, 87–105. [Google Scholar] [CrossRef]
- Drake, J.W. The Molecular Basis of Mutation; Holden-Day: San Francisco, CA, USA, 1970; ISBN 978-0-816-22450-0. [Google Scholar]
- Ludwig, V.; Amaral, M.S.D.; da Costa, Z.M.; Borin, A.C.; Canuto, S.; Serrano-Andrés, L. 2-Aminopurine non-radiative decay and emission in aqueous solution: A theoretical study. Chem. Phys. Lett. 2008, 463, 201–205. [Google Scholar] [CrossRef]
- Glickman, B.W. 2-Aminopurine Mutagenesis in Escherichia coli. In Genetic Consequences of Nucleotide Pool Imbalance, Basic Life Sciences; de Serres, F.J., Ed.; Springer: Boston, MA, USA, 1985; Volume 31, pp. 353–379. ISBN 978-1-4612-9488-7. [Google Scholar]
- Ronen, A. 2-Aminopurine. Mutat. Res./Rev. Mutat. Res. 1980, 75, 1–47. [Google Scholar] [CrossRef]
- de Serres, F.J.; Brockman, H.E.; Hung, C.Y.; Overton, L.K. Mutagenicity of 2-aminopurine, 6-N-hydroxylaminopurine, and 2-amino-N6-hydroxyadenine in Neurospora crassa. In Genetic Consequences of Nucleotide Pool Imbalance, Basic Life Sciences; de Serres, F.J., Ed.; Springer: Boston, MA, USA, 1985; Volume 31, pp. 381–389. ISBN 978-1-4612-9488-7. [Google Scholar]
- Laufer, S.A.; Domeyer, D.M.; Scior, T.R.F.; Albrecht, W.; Hauser, D.R.J. Synthesis and Biological Testing of Purine Derivatives as Potential ATP-Competitive Kinase Inhibitors. J. Med. Chem. 2005, 48, 710–722. [Google Scholar] [CrossRef]
- Baker, B.; Hendrickson, J. Irreversible enzyme inhibitors XCII. Inhibition of xanthine oxidase by some purines and pyr midines. J. Pharm. Sci. 1967, 56, 955–959. [Google Scholar] [CrossRef]
- Vanotti, E.; Bani, M.; Favara, D.; Gobetti, M.; Lombroso, M.; Magnetti, S.; Olgiati, V.; Palladino, M.; Tonon, G. 8-Substituted purine derivatives: A new class of lipid-lowering agents. Eur. J. Med. Chem. 1994, 29, 287–294. [Google Scholar] [CrossRef]
- Huryn, D.M.; Okabe, M. AIDS-driven nucleoside chemistry. Chem. Rev. 1992, 92, 1745–1768. [Google Scholar] [CrossRef]
- Seela, F.; Winter, H. N7-DNA: Synthesis and Base Pairing of Oligonucleotides containing 7-(2-Deoxy-D-erythro-pentofuranosyl)adenine (N7Ad). Helv. Chim. Acta 1994, 77, 597–607. [Google Scholar] [CrossRef]
- Boryski, J. Transglycosylation Reactions of Purine Nucleosides. A Review. Nucleosides Nucleotides 1996, 15, 771–791. [Google Scholar] [CrossRef]
- Marek, R.; Brus, J.; Toušek, J.; Kovács, L.; Hocková, D. N7- and N9-substituted purine derivatives: A 15n NMR study. Org. Magn. Reson. 2002, 40, 353–360. [Google Scholar] [CrossRef]
- Arico, J.W. 3-Substituted Purines: Methodology, Synthesis, and Studies of DNA Hydration in the Minor Groove. Ph.D. Thesis, Department of Chemistry, The Graduate School of Arts and Sciences, Boston College, Boston, MA, USA, 2010. Available online: http://hdl.handle.net/2345/1824 (accessed on 20 March 2023).
- Raczyńska, E.D.; Makowski, M. Geometric consequences of electron delocalization for adenine tautomers in aqueous solution. J. Mol. Model. 2014, 20, 2234. [Google Scholar] [CrossRef] [PubMed]
- Mburu, E.; Matsika, S. An Ab Initio Study of Substituent Effects on the Excited States of Purine Derivatives. J. Phys. Chem. A 2008, 112, 12485–12491. [Google Scholar] [CrossRef] [PubMed]
- Palamarchuk, G.V.; Shishkin, O.V.; Gorb, L.; Leszczynski, J. Nucleic Acid Bases in Anionic 2′-Deoxyribonucleotides: A DFT/B3LYP Study of Structures, Relative Stability, and Proton Affinities. J. Phys. Chem. B 2013, 117, 2841–2849. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Miertuš, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Hammett, L.P. Physical Organic Chemistry, Table II; McGraw–Hill: New York, NY, USA, 1940; p. 189. [Google Scholar]
- Hammett, L.P. The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J. Am. Chem. Soc. 1937, 59, 96–103. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Exner, O. Theory of Substituent Effects: Recent Advances. Curr. Org. Chem. 2006, 10, 763–778. [Google Scholar] [CrossRef]
- Galabov, B.; Ilieva, S.; Schaefer, I.H.F. An Efficient Computational Approach for the Evaluation of Substituent Constants. J. Org. Chem. 2006, 71, 6382–6387. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Zachara, J.E.; Szatylowicz, H. Molecular Geometry as a Source of Chemical Information. 3. How H-Bonding Affects Aromaticity of the Ring in the Case of Phenol and p-Nitrophenol Complexes: A B3LYP/6-311+G** Study. J. Org. Chem. 2004, 69, 7038–7043. [Google Scholar] [CrossRef] [PubMed]
- Sadlej-Sosnowska, N. On the way to physical interpretation of Hammett constants: How substituent active space impacts on acidity and electron distribution in p-substituted benzoic acid molecules. Polish J. Chem. 2007, 81, 1123–1134. [Google Scholar]
- Sadlej-Sosnowska, N. Substituent active region—A gate for communication of substituent charge with the rest of a molecule: Monosubstituted benzenes. Chem. Phys. Lett. 2007, 447, 192–196. [Google Scholar] [CrossRef]
- Stasyuk, O.A.; Szatylowicz, H.; Guerra, C.F.; Krygowski, T.M. Theoretical study of electron-attracting ability of the nitro group: Classical and reverse substituent effects. Struct. Chem. 2015, 26, 905–913. [Google Scholar] [CrossRef]
- Szatylowicz, H.; Siodla, T.; Stasyuk, O.A.; Krygowski, T.M. Towards physical interpretation of substituent effects: The case of meta- and para-substituted anilines. Phys. Chem. Chem. Phys. 2015, 18, 11711–11721. [Google Scholar] [CrossRef] [PubMed]
- Szatyłowicz, H.; Krygowski, T.M.; Hobza, P. How the Shape of the NH2 Group Depends on the Substituent Effect and H-Bond Formation in Derivatives of Aniline. J. Phys. Chem. A 2006, 111, 170–175. [Google Scholar] [CrossRef]
- Szatylowicz, H.; Jezuita, A.; Siodła, T.; Varaksin, K.S.; Ejsmont, K.; Madura, I.D.; Krygowski, T.M. Dependence of the Substituent Effect on Solvent Properties. J. Phys. Chem. A 2018, 122, 1896–1904. [Google Scholar] [CrossRef]
- Iii, G.L.D.; Je, L.; Tanski, J.M. Crystallographic and spectroscopic characterization of 4-nitro-2-(trifluoromethyl)benzoic acid and 4-nitro-3-(trifluoromethyl)benzoic acid. Acta Crystallogr. Sect. E Crystallogr. Commun. 2019, 75, 524–528. [Google Scholar] [CrossRef]
- Hanson, J.R.; Hitchcock, P.B.; Toche, F. The Rotation of the Nitro and Formyl Groups Relative to the Aromatic Ring in Some ortho-nitroarylaldehydes. J. Chem. Res. 2008, 2008, 476–478. [Google Scholar] [CrossRef]
- Dobrowolski, M.A.; Krygowski, T.M.; Cyrański, M.K. Substituent Constants (σp−) of the Rotated Nitro Group. The In-terplay Between the Substituent Effect of a Rotated−NO2 Group and H-Bonds Affecting π-Electron Delocalization in 4-Nitrophenol and 4-Nitrophenolate Complexes: A B3LYP/6-311+ G** Study. Croat. Chem. Acta 2009, 82, 139–147. [Google Scholar]
- Jezuita, A.; Wieczorkiewicz, P.; Szatylowicz, H.; Krygowski, T. Solvent Effect on the Stability and Reverse Substituent Effect in Nitropurine Tautomers. Symmetry 2021, 13, 1223. [Google Scholar] [CrossRef]
- Jezuita, A.; Wieczorkiewicz, P.A.; Szatylowicz, H.; Krygowski, T.M. Effect of the Solvent and Substituent on Tautomeric Preferences of Amine-Adenine Tautomers. ACS Omega 2021, 6, 18890–18903. [Google Scholar] [CrossRef] [PubMed]
- Wieczorkiewicz, P.A. Relationships between the Substituent and Solvent Effects in Selected Pyrimidine and Purine Derivatives. Master’s Thesis, Faculty of Chemistry, Warsaw University of Technology,, Warsaw, Poland, 2022. Available online: https://repo.pw.edu.pl/info/master/WUTbdff88096cd14b41a128a1d9253761b7/ (accessed on 20 March 2023).
- Desiraju, G.R. A Bond by Any Other Name. Angew. Chem. Int. Ed. 2010, 50, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Raczyńska, E.D.; Hallman, M.; Kolczynska, K.; Stępniewski, T.M. On the Harmonic Oscillator Model of Electron Delocalization (HOMED) Index and its Application to Heteroatomic π-Electron Systems. Symmetry 2010, 2, 1485–1509. [Google Scholar] [CrossRef]
- Szczȩśniak, M.; Nowak, M.; Szczepaniak, K. Infrared matrix isolation studies on tautomerism of cytosine and isocytosine methyl-derivatives. J. Mol. Struct. 1984, 115, 221–224. [Google Scholar] [CrossRef]
- Jezuita, A.; Szatylowicz, H.; Marek, P.H.; Krygowski, T.M. Aromaticity of the most stable adenine and purine tautomers in terms of Hückel’s 4n+2 principle. Tetrahedron 2019, 75, 130474. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Grimme, S.; Antony, J.; Schwabe, T.; Mück-Lichtenfeld, C. Density functional theory with dispersion corrections for supramolecular structures, aggregates, and complexes of (bio)organic molecules. Org. Biomol. Chem. 2007, 5, 741–758. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Marek, P.H.; Szatylowicz, H.; Krygowski, T.M. Stacking of nucleic acid bases: Optimization of the computational approach—the case of adenine dimers. Struct. Chem. 2018, 30, 351–359. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Kruszewski, J.; Krygowski, T. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 1972, 13, 3839–3842. [Google Scholar] [CrossRef]
- Krygowski, T.M. Crystallographic studies of inter- and intramolecular interactions reflected in aromatic character of .pi.-electron systems. J. Chem. Inf. Comput. Sci. 1993, 33, 70–78. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
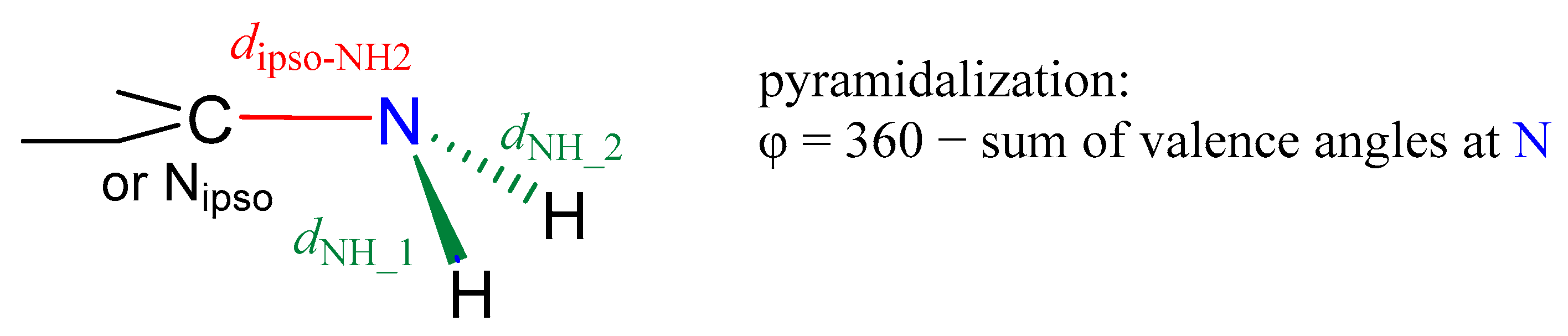{kind=link}
| Solvent/Medium | Acronym | ε | 1/ε |
|---|---|---|---|
| Formamide | FA | 108.94 | 0.0092 |
| Water | H2O | 78.36 | 0.0128 |
| Dimethyl sulfoxide | DMSO | 46.83 | 0.0214 |
| Ethanol | EtOH | 24.85 | 0.0402 |
| Pyridine | Py | 12.98 | 0.0771 |
| Tetrahydrofuran | THF | 7.43 | 0.1347 |
| o-cresol | o-Cr | 6.76 | 0.1479 |
| Chloroform | Chf | 4.71 | 0.2123 |
| Toluene | Tol | 2.37 | 0.4212 |
| Gas phase | GP | 1.00 | 1.00 |
| ΔcSAR(NH2) | a | R2 | Proximity Type | ||
|---|---|---|---|---|---|
| C2 | 9H | 0.0102 | −0.0104 | 0.9964 | 2× att |
| 7H | −0.0007 | 0.0008 | 0.0585 | 2× att | |
| 3H | 0.0956 | −0.0979 | 0.9744 | mix | |
| 1H | 0.0919 | −0.0947 | 0.9691 | mix | |
| C6 | 9H | 0.0238 | −0.0244 | 0.9989 | 2× att |
| 7H | 0.0795 | −0.0818 | 0.9415 | mix | |
| 3H | 0.0359 | −0.0367 | 0.9877 | 2× att | |
| 1H | 0.1163 | −0.1189 | 0.9668 | mix | |
| C8 | 9H | 0.0747 | −0.0766 | 0.9703 | mix |
| 7H | 0.0823 | −0.0846 | 0.9692 | mix | |
| 3H | 0.0132 | −0.0134 | 0.9947 | 2× att | |
| 1H | −0.0032 | 0.0035 | 0.3025 | 2× att | |
| N | N9 | 0.0449 | −0.0460 | 0.9780 | mix |
| N7 | 0.0623 | −0.0638 | 0.9725 | 2× rep | |
| N3 | 0.0510 | −0.0523 | 0.9795 | mix | |
| N1 | 0.0789 | −0.0808 | 0.9121 | 2× rep |
| X = NH2 | X = NO2 * | ||||||
|---|---|---|---|---|---|---|---|
| GP | H2O | ∆ | GP | H2O | ∆ | ||
| C2 | 9H | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 7H | 3.59 | 0.71 | −2.88 | 3.33 | −0.31 | −3.64 | |
| 3H | 12.25 | 7.80 | −4.45 | 8.17 | 11.60 | 3.43 | |
| 1H | 15.92 | 7.47 | −8.45 | 9.69 | 9.36 | −0.33 | |
| C6 | 9H | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 7H | 7.18 | 1.91 | −5.27 | −4.06 | −2.12 | 1.94 | |
| 3H | 7.09 | 4.74 | −2.35 | 10.16 | 7.99 | −2.17 | |
| 1H | 16.94 | 7.69 | −9.25 | 10.34 | 9.81 | −0.53 | |
| C8 | 9H | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 7H | 2.62 | −0.69 | −3.31 | 3.67 | 0.90 | −2.77 | |
| 3H | 2.95 | 3.50 | 0.55 | 8.96 | 2.10 | −6.86 | |
| 1H | 4.93 | 1.80 | −3.13 | 11.55 | −0.08 | −11.63 | |
| N | N9 | 0.00 | 0.00 | 0.00 | |||
| N7 | 4.98 | 0.90 | −4.07 | ||||
| N3 | 7.11 | 5.86 | −1.25 | ||||
| N1 | 13.03 | 6.14 | −6.89 | ||||
| PU | 9H | 0.00 ** | 0.00 | 0.00 | |||
| 7H | 3.29 ** | 0.15 | −3.14 | ||||
| 3H | 8.69 ** | 6.70 | −1.99 | ||||
| 1H | 11.50 ** | 4.98 | −6.52 | ||||
| a | R2 | μ/D | Δμ/D | Proximity Type | ||
|---|---|---|---|---|---|---|
| 9H | C2 | 8.905 | 0.979 | 3.2 | 1.3 | 2× att |
| C6 | 8.949 | 0.981 | 2.4 | 1.0 | 2× att | |
| C8 | 11.211 | 0.976 | 5.4 | 2.8 | mix | |
| N9 | 7.585 | 0.982 | 3.1 | 1.2 | mix | |
| 7H | C2 | 11.869 | 0.970 | 4.1 | 2.0 | 2× att |
| C6 | 14.364 | 0.964 | 6.8 | 3.6 | mix | |
| C8 | 14.611 | 0.968 | 6.9 | 3.6 | mix | |
| N7 | 11.765 | 0.973 | 6.2 | 2.4 | 2× rep | |
| 3H | C2 | 13.466 | 0.976 | 6.0 | 3.3 | mix |
| C6 | 11.353 | 0.981 | 3.9 | 1.8 | 2× att | |
| C8 | 10.645 | 0.978 | 3.0 | 1.3 | 2× att | |
| N3 | 8.855 | 0.977 | 3.1 | 2.4 | mix | |
| 1H | C2 | 17.581 | 0.969 | 7.7 | 4.5 | mix |
| C6 | 18.453 | 0.967 | 8.2 | 4.8 | mix | |
| C8 | 14.432 | 0.972 | 4.1 | 2.4 | 2× att | |
| N1 | 14.650 | 0.974 | 7.0 | 3.1 | 2× rep |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jezuita, A.; Wieczorkiewicz, P.A.; Krygowski, T.M.; Szatylowicz, H. Influence of the Solvent on the Stability of Aminopurine Tautomers and Properties of the Amino Group. Molecules 2023, 28, 2993. https://doi.org/10.3390/molecules28072993
Jezuita A, Wieczorkiewicz PA, Krygowski TM, Szatylowicz H. Influence of the Solvent on the Stability of Aminopurine Tautomers and Properties of the Amino Group. Molecules. 2023; 28(7):2993. https://doi.org/10.3390/molecules28072993
Chicago/Turabian StyleJezuita, Anna, Paweł A. Wieczorkiewicz, Tadeusz M. Krygowski, and Halina Szatylowicz. 2023. "Influence of the Solvent on the Stability of Aminopurine Tautomers and Properties of the Amino Group" Molecules 28, no. 7: 2993. https://doi.org/10.3390/molecules28072993
APA StyleJezuita, A., Wieczorkiewicz, P. A., Krygowski, T. M., & Szatylowicz, H. (2023). Influence of the Solvent on the Stability of Aminopurine Tautomers and Properties of the Amino Group. Molecules, 28(7), 2993. https://doi.org/10.3390/molecules28072993