

Experimental, Spectroscopic, and Computational Insights into the Reactivity of “Methanal” with 2-Naphthylamines
 , , ,
, , ,
Abstract
:1. Introduction
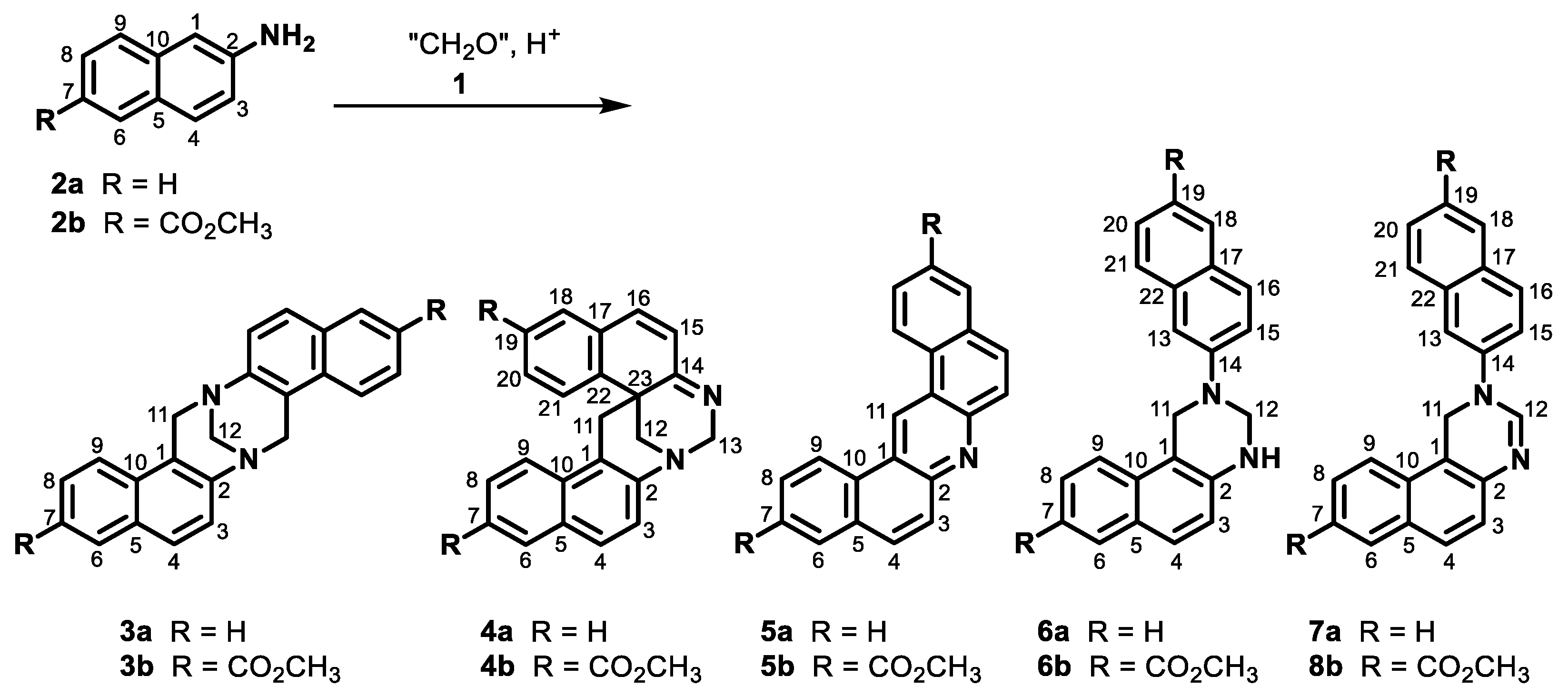
2. Results and Discussion
2.1. Studies of Formalin and Paraformaldehyde
2.2. Calculations on the Reactivity of Methanal
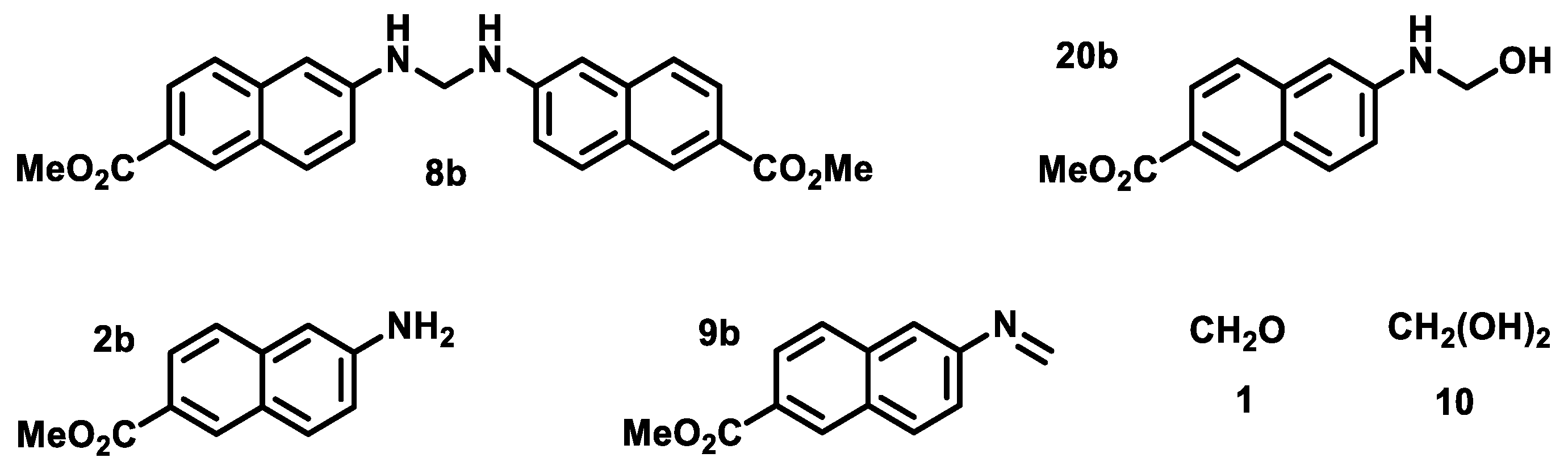
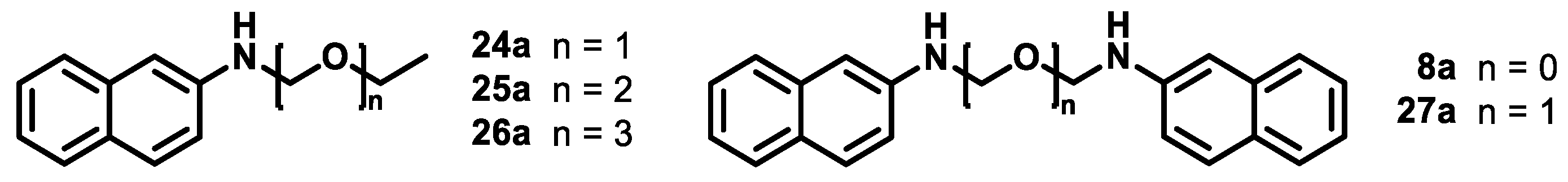
2.3. Attempts to Prepare Aminal 8a
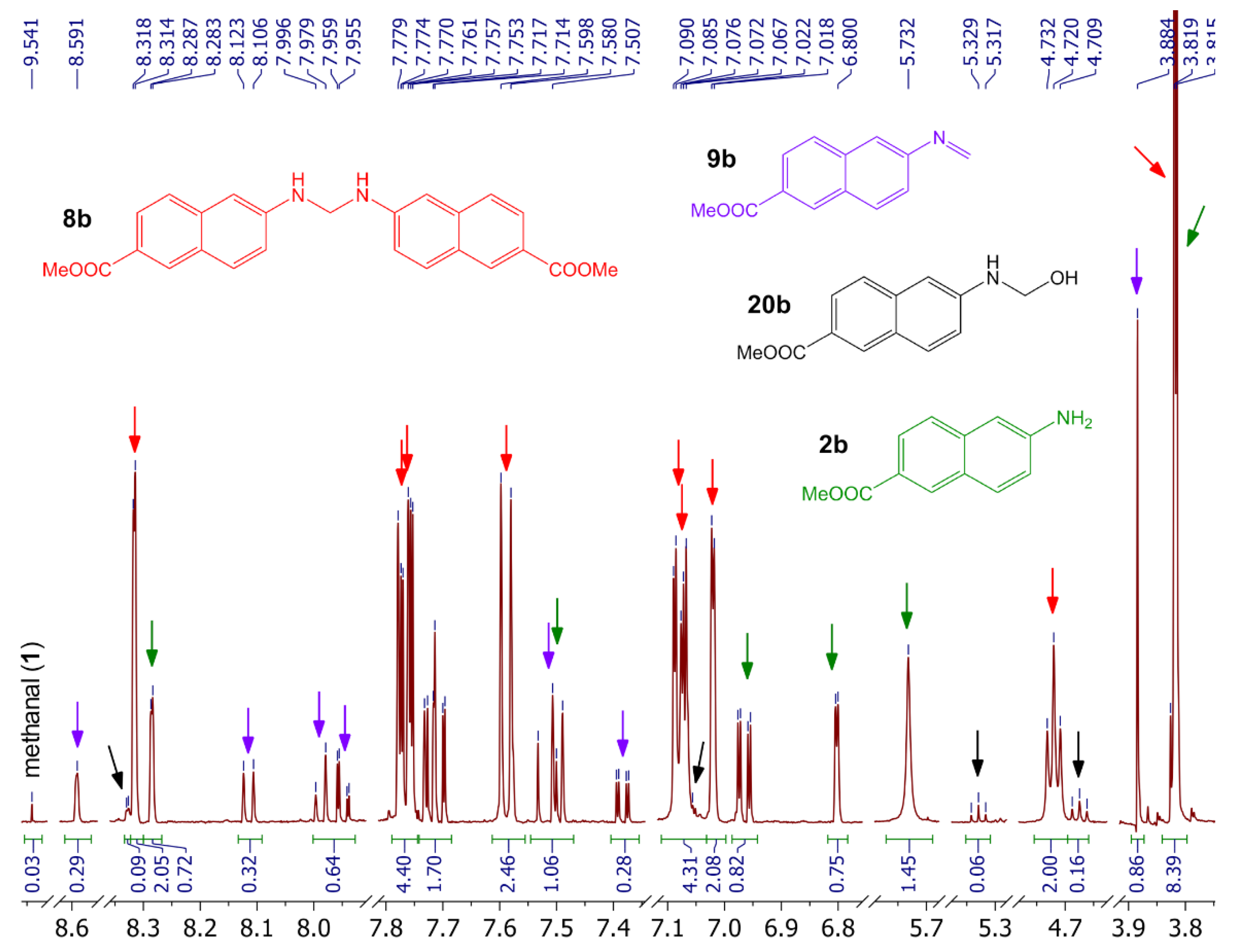
2.4. Attempts to Prepare Aminal 8b
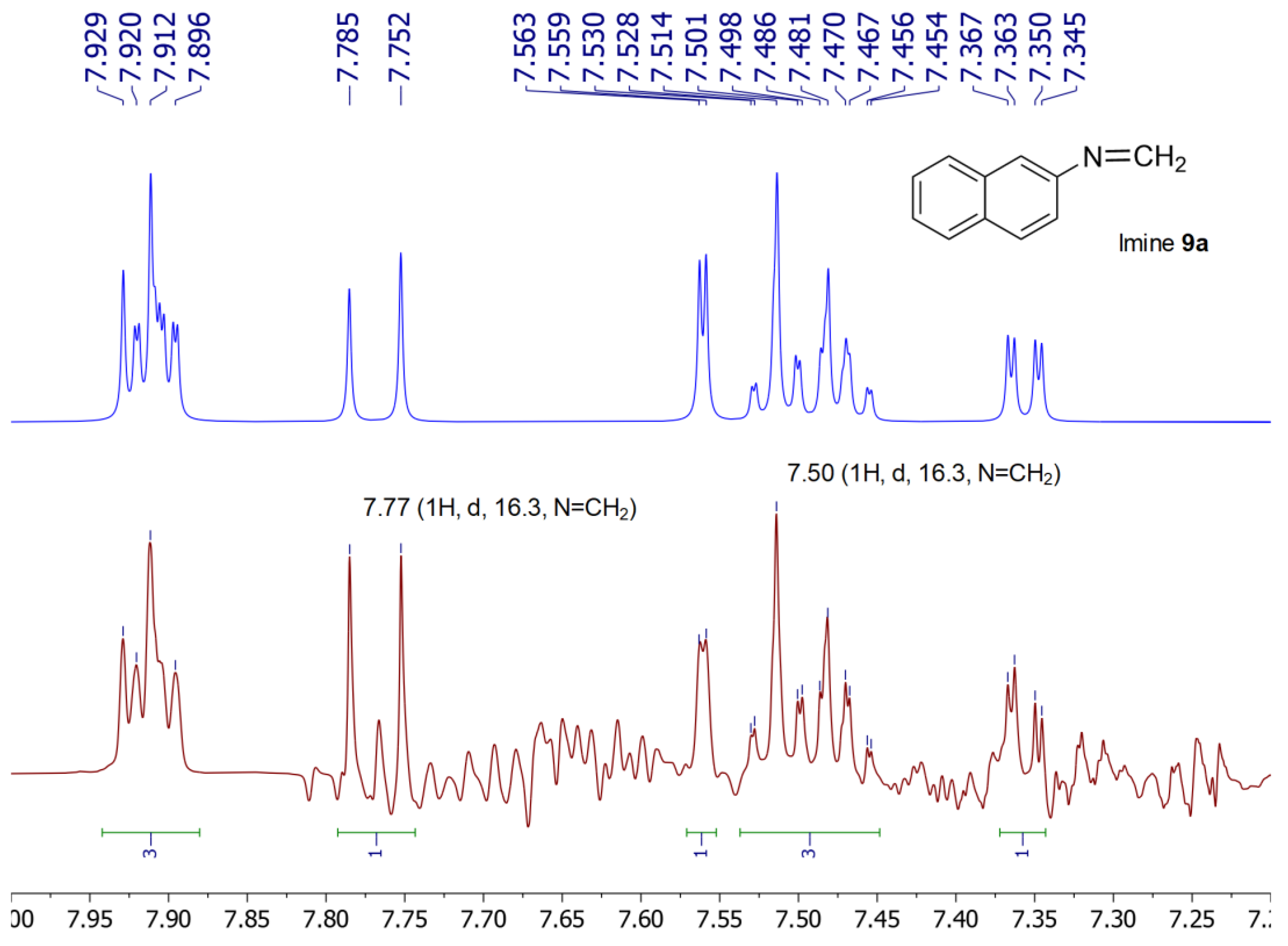
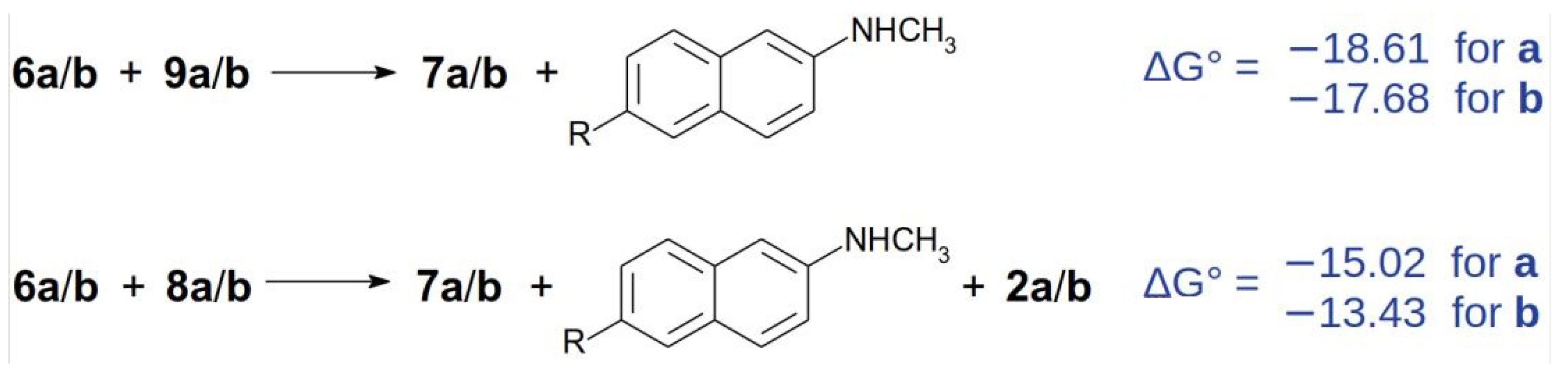
2.5. Preparation of Imine 9a under Acidic Conditions
2.6. Preparation of Imine 9a under Basic Conditions
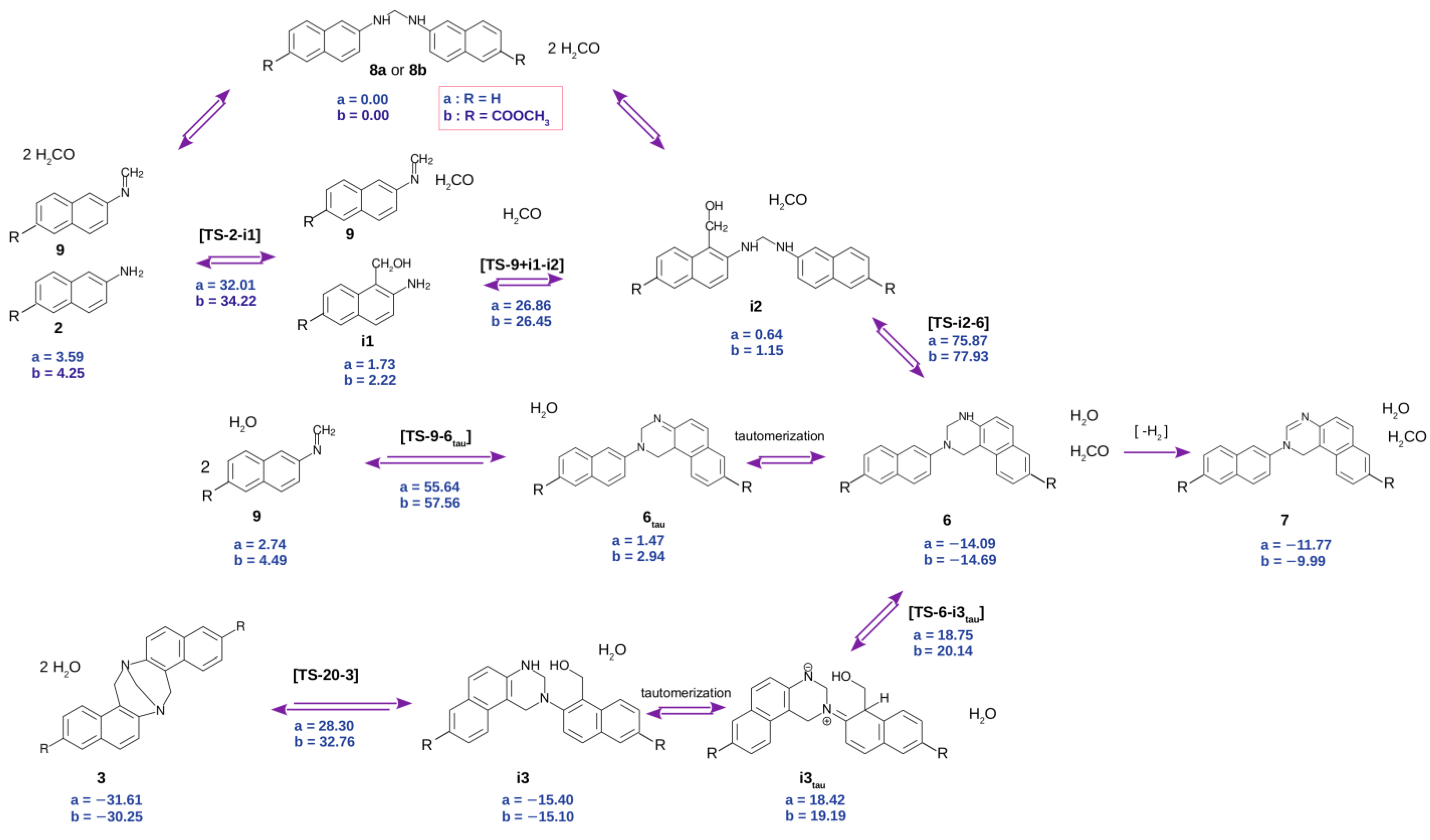
2.7. Density Functional Theory Calculations
3. Materials and Methods
3.1. Measurements and Materials
3.2. Computational Procedure
3.3. Studies of Formalin and Paraformaldehyde
- (a)
- An NMR tube was charged with 500 μL of DMSO-d6 (standard quality) and 5 μL of formalin (ASC reagent, formaldehyde solution, 37% m/m in H2O, containing 10–15% methanol as a stabilizer) and closed with a gas-tight cup. The solution was monitored by 1H NMR spectra at 25 °C. Equilibrium was reached within a few hours. The sample composition was determined by 1D and 2D NMR (Table 1). The sample was heated to 50 °C, left to equilibrate (1–2 h), and analyzed by 1H NMR. The same was done at temperatures of 75, 100, and 115 °C. After cooling back to 25 °C, the compositions were slowly returned to equilibrium (two days).
- (b)
- An NMR tube was charged with paraformaldehyde (1.0 mg, 33 μmol), DMSO-d6 (0.5 mL), and water (10 μL, 555 μmol) and closed with a gas-tight cup. The mixture was shaken until the paraformaldehyde dissolved and then monitored by 1H NMR at 25 °C. Equilibrium was reached within several hours. The sample composition was determined by 1D and 2D NMR (Table 2); the higher diols contents did not exceed 0.5‰ (n/n). The sample was heated following the procedure described in (a).
- (c)
- An NMR tube was charged with paraformaldehyde (1.0 mg, 33 μmol) and DMSO-d6 (0.5 mL) and closed with a gas-tight cup. The mixture was shaken, but part of the paraformaldehyde remained undissolved. The sample was heated following the procedure described in (a).
3.4. Reaction of Naphthylamine 2a with Formalin under Neutral Condition
- (a)
- Aqueous formaldehyde (37%, 0.1 mL, 1.23 mmol) was added to a solution of naphthylamine 2a (352 mg, 2.46 mmol) in acetone (20 mL). The mixture was refluxed for five hours. The reaction mixture was evaporated to dryness in vacuo, and the residue was analyzed using 1D and 2D NMR experiments. The residue was purified by crystallization from ethanol. An insoluble fraction (18 mg) was obtained, which contained quinazoline 6a (7 mg, 2% yield) and bisquinazoline 16a (11 mg, 3% yield), as well as crystals of pure quinazoline 6a (133 mg, 35% yield).
- (b)
- Aqueous formaldehyde (37%, 0.1 mL, 1.23 mmol) was added to the solution of naphthylamine 2a (352 mg, 2.46 mmol) in acetone (20 mL). The mixture was refluxed for five hours and then evaporated to dryness in vacuo. The obtained solid (387 mg) contained mostly naphthylamine 2a, quinazoline 6a, and dinaphthylamine 17a in a molar ratio of 41:45:14, according to NMR. The solid was purified by column chromatography on silica (dichloromethane/methanol from 1:0 to 4:1) to produce four fractions of various compositions. The yields were calculated based on the 1H NMR spectra: 105 mg (30% recovered) of naphthylamine 2a, 196 mg (51% yield) of quinazoline 6a, 3 mg (1% yield) of dinaphthylamine 17a, 31 mg (9% yield) of acridine 5a, 31 mg (8% yield) of bisnaphthylamine 18a, and 5 mg (1% yield) of TB 3a.
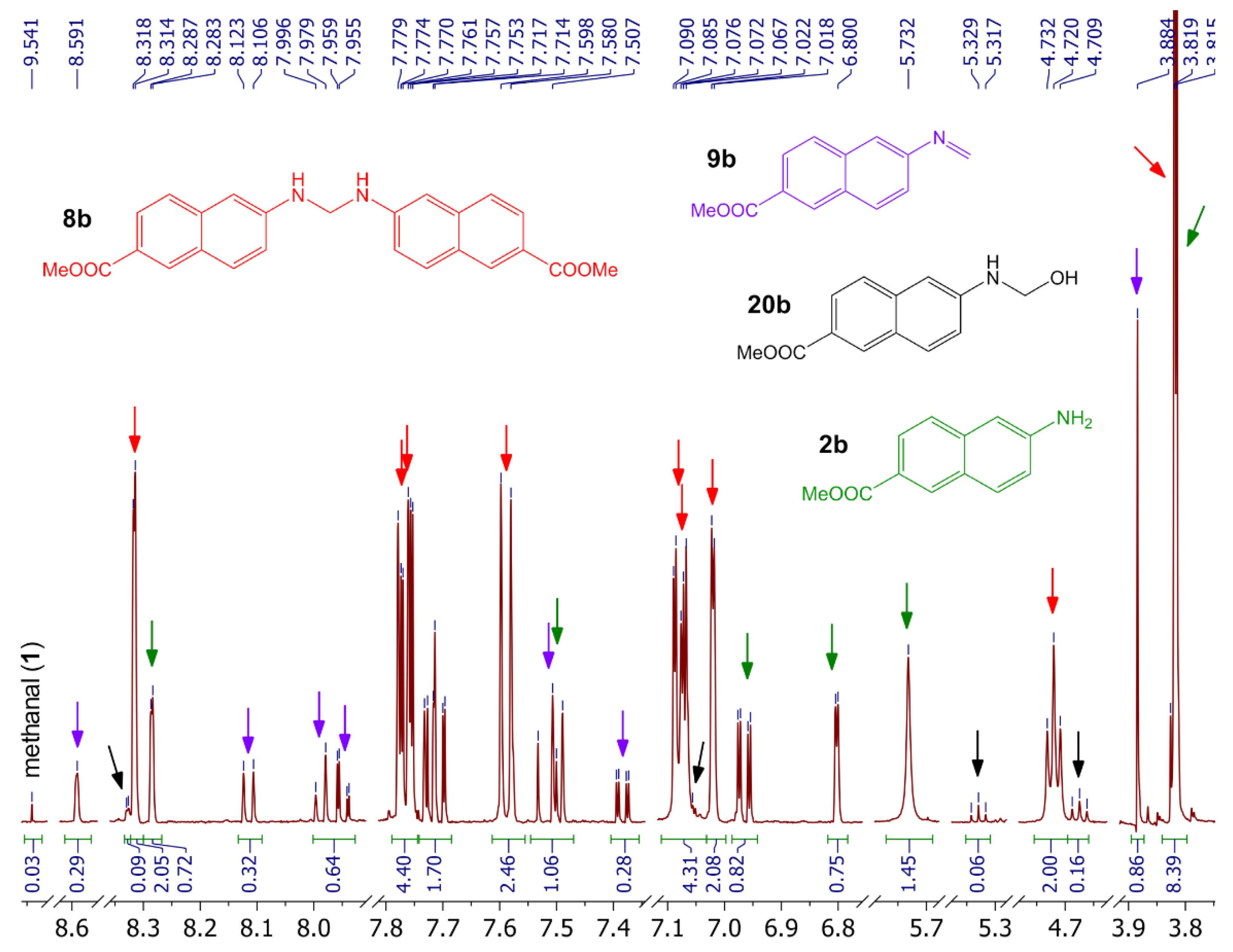
3.5. Reaction of Naphthylamine 2b with Formalin under Neutral Conditions
3.6. Reaction of Naphthylamine 2a with Formalin under Acidic Conditions
- (a)
- Aqueous formaldehyde (37%, 3.8 mL, 50 mmol) in acetic acid (99%, 7 mL) was added dropwise (1 min) to the solution of naphthylamine 2a (7.2 g, 50 mmol) in acetic acid (99%, 110 mL) at room temperature. The mixture was stirred for the next 2 min and then poured into a 1% brine solution (20 mL) and stirred for 30 min. The solid was filtered off, washed with water, and dried in vacuo to obtain 9.1 g of the crude product exhibiting no signals for imine 9a in the 1H NMR spectrum. The crystallization of the crude product from ethanol produced white crystals consisting of a mixture of quinazoline 6a and bisquinazoline 16a in a molar ratio of 94:6. Repeating the crystallization procedure produced pure quinazoline 6a (4.70 g, 60% yield) and pure bisquinazoline 16a (0.24 g, 3% yield).
- (b)
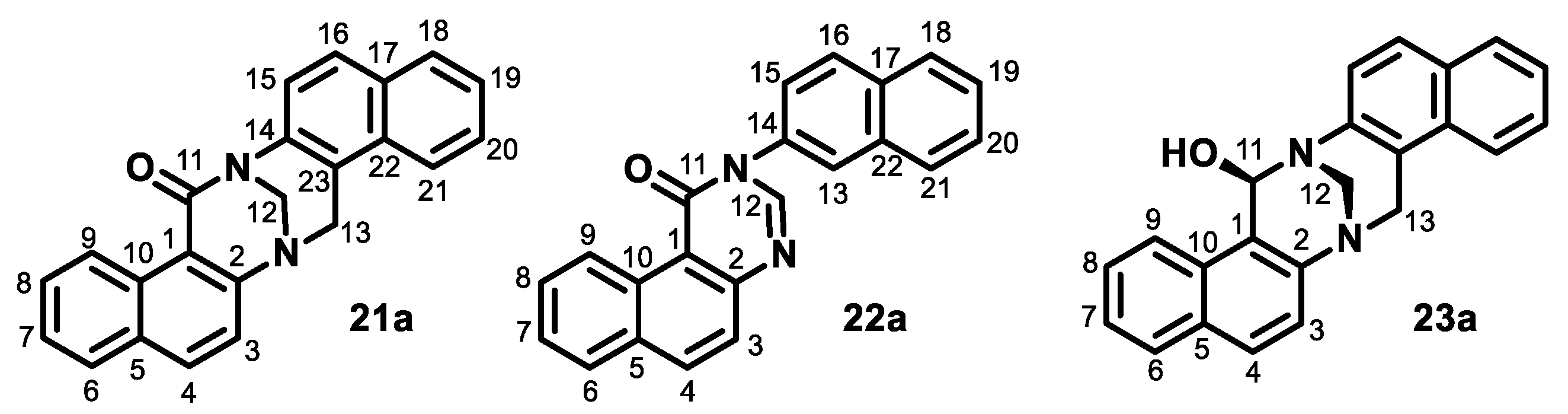
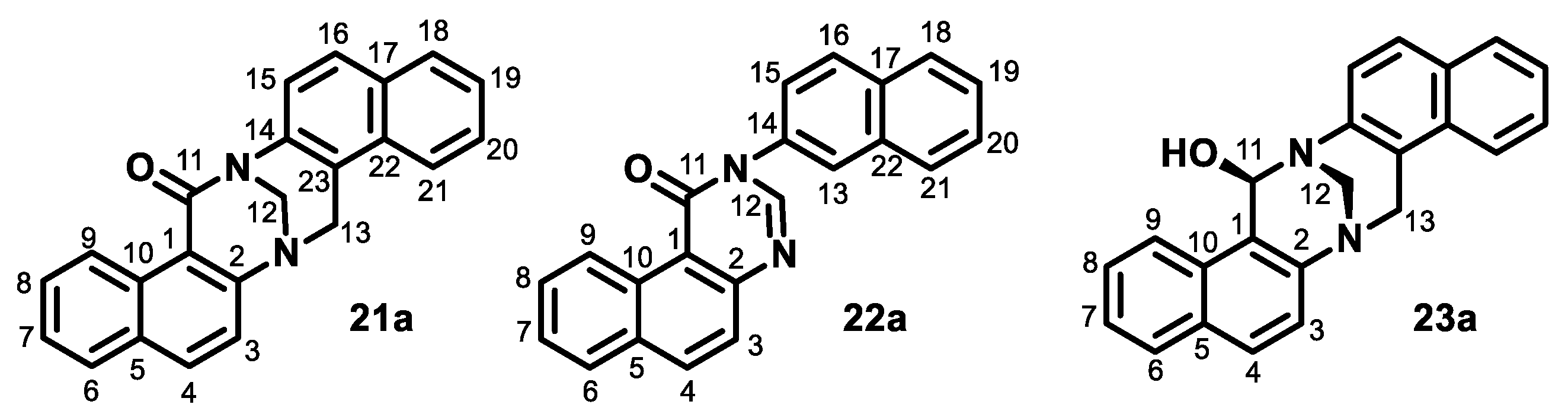
- Aqueous formaldehyde (37%, 0.38 mL, 5 mmol) in acetic acid (99%, 1 mL) was added dropwise (1 min) to the solution of naphthylamine 2a (0.72 g, 5 mmol) in acetic acid (99%, 11 mL) at room temperature. The mixture was stirred for the next 2 min. The mixture was then diluted with water (20 mL) and alkalized with aqueous NH3, and the product was extracted with dichloromethane. The organic solution was washed sequentially with water and brine, dried over anhydrous sodium sulfate, and evaporated to dryness in vacuo. The residue was purified by column chromatography on silica (dichloromethane/methanol from 1:0 to 8:2) to produce quinazoline 6a (390 mg, 50%), TB 3a (81 mg, 10%), acridine 5a (23 mg, 3%), starting naphthylamine 2a (150 mg, 21%), oxo-TB 21a (17 mg, 2%), dihydroquinazoline 7a (25 mg, 3%), and oxo-quinazoline 22a (5 mg, under 1%). Those compounds were followed by a polar fraction that contained spiroTB 4a and a diastereomer of unidentified hydroxy-TB 23a.
3.7. Reaction of Naphthylamine 2a with Formalin under Basic Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Tatar, A.; Čejka, J.; Král, V.; Dolenský, B. Spiro Tröger’s Base Derivatives: Another Structural Phoenix? Org. Lett. 2010, 12, 1872–1875. [Google Scholar] [CrossRef] [PubMed]
- Tröger, J. Ueber einige mittelst nascirenden Formaldehydes entstehende Basen. J. Prakt. Chem. 1887, 36, 225–245. [Google Scholar] [CrossRef]
- Spielman, M.A. The Structure of Troeger’s Base. J. Am. Chem. Soc. 1935, 57, 583–585. [Google Scholar] [CrossRef]
- Vögtle, F. Heterocycles and Biologically Active Compounds. In Fascinating Molecules in Organic Chemistry, 1st ed.; John Wiley & Sons: Chichester, UK, 1992; pp. 237–290. [Google Scholar]
- Dolenský, B.; Elguero, J.; Král, V.; Pardo, C.; Valík, M. Current Tröger’s Base Chemistry. Adv. Heterocycl. Chem. 2007, 93, 1–56. [Google Scholar]
- Sergeyev, S. Recent Developments in Synthetic Chemistry, Chiral Separations, and Applications of Tröger’s Base Analogues. Helv. Chim. Acta 2009, 92, 415–444. [Google Scholar] [CrossRef]
- Reed, J.H. Ueber β-Naphtoacridin. Vorläufige Mittheilung. J. Prakt. Chem. 1886, 34, 160–161. [Google Scholar] [CrossRef]
- Reed, J.H., VIII. Ueber Methylderivate der Naphtochinoline und über β-Naphtoacridin. J. Prakt. Chem. 1887, 35, 298–322. [Google Scholar] [CrossRef]
- Morgan, G.T. LVI—Action of formaldehyde on amines of the naphthalene series. Part I. J. Chem. Soc., Trans. 1898, 73, 536–554. [Google Scholar] [CrossRef]
- Farrar, W.V. Reactions of formaldehyde with aromatic amines. J. Appl. Chem. 1964, 14, 389–399. [Google Scholar] [CrossRef]
- Tálas, E.; Margitfalvi, J.; Machytka, D.; Czugler, M. Synthesis and resolution of naphthyl-Tröger’s base. Tetrahedron Asymmetry 1998, 9, 4151–4156. [Google Scholar] [CrossRef]
- Paleta, O.; Dolenský, B.; Paleček, J.; Kvíčala, J. Three-Component (Domino) Reaction Affording Substituted Pyrroloquinazolines: Cyclization Regioselectivity and Stereoselectivity. Eur. J. Org. Chem. 2013, 2013, 1262–1270. [Google Scholar] [CrossRef]
- Valenta, P. Oszillographische strom-spannungs-kurven III. Untersuchung des formaldehyds in gepuffertem milieu. Collect. Czech. Chem. Commun. 1960, 25, 853–861. [Google Scholar] [CrossRef]
- Albert, K.; Peters, B.; Bayer, E.; Treiber, U.; Zwilling, M. Crosslinking of Gelatin with Formaldehyde; a 13C NMR Study. Z. Naturforsch. B J. Chem. Sci. 1986, 41b, 351–358. [Google Scholar] [CrossRef]
- Cluşaru, A.; Crişan, I.; Kůta, J. Equilibrium constants and rates of dehydration of formaldehyde in buffered solutions of light and heavy water studied at DME. J. Electroanal. Chem. Interfacial Electrochem. 1973, 46, 51–62. [Google Scholar] [CrossRef]
- Walker, F. Some Properties of Anhydrous Formaldehyde. J. Am. Chem. Soc. 1933, 55, 2821–2826. [Google Scholar] [CrossRef]
- Lanza, P.A.; Dusso, D.; Ramírez, C.L.; Parise, A.R.; Chesta, C.A.; Moyano, E.L.; Vera, D.M.A. Uncovering the mechanism leading to the synthesis of symmetric and asymmetric Tröger’s bases. Eur. J. Org. Chem. 2019, 2019, 7644–7655. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Shubin, L. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Puiatti, M.; Vera, D.M.A.; Pierini, A.B. In search for an optimal methodology to calculate the valence electron affinities of temporary anions. Phys. Chem. Chem. Phys. 2009, 11, 9013–9024. [Google Scholar] [CrossRef]
- Borioni, L.; Puiatti, M.; Vera, D.M.A.; Pierini, A.B. In search of the best DFT functional for dealing with organic anionic species. Phys. Chem. Chem. Phys. 2017, 19, 9189–9198. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Möhlau, R.; Haase, O. Ueber Naphtacrihydridin. Ber. Dtsch. Chem. Ges. 1902, 35, 4164–4172. [Google Scholar] [CrossRef]
- Wagner, E.C. A rationalization of acid-induced reactions of methylene-bis-amines, methylene-amines, and of formaldehyde and amines. J. Org. Chem. 1954, 19, 1863–1881. [Google Scholar] [CrossRef]
- Mahon, A.B.; Craig, D.C.; Try, A.C. Synthesis of 5,6,11,12-tetrahydrodibenzo[b,f][1,5]diazocines and a demonstration of their reactivity to afford methano strap-modified Tröger’s base analogues. ARKIVOC 2008, 13, 148–163. [Google Scholar] [CrossRef]
- Artacho, J.; Ascic, E.; Rantanen, T.; Karlsson, J.; Wallentin, C.-J.; Wang, R.; Wendt, O.F.; Harmata, M.; Snieckus, V.; Wärnmark, K. Twisted Amide Analogues of Tröger’s Base. Chem. Eur. J. 2012, 18, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, N.G.; Kadutskii, A.P. Synthesis of Azaphenanthrene Derivatives by Condensation of 2-Methyleneaminonaphthalene with Cyclic β-Diketones. Russ. J. Org. Chem. 2002, 38, 129–133. [Google Scholar] [CrossRef]
- Kadutskii, A.P.; Kozlov, N.G. One-stage synthesis of benzoacridine and benzophenanthroline functionalized derivatives. Russ. J. Org. Chem. 2006, 42, 748–751. [Google Scholar] [CrossRef]
- Kozlov, N.G.; Kadutskii, A.P.; Baranovskii, A.V. Condensation of 2-naphthylamine or N-benzyl-2-naphthylamines with formaldehyde and methyl 2,2-dimethyl-4,6-dioxocyclohexane-3-carboxylate. Russ. J. Org. Chem. 2012, 48, 1456–1463. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. Chem. Phys. 2010, 132, 114110–114115. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, G. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Colomer, J.P.; Sciú, M.-L.; Ramirez, C.L.; Vera, D.M.A.; Moyano, E.L. Thermal ring opening of pyrazolo[3,4-d][1,2,3]triazin-4-ones: An experimental and theoretical study. Eur. J. Org. Chem. 2018, 13, 1514–1524. [Google Scholar] [CrossRef]

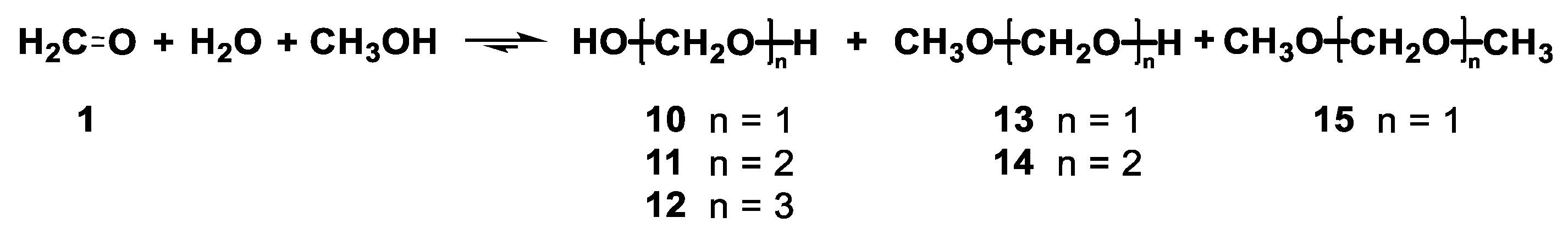


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature | Methanal (1) | Methandiol (10) | Hemiacetal 13 |
|---|---|---|---|
| 25 °C | 3 | 69 | 28 |
| 50 °C | 6 | 59 | 35 |
| 75 °C | 13 | 47 | 40 |
| 100 °C | 40 | 31 | 29 |
| 115 °C | 58 | 23 | 19 |
| Temperature | Methanal (1) | Methandiol (10) | Diol 11 | Diol 12 |
|---|---|---|---|---|
| 25 °C | 1 | 87 | 10 | 2 |
| 50 °C | 1 | 78 | 19 | 2 |
| 75 °C | 5 | 78 | 16 | 1 |
| 100 °C | 17 | 74 | 8 | 1 |
| 115 °C | 36 | 59 | 5 | 0 |
| Species | Gap HOMO/LUMO (eV) |
|---|---|
| methanal | 9.24 |
| ethanal | 9.57 |
| acetone | 9.59 |
| trifluoroethanal | 9.39 |
| hexafluoroacetone | 9.29 |
| Species | VIP (eV) | VEA (eV) | ω (eV) |
|---|---|---|---|
| methanal | 11.10 | −1.01 | 4.20 |
| ethanal | 10.46 | −1.60 | 3.25 |
| acetone | 9.95 | −1.47 | 2.87 |
| trifluoroethanal | 11.93 | −0.16 | 5.72 |
| ΔG°hyd (kcal/mol) | |
|---|---|
| methanal | −19.27 |
| ethanal | −13.93 |
| trifluoroethanal | −20.54 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Havlík, M.; Navrátilová, T.; Drozdová, M.; Tatar, A.; Lanza, P.A.; Dusso, D.; Moyano, E.L.; Chesta, C.A.; Vera, D.M.A.; Dolenský, B. Experimental, Spectroscopic, and Computational Insights into the Reactivity of “Methanal” with 2-Naphthylamines. Molecules 2023, 28, 1549. https://doi.org/10.3390/molecules28041549
Havlík M, Navrátilová T, Drozdová M, Tatar A, Lanza PA, Dusso D, Moyano EL, Chesta CA, Vera DMA, Dolenský B. Experimental, Spectroscopic, and Computational Insights into the Reactivity of “Methanal” with 2-Naphthylamines. Molecules. 2023; 28(4):1549. https://doi.org/10.3390/molecules28041549
Chicago/Turabian StyleHavlík, Martin, Tereza Navrátilová, Michaela Drozdová, Ameneh Tatar, Priscila A. Lanza, Diego Dusso, Elizabeth Laura Moyano, Carlos A. Chesta, Domingo Mariano A. Vera, and Bohumil Dolenský. 2023. "Experimental, Spectroscopic, and Computational Insights into the Reactivity of “Methanal” with 2-Naphthylamines" Molecules 28, no. 4: 1549. https://doi.org/10.3390/molecules28041549
APA StyleHavlík, M., Navrátilová, T., Drozdová, M., Tatar, A., Lanza, P. A., Dusso, D., Moyano, E. L., Chesta, C. A., Vera, D. M. A., & Dolenský, B. (2023). Experimental, Spectroscopic, and Computational Insights into the Reactivity of “Methanal” with 2-Naphthylamines. Molecules, 28(4), 1549. https://doi.org/10.3390/molecules28041549