
In Silico Computational Studies of Bioactive Secondary Metabolites from Wedelia trilobata against Anti-Apoptotic B-Cell Lymphoma-2 (Bcl-2) Protein Associated with Cancer Cell Survival and Resistance

 , , , , ,
, , , , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
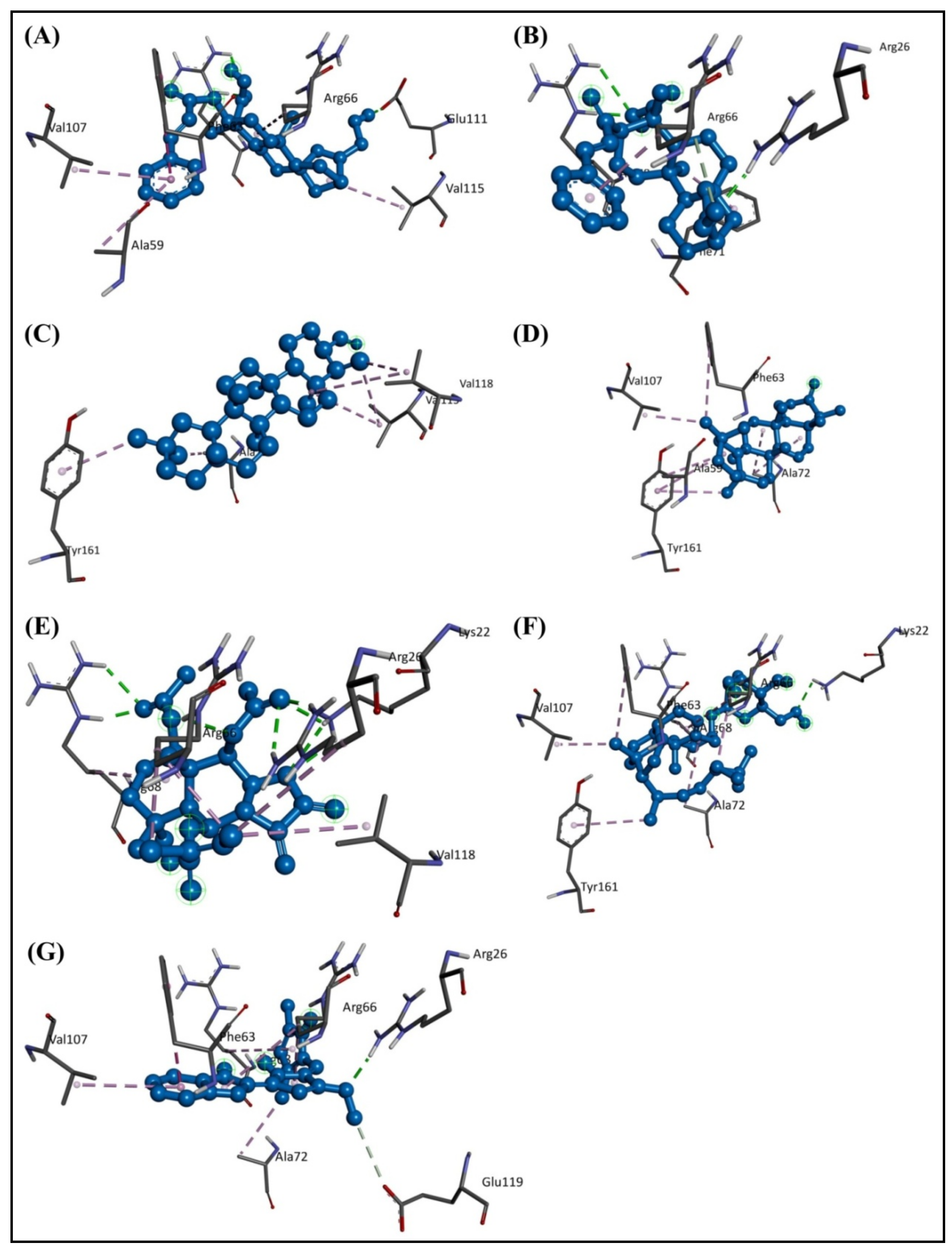
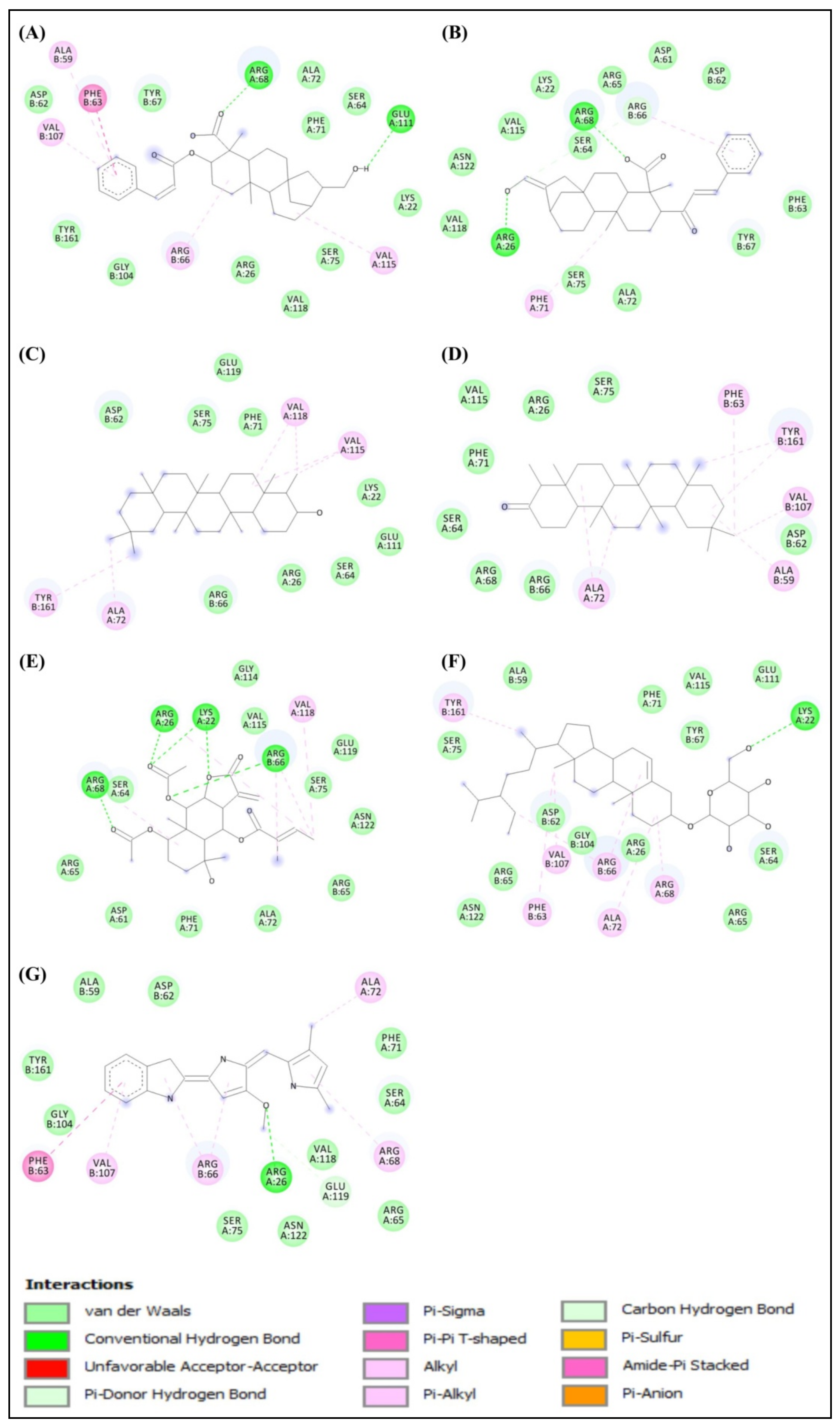
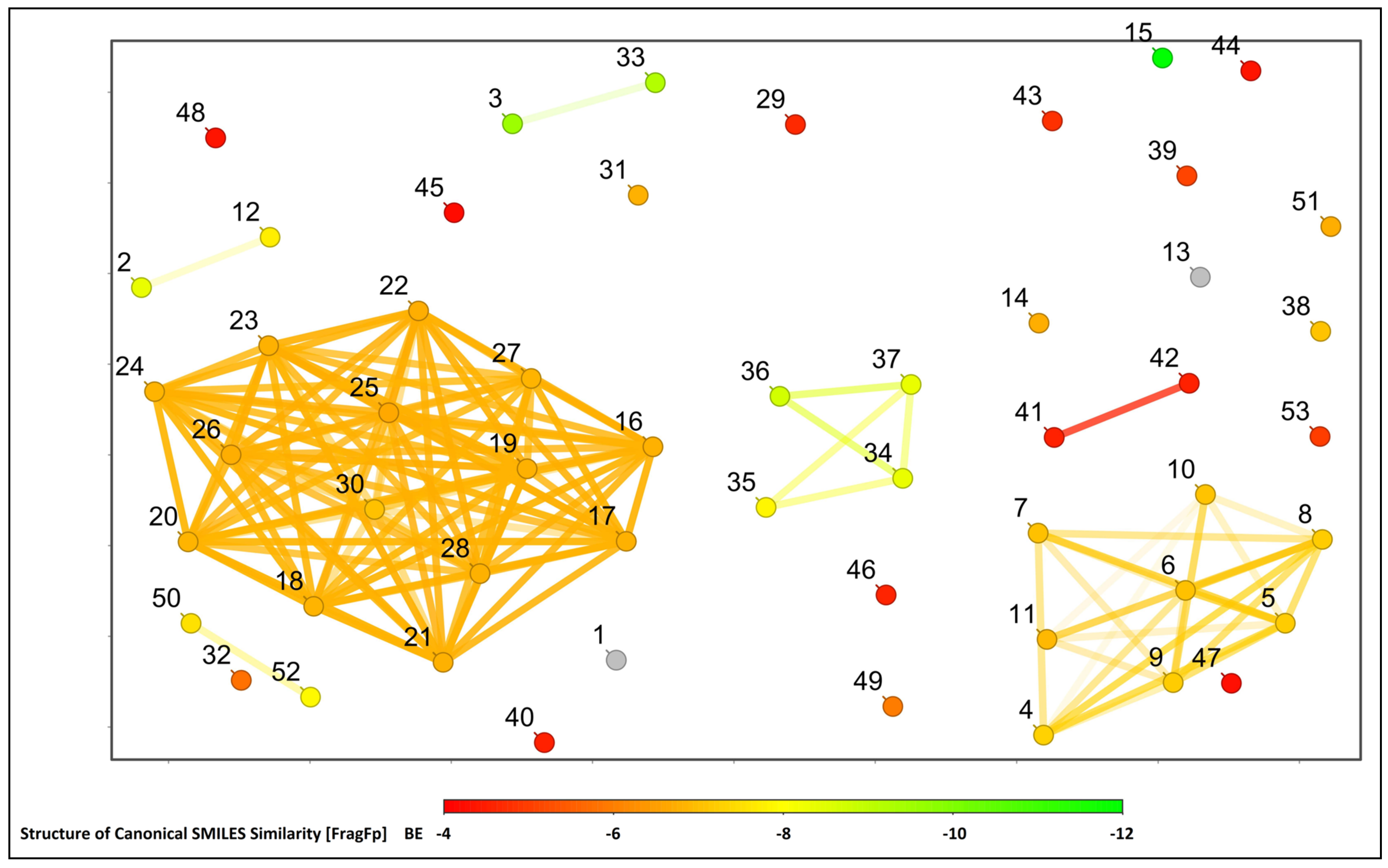
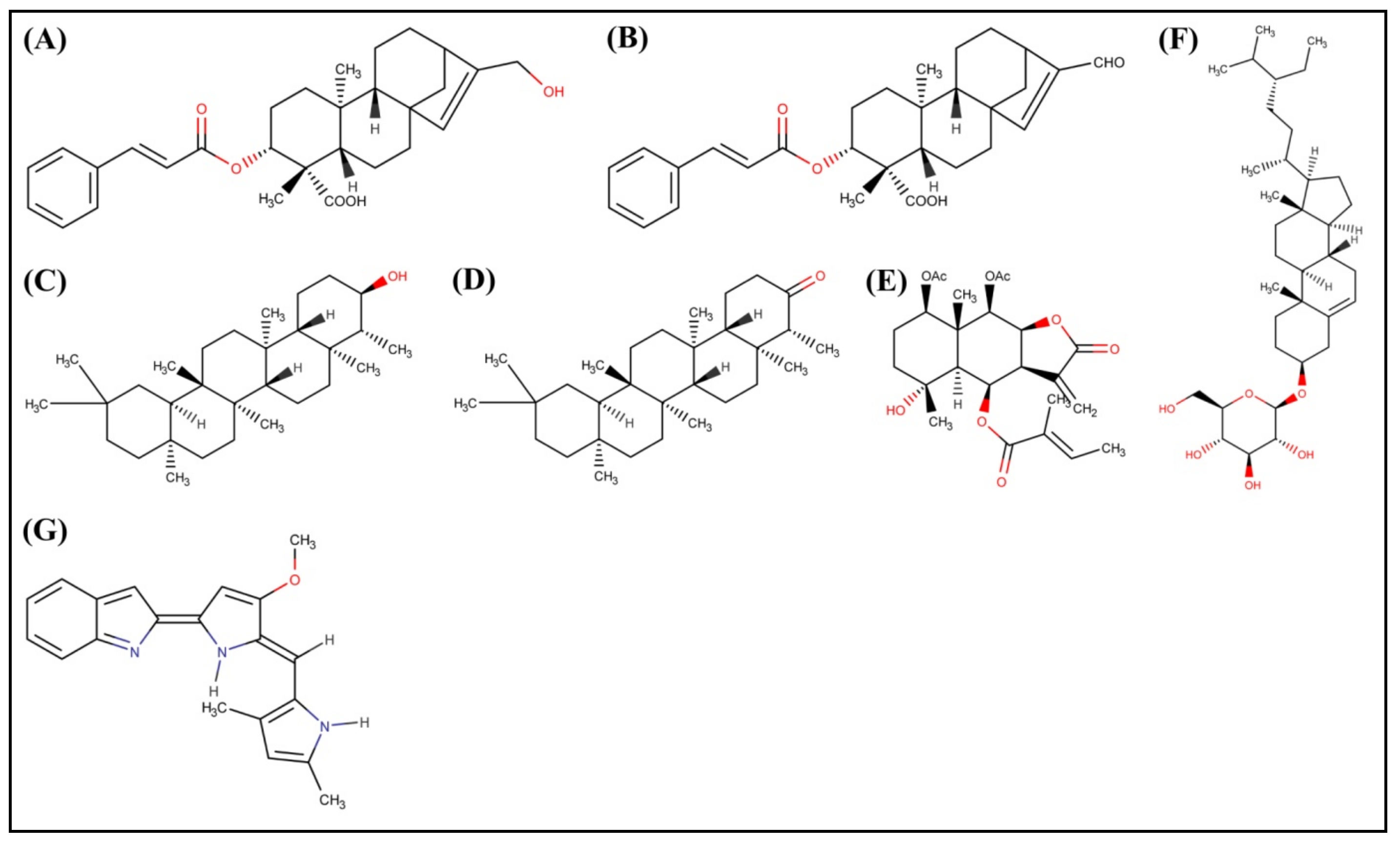
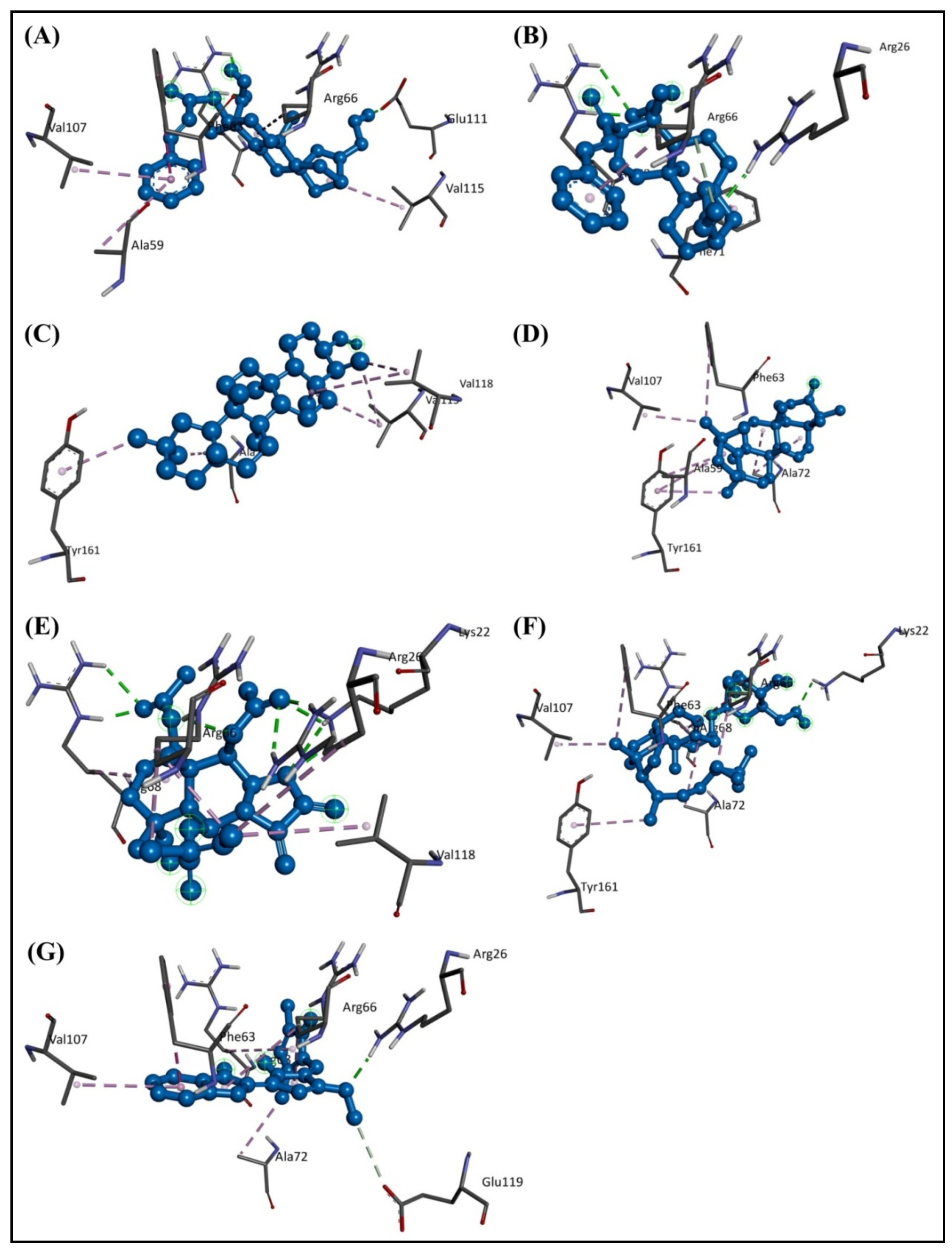
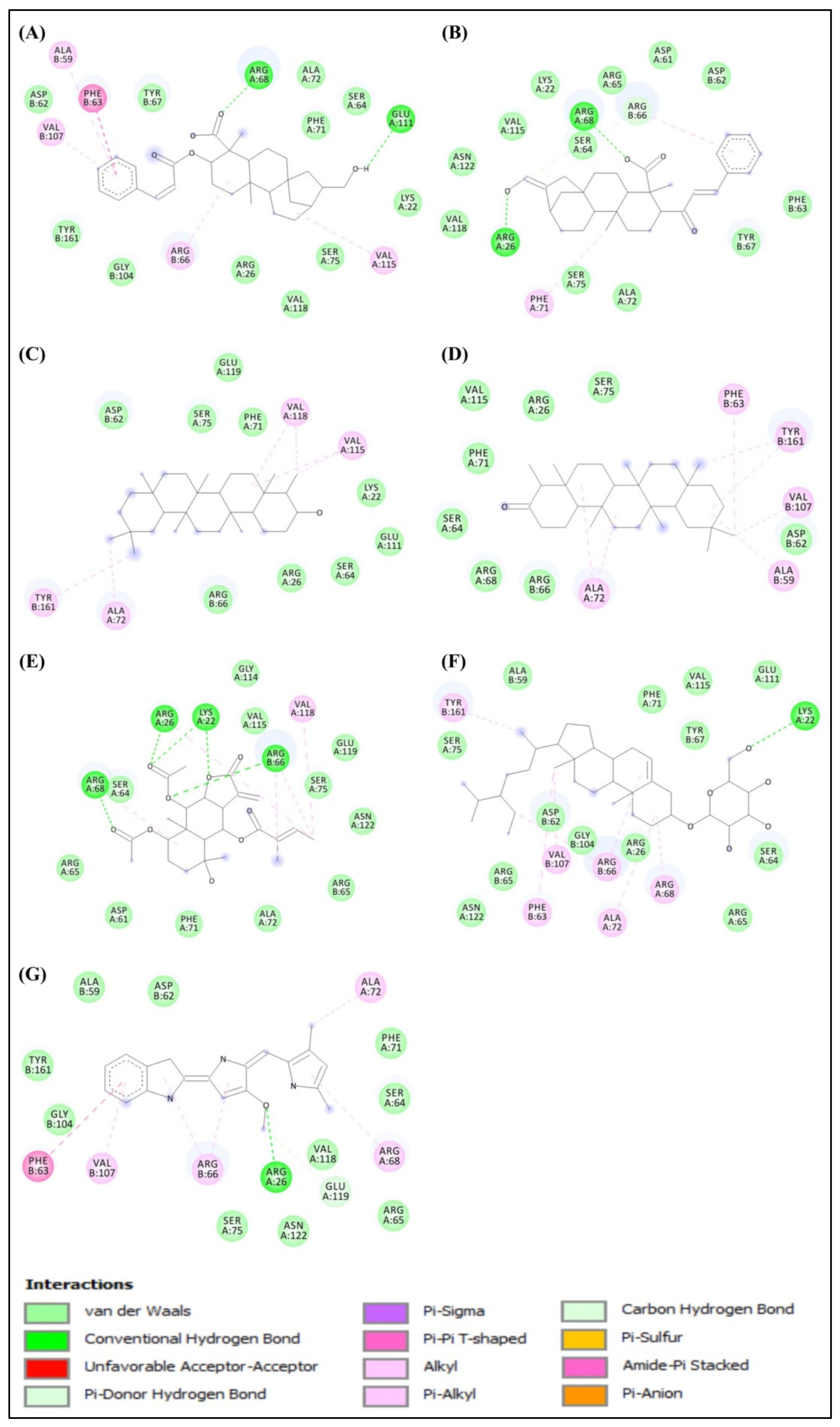
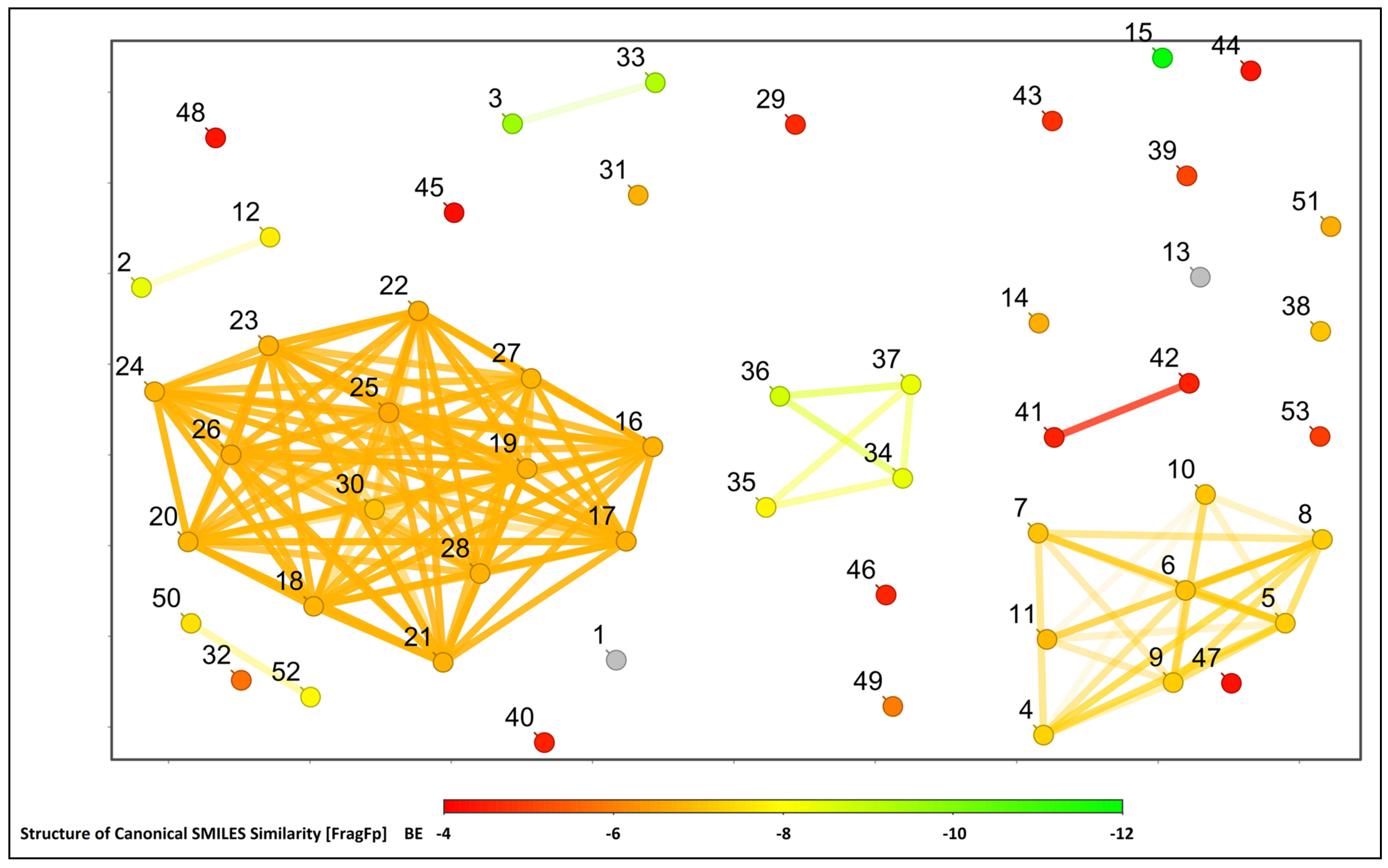
2.1. Molecular Docking Study
2.2. Structural Activity Relationship Analysis
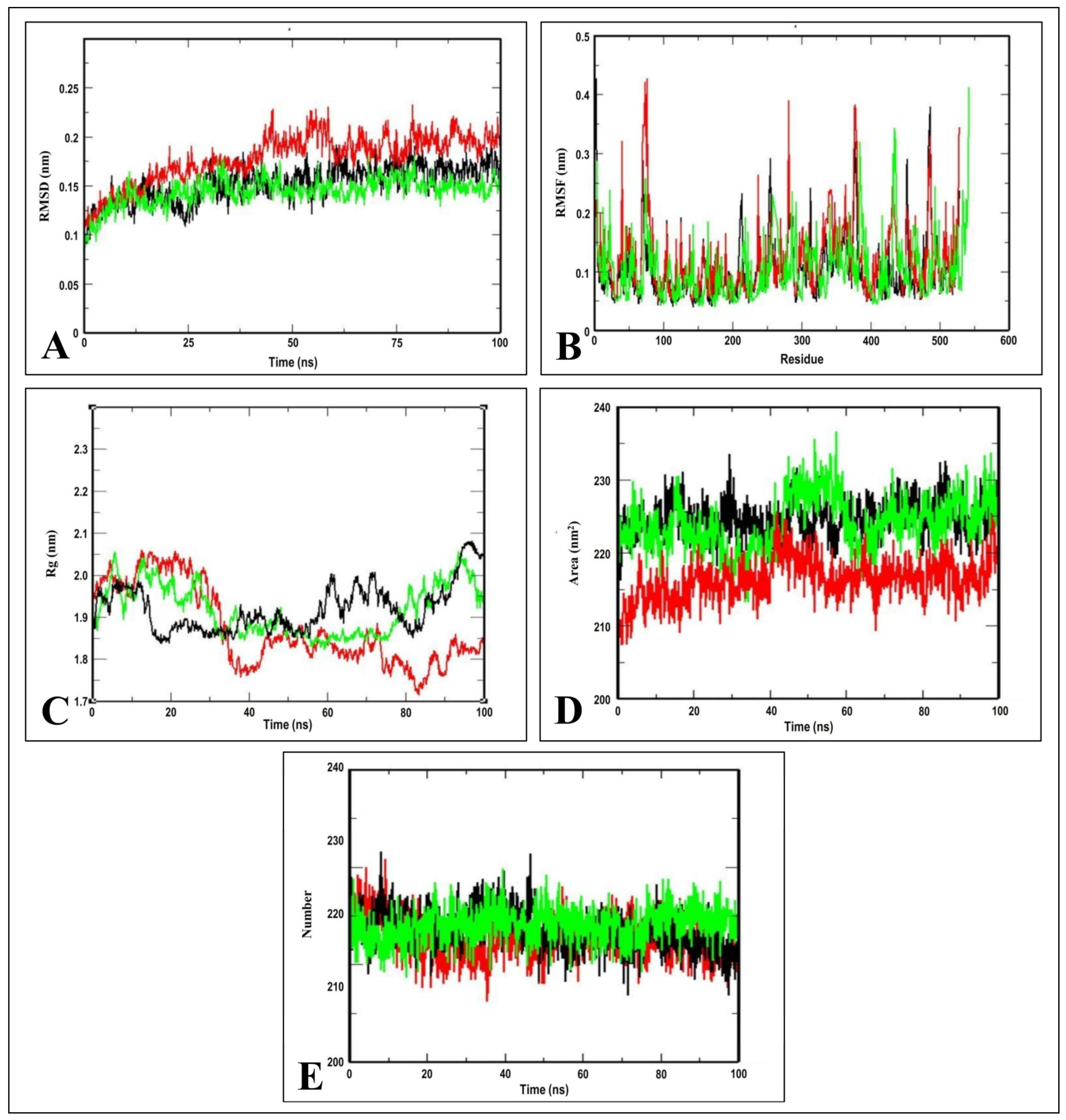
2.3. Molecular Dynamics Simulation Studies
2.4. Binding Free Energy Calculation
2.5. Prediction of ADMET Properties of Ligand Molecules
3. Materials and Methods
3.1. Preparation of Ligands
3.2. Preparation of Bcl-2 Protein Structure
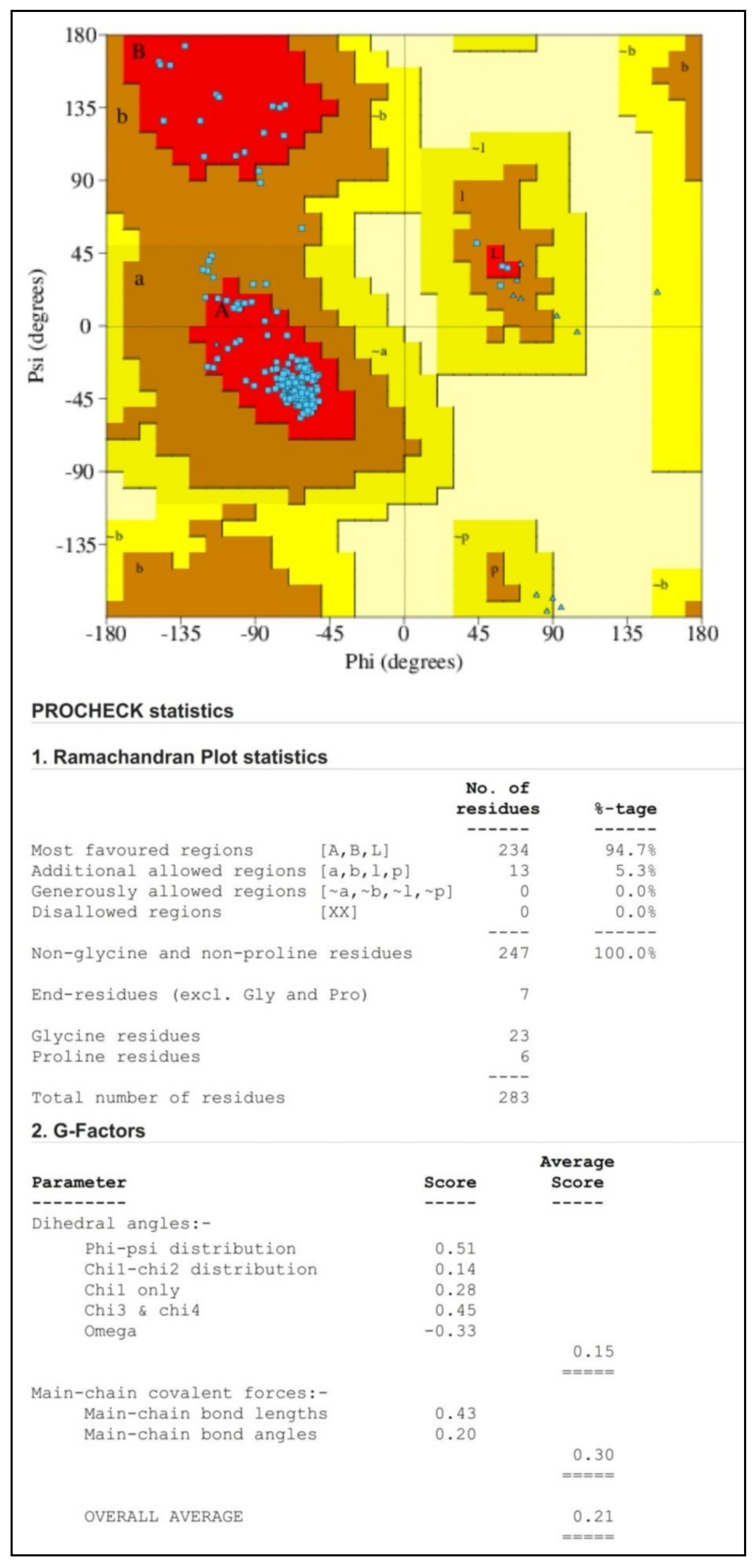
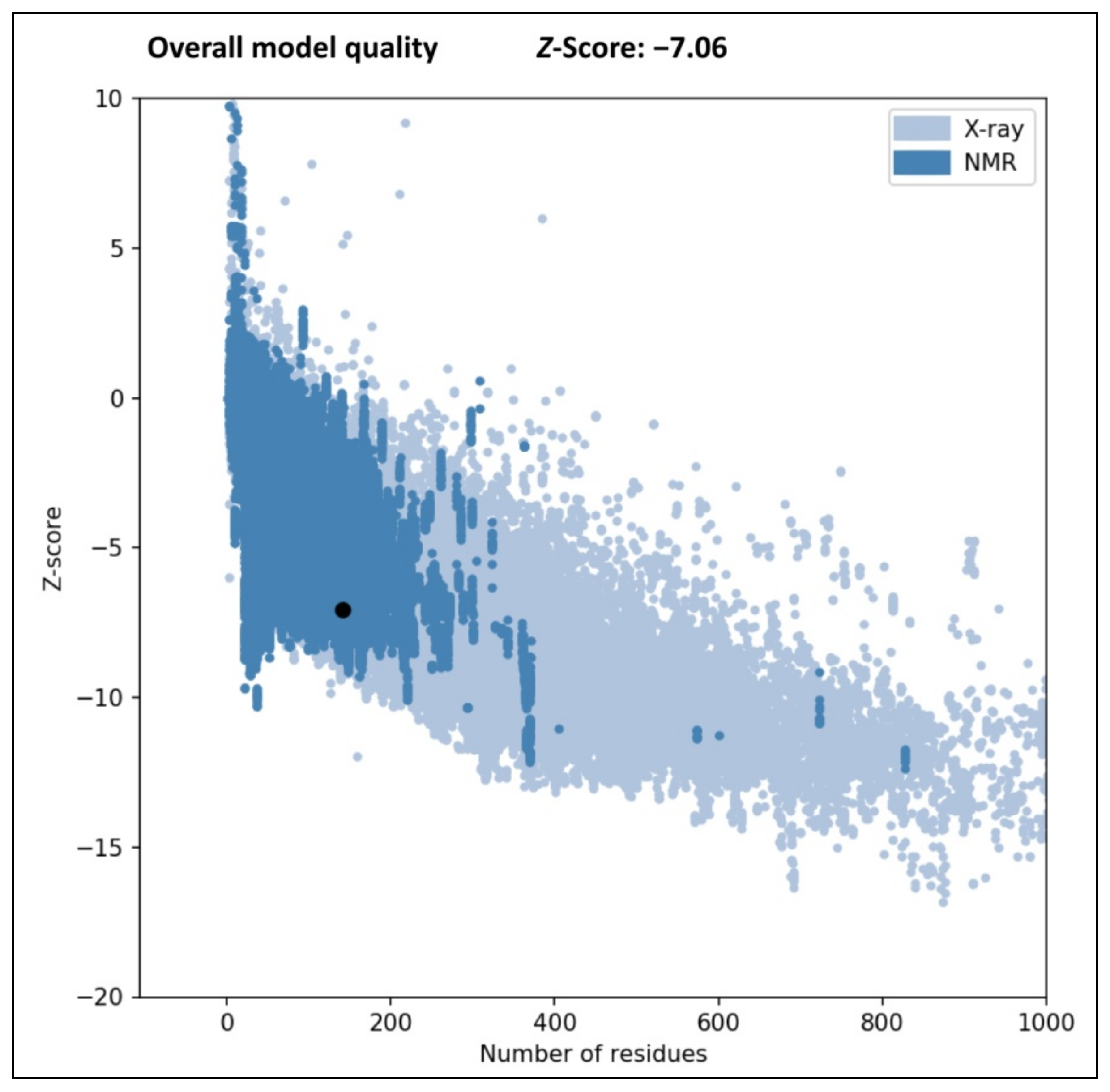
3.3. Validation of Protein Structure
3.4. Molecular Docking Analysis
3.5. Structural Activity Relationship Analysis
3.6. Molecular Dynamics Simulation
3.7. Binding Free Energy Calculation
3.8. Prediction of ADMET Properties of Ligands
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Belka, C.; Budach, W. Anti-apoptotic Bcl-2 proteins: Structure, function and relevance for radiation biology. Int. J. Radiat. Biol. 2002, 78, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Ali, S.; Mohammad, T.; Hasan, G.M.; Yadav, D.K.; Hassan, M.I. B Cell lymphoma 2: A potential therapeutic target for cancer therapy. Int. J. Mol. Sci. 2021, 22, 10442. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.; Payne, A.; de Candole, B.; Ford, D.; Hutchinson, B.; Trevitt, G.; Turner, J.; Edwards, C.; Watkins, C.; Whitcombe, I.; et al. Tetrahydroisoquinoline amide substituted phenyl pyrazoles as selective Bcl-2 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Lichota, A.; Gwozdzinski, K. Anticancer Activity of Natural Compounds from Plant and Marine Environment. Int. J. Mol. Sci. 2018, 19, 3533. [Google Scholar] [CrossRef]
- García-Aranda, M.; Pérez-Ruiz, E.; Redondo, M. Bcl-2 Inhibition to Overcome Resistance to Chemo- and Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3950. [Google Scholar] [CrossRef]
- Kapoor, I.; Bodo, J.; Hill, B.T.; His, E.D.; Almasan, A. Targeting BCL-2 in B-cell malignancies and overcoming therapeutic resistance. Cell Death Dis. 2020, 11, 941. [Google Scholar] [CrossRef]
- Puttaswamy, H.; Gowtham, H.G.; Ojha, M.D.; Yadav, A.; Choudhir, G.; Raguraman, V.; Kongkham, B.; Selvaraju, K.; Shareef, S.; Gehlot, P.; et al. In silico studies evidenced the role of structurally diverse plant secondary metabolites in reducing SARS-CoV-2 pathogenesis. Sci. Rep. 2020, 10, 20584. [Google Scholar] [CrossRef]
- Murali, M.; Gowtham, H.G.; Ansari, M.A.; Alomary, M.N.; Alghamdi, S.; Almehmadi, M.; Singh, S.B.; Shilpa, N.; Aiyaz, M.; Kalegowda, N.; et al. Repositioning Therapeutics for SARS-CoV-2: Virtual Screening of Plant-based Anti-HIV Compounds as Possible Inhibitors against COVID-19 Viral RdRp. Curr. Pharm. Des. 2022, 28, 969–980. [Google Scholar]
- Murali, M.; Gowtham, H.G.; Shilpa, N.; Krishnappa, H.K.N.; Ledesma, A.E.; Jain, A.S.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I.; Achar, R.R.; et al. Exploration of Anti-HIV Phytocompounds against SARS-CoV-2 Main Protease: Structure-Based Screening, Molecular Simulation, ADME Analysis and Conceptual DFT Studies. Molecules 2022, 27, 8288. [Google Scholar] [CrossRef]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell Dev. Biol. 2020, 8, 401. [Google Scholar] [CrossRef]
- Borghi, S.M.; Mizokami, S.S.; Carvalho, T.T.; Rasquel-Oliveira, F.S.; Ferraz, C.R.; Fattori, V.; Hayashida, T.H.; Peron, J.P.S.; Camilios-Neto, D.; Ambrosio, S.R.; et al. The diterpene from Sphagneticola trilobata (L.) Pruski, kaurenoic acid, reduces lipopolysaccharide-induced peritonitis and pain in mice. J. Ethnopharmacol. 2021, 273, 113980. [Google Scholar] [CrossRef]
- Mizokami, S.S.; Arakawa, N.S.; Ambrosio, S.R.; Zarpelon, A.C.; Casagrande, R.; Cunha, T.M.; Ferreira, S.H.; Cunha, F.Q.; Verri, W.A., Jr. Kaurenoic acid from Sphagneticola trilobata inhibits inflammatory pain: Effect on cytokine production and activation of the NO–cyclic GMP–protein kinase G–ATP-sensitive potassium channel signaling pathway. J. Nat. Prod. 2012, 75, 896–904. [Google Scholar] [CrossRef]
- Buddhakala, N.; Talubmook, C. Toxicity and antidiabetic activity of ethanolic extract of Sphagneticola trilobata (L.) Pruski flower in rats. J. Ethnopharmacol. 2020, 262, 113128. [Google Scholar] [CrossRef]
- Mardina, V.; Mastura; Hamdani; Sufriadi, E. Flower of Sphagneticola trilobata (L.) J.F Pruski from Aceh, Indonesia: Antioxidant and cytotoxic activity on HeLa cells. IOP Conf. Ser.: Mater. Sci. Eng. 2020, 1007, 012182. [Google Scholar] [CrossRef]
- Coimbra, J.T.S.; Feghali, R.; Ribeiro, R.P.; Ramos, M.J.; Fernandes, P.A. The Importance of Intramolecular Hydrogen Bonds on the Translocation of the Small Drug Piracetam through a Lipid Bilayer. RSC Adv. 2021, 11, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Adewole, K.E.; Ishola, A.A. Phytosterols and triterpenes from Morinda lucida Benth (Rubiaceae) as potential inhibitors of anti-apoptotic BCL-XL, BCL-2, and MCL-1: An in-silico study. J. Recept. Signal Transduct. 2019, 39, 87–97. [Google Scholar] [CrossRef]
- Mohamad Rosdi, M.N.; Mohd Arif, S.; Abu Bakar, M.H.; Razali, S.A.; Zulkifli, R.M.; Ya’akob, H. Molecular docking studies of bioactive compounds from Annona muricata Linn as potential inhibitors for Bcl-2, Bcl-w and Mcl-1 antiapoptotic proteins. Apoptosis 2018, 23, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.K.; Pradeep, S.; Shimavallu, C.; Kollur, S.P.; Syed, A.; Marraiki, N.; Egbuna, C.; Gaman, M.-A.; Kosakowska, O.; Cho, W.C.; et al. Evaluation of Annona muricata Acetogenins as Potential Anti-SARS-CoV-2 Agents Through Computational Approaches. Front. Chem. 2021, 8, 624716. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Processes 2021, 9, 71. [Google Scholar] [CrossRef]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef]
- Boroujeni, M.B.; Dastjerdeh, M.S.; Shokrgozar, M.A.; Rahimi, H.; Omidinia, E. Computational driven molecular dynamics simulation of keratinocyte growth factor behavior at different pH conditions. Inform. Med. Unlocked 2021, 23, 100514. [Google Scholar] [CrossRef]
- Sharma, A.; Magotra, A.; Dogra, A.; Rath, S.K.; Rayees, S.; Wazir, P.; Sharma, S.; Sangwan, P.L.; Singh, S.; Singh, G.; et al. Pharmacokinetics, pharmacodynamics and safety profiling of IS01957, a preclinical candidate possessing dual activity against inflammation and nociception. Regul. Toxicol. Pharmacol. 2017, 91, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Schaftenaar, G.; de Vlieg, J. Quantum mechanical polar surface area. J. Comput. Aided Mol. Des. 2012, 26, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert. Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Balekar, N.; Nakpheng, T.; Srichana, T. Wedelia trilobata L.: A phytochemical and pharmacological review. Chiang Mai J. Sci. 2014, 41, 590–605. [Google Scholar]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2019, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Costanzo, L.D.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Pradeep, S.; Jain, A.S.; Dharmashekara, C.; Prasad, S.K.; Akshatha, N.; Pruthvish, R.; Amachawadi, R.G.; Srinivasa, C.; Syed, A.; Elgorban, A.M.; et al. Synthesis, Computational Pharmacokinetics Report, Conceptual DFT-Based Calculations and Anti-Acetylcholinesterase Activity of Hydroxyapatite Nanoparticles Derived From Acorus Calamus Plant Extract. Front. Chem. 2021, 9, 741037. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Uppar, V.; Chandrashekharappa, S.; Shivamallu, C.; Sushma, P.; Kollur, S.P.; Ortega-Castro, J.; Frau, J.; Flores-Holguín, N.; Basarikatti, A.I.; Chougala, M.; et al. Investigation of Antifungal Properties of Synthetic Dimethyl-4-Bromo-1-(Substituted Benzoyl) Pyrrolo [1,2-a] Quinoline-2,3-Dicarboxylates Analogues: Molecular Docking Studies and Conceptual DFT-Based Chemical Reactivity Descriptors and Pharmacokinetics Evaluation. Molecules 2021, 26, 2722. [Google Scholar] [PubMed]
- Gowtham, H.G.; Murali, M.; Singh, S.B.; Shivamallu, C.; Pradeep, S.; Shivakumar, C.S.; Anandan, S.; Thampy, A.; Achar, R.R.; Silina, E.; et al. Phytoconstituents of Withania somnifera unveiled Ashwagandhanolide as a potential drug targeting breast cancer: Investigations through computational, molecular docking and conceptual DFT studies. PLoS ONE 2022, 17, e0275432. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Ramu, R.; Prithvi, S.; Shirahatti, V.B.; Kumari, C.; Sushma, P.; Mandal, S.P.; Patil, S.M. α-Glucosidase, α-Amylase Inhibition, Kinetics and Docking Studies of Novel (2-Chloro-6-(trifluoromethyl)benzyloxy)arylidene) Based Rhodanine and Rhodanine Acetic Acid Derivatives. Chemistry Select. 2021, 6, 9637–9644. [Google Scholar] [CrossRef]
- Modak, R.; Basha, J.; Bharathy, N.; Maity, K.; Mizar, P.; Bhat, A.V.; Vasudevan, M.; Rao, V.K.; Kok, W.K.; Natesh, N.; et al. Probing p300/CBP associated factor (PCAF)-dependent pathways with a small molecule inhibitor. ACS Chem. Biol. 2013, 8, 1311–1323. [Google Scholar] [CrossRef]
- Krishna, S.; Kumar, S.B.; Murthy, T.P.K.; Murahari, M. Structure-based design approach of potential BCL-2 inhibitors for cancer chemotherapy. Comput. Biol. Med. 2021, 134, 104455. [Google Scholar] [CrossRef]
- Pal, P.; Thummuri, D.; Lv, D.; Liu, X.; Zhang, P.; Hu, W.; Poddar, S.K.; Hua, N.; Khan, S.; Yuan, Y.; et al. Discovery of a Novel BCL-XL PROTAC Degrader with Enhanced BCL-2 Inhibition. J. Med. Chem. 2021, 64, 14230–14246. [Google Scholar] [CrossRef]
- Prasad, A.; Shruthi, G.; Sushma, P.; Jain, A.S.; Chandan, D.; Prasad, M.N.N.; Kollur, S.P.; Srinivasa, C.; Shivamallu, C. Helicobacter pylori Infection: A Bioinformatic Approach. Int. J. Pharm. Sci. Res. 2020, 11, 5469–5483. [Google Scholar]
- Dharmashekara, C.; Pradeep, S.; Prasad, S.K.; Jain, A.S.; Syed, A.; Prasad, K.S.; Patil, S.S.; Beelagi, M.S.; Srinivasa, C.; Shivamallu, C. Virtual screening of potential phyto-candidates as therapeutic leads against SARS-CoV-2 infection. Environ. Chall. 2021, 4, 100136. [Google Scholar] [CrossRef]
- Pradeep, S.; Patil, S.M.; Dharmashekara, C.; Jain, A.; Ramu, R.; Shirahatti, P.S.; Mandal, S.P.; Reddy, P.; Srinivasa, C.; Patil, S.S.; et al. Molecular insights into the in silico discovery of corilagin from Terminalia chebula as a potential dual inhibitor of SARS-CoV-2 structural proteins. J. Biomol. Struct. Dyn. 2022, 2022, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chadha, N.; Tiwari, A.K.; Kumar, V.; Milton, M.D.; Mishra, A.K. In Silico Thermodynamics Stability Change Analysis Involved in BH4 Responsive Mutations in Phenylalanine hydroxylase: QM/MM and MD Simulations Analysis. J. Biomol. Struct. Dyn. 2015, 33, 573–583. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Anandan, S.; Gowtham, H.G.; Shivakumara, C.S.; Thampy, A.; Brijesh Singh, S.; Murali, M.; Shivamallu, C.; Pradeep, S.; Shilpa, N.; Shati, A.A.; et al. Integrated approach for studying bioactive compounds from Cladosporium spp. against estrogen receptor alpha as breast cancer drug target. Sci. Rep. 2022, 22038. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No. | Compound Name | Binding Energy (kcal/mol) |
|---|---|---|
| 1. | Kaurenoic acid | −8.5 |
| 2. | Grandiflorenic acid | −7.7 |
| 3. | ent-kaura-9(11), 16-dien-19 oic acid methyl ester | −7.6 |
| 4. | 3alpha-(senecioyloxy)-ent-kaur-16-en-19 oic acid | −8.6 |
| 5. | (3alpha)-3-(angeloyloxy)-ent-kaur-16-en-19 oic acid | −8.0 |
| 6. | 3alpha-(angeloyloxy) 9beta-hydroxy-ent-kaur-16- en-19 oic acid | −8.1 |
| 7. | Methyl-3alpha-(angeloyloxy) 9beta-hydroxy-entkaurenoate | −8.4 |
| 8. | (3alpha)-3-(tiglinoyloxy)-ent-kaur-16-en-19 oic acid | −7.9 |
| 9. | (3alpha)-3-(cinnamoyloxy)-entkaur-16-en-19 oic acid | −8.7 |
| 10. | 15alpha-(cinnamoyloxy)-ent-kaur-16-en-19 oic acid | −8.6 |
| 11. | Wedelidin A | −9.4 |
| 12. | Wedelidin B | −9.2 |
| 13. | Wedelia-seco-kaurenolide | −7.7 |
| 14. | Friedelinol | −9.6 |
| 15. | Friedelin | −10.1 |
| 16. | 1beta, 4alpha-dihydroxy-6beta-isobutyryloxy-9alpha-(tigloyloxy) prostatolide | −8.8 |
| 17. | 9alpha-(angeloyloxy)-1beta, 4alpha-dihydroxy-6 beta-isobutyryloxyprostatolide | −8.6 |
| 18. | 1beta, 9alpha-diacetoxy-4alpha hydroxy-6beta-isobutyryloxy prostatolide | −8.5 |
| 19. | 1beta, 9alpha-diacetoxy-4alpha hydroxy-6beta-methacryloxy prostatolide | −8.3 |
| 20. | Wedeliatrilolactone A | −8.4 |
| 21. | Wedeliatrilolactone B | −8.0 |
| 22. | Trilobolide 6-O-angelate | −9.1 |
| 23. | Trilobolide 6-O-methacrylate | −8.5 |
| 24. | Oxidoisotrilobolide 6-O-isobutyrate | −7.5 |
| 25. | Oxidoisotrilobolide 6-O-angelate | −7.8 |
| 26. | Oxidoisotrilobolide 6-O-methacrylate | −7.7 |
| 27. | Wedelolide A | −8.2 |
| 28. | Wedelolide B | −8.3 |
| 29. | Wedelolactone | −8.1 |
| 30. | Ivalin | −7.0 |
| 31. | Germacrene D | −6.4 |
| 32. | alpha-humulene | −6.8 |
| 33. | Caryophyllene | −6.8 |
| 34. | Stigmasterol | −8.5 |
| 35. | (7alpha)-7-hydroxystigmasterol | −8.3 |
| 36. | (3beta)-3-hydroxy stigmasta-5, 22-dien7-one | −8.3 |
| 37. | Sitosterol | −8.0 |
| 38. | Daucosterol | −9.0 |
| 39. | Squalene | −7.6 |
| 40. | 3-hydroxy-6-methoxychromen-4-one | −5.8 |
| 41. | Apigenin | −7.7 |
| 42. | Diosmentin | −7.3 |
| 43. | Benzeneacetic acid 2-phenylethenyl ester | −7.1 |
| 44. | Isocinnamic acid | −5.9 |
| 45. | 4-methoxy catechol | −5.3 |
| 46. | p-cymene | −6.1 |
| 47. | Protocatechualdehyde | −5.6 |
| 48. | Caffeic acid | −6.6 |
| 49. | alpha-phellandrene | −5.5 |
| 50. | alpha-pinene | −5.7 |
| 51. | D-limonene | −5.5 |
| 52. | Bicyclogermacrene | −6.6 |
| 53. | Obatoclax [Standard drug] | −8.4 |
| Categories | 2W3L—Friedelin Complex | 2W3L—Obatoclax Complex | ||
|---|---|---|---|---|
| Values (kcal/mol) | Standard Deviation (kcal/mol) | Values (kcal/mol) | Standard Deviation (kcal/mol) | |
| Van der Waal’s energy | −138.486 | ±36.214 | −210.986 | ±62.264 |
| Electrostatic energy | −14.698 | ±9.246 | −58.677 | ±17.622 |
| Polar salvation energy | 82.732 | ±33.218 | 89.918 | ±29.725 |
| SASA energy | −16.140 | ±6.982 | 56.809 | ±18.264 |
| Binding energy | −106.362 | ±36.542 | −229.323 | ±58.825 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gowtham, H.G.; Ahmed, F.; Anandan, S.; Shivakumara, C.S.; Bilagi, A.; Pradeep, S.; Shivamallu, C.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I.; et al. In Silico Computational Studies of Bioactive Secondary Metabolites from Wedelia trilobata against Anti-Apoptotic B-Cell Lymphoma-2 (Bcl-2) Protein Associated with Cancer Cell Survival and Resistance. Molecules 2023, 28, 1588. https://doi.org/10.3390/molecules28041588
Gowtham HG, Ahmed F, Anandan S, Shivakumara CS, Bilagi A, Pradeep S, Shivamallu C, Shati AA, Alfaifi MY, Elbehairi SEI, et al. In Silico Computational Studies of Bioactive Secondary Metabolites from Wedelia trilobata against Anti-Apoptotic B-Cell Lymphoma-2 (Bcl-2) Protein Associated with Cancer Cell Survival and Resistance. Molecules. 2023; 28(4):1588. https://doi.org/10.3390/molecules28041588
Chicago/Turabian StyleGowtham, Hittanahallikoppal Gajendramurthy, Faiyaz Ahmed, Satish Anandan, C. S. Shivakumara, Ashween Bilagi, Sushma Pradeep, Chandan Shivamallu, Ali A. Shati, Mohammad Y. Alfaifi, Serag Eldin I. Elbehairi, and et al. 2023. "In Silico Computational Studies of Bioactive Secondary Metabolites from Wedelia trilobata against Anti-Apoptotic B-Cell Lymphoma-2 (Bcl-2) Protein Associated with Cancer Cell Survival and Resistance" Molecules 28, no. 4: 1588. https://doi.org/10.3390/molecules28041588
APA StyleGowtham, H. G., Ahmed, F., Anandan, S., Shivakumara, C. S., Bilagi, A., Pradeep, S., Shivamallu, C., Shati, A. A., Alfaifi, M. Y., Elbehairi, S. E. I., Achar, R. R., Silina, E., Stupin, V., Murali, M., & Kollur, S. P. (2023). In Silico Computational Studies of Bioactive Secondary Metabolites from Wedelia trilobata against Anti-Apoptotic B-Cell Lymphoma-2 (Bcl-2) Protein Associated with Cancer Cell Survival and Resistance. Molecules, 28(4), 1588. https://doi.org/10.3390/molecules28041588