

Molecular Toxicity Virtual Screening Applying a Quantized Computational SNN-Based Framework



Abstract
1. Introduction
2. Results
2.1. Classification Outcomes
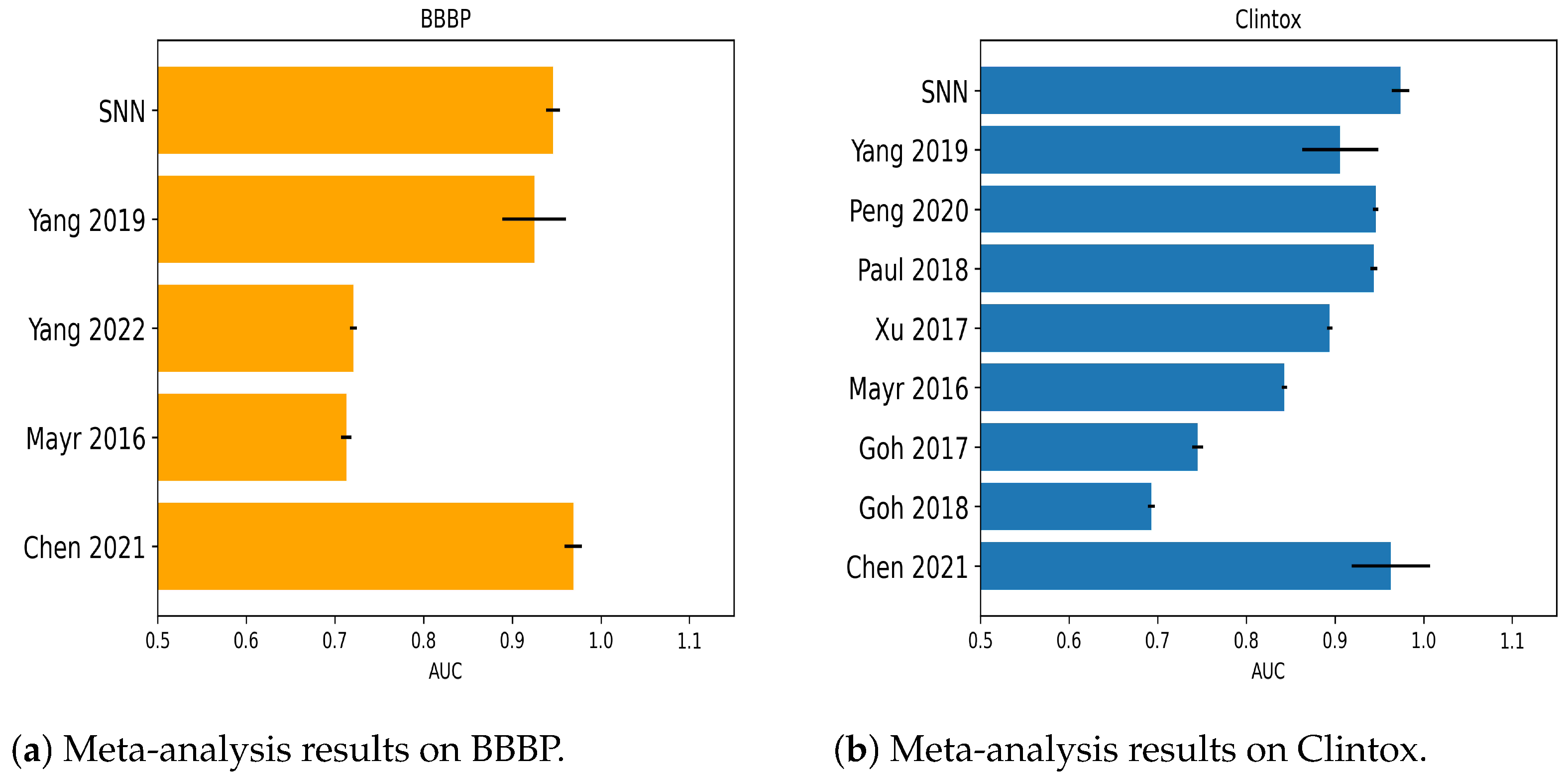
2.1.1. Numerical Experiments on Clintox
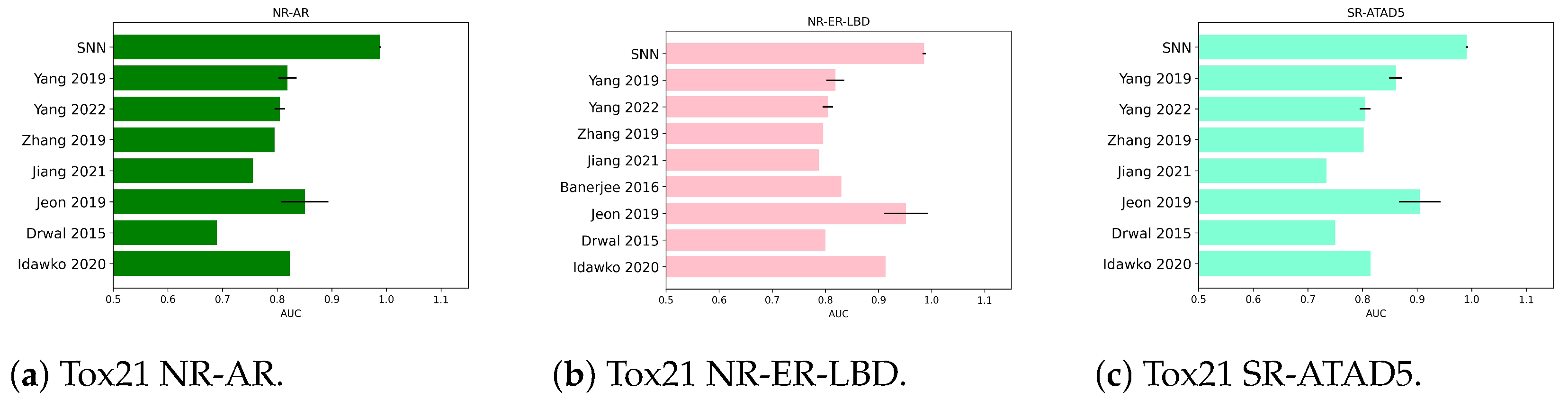
2.1.2. Numerical Experiments on Tox21 NR-AR
2.1.3. Numerical Experiments on Tox21 NR-ER-LBD
2.1.4. Numerical Experiments on Tox21 SR-ATAD5
2.1.5. Numerical Experiments on TOXCAST TR-LUC-GH3-Ant
2.1.6. Numerical Experiments on BBBP
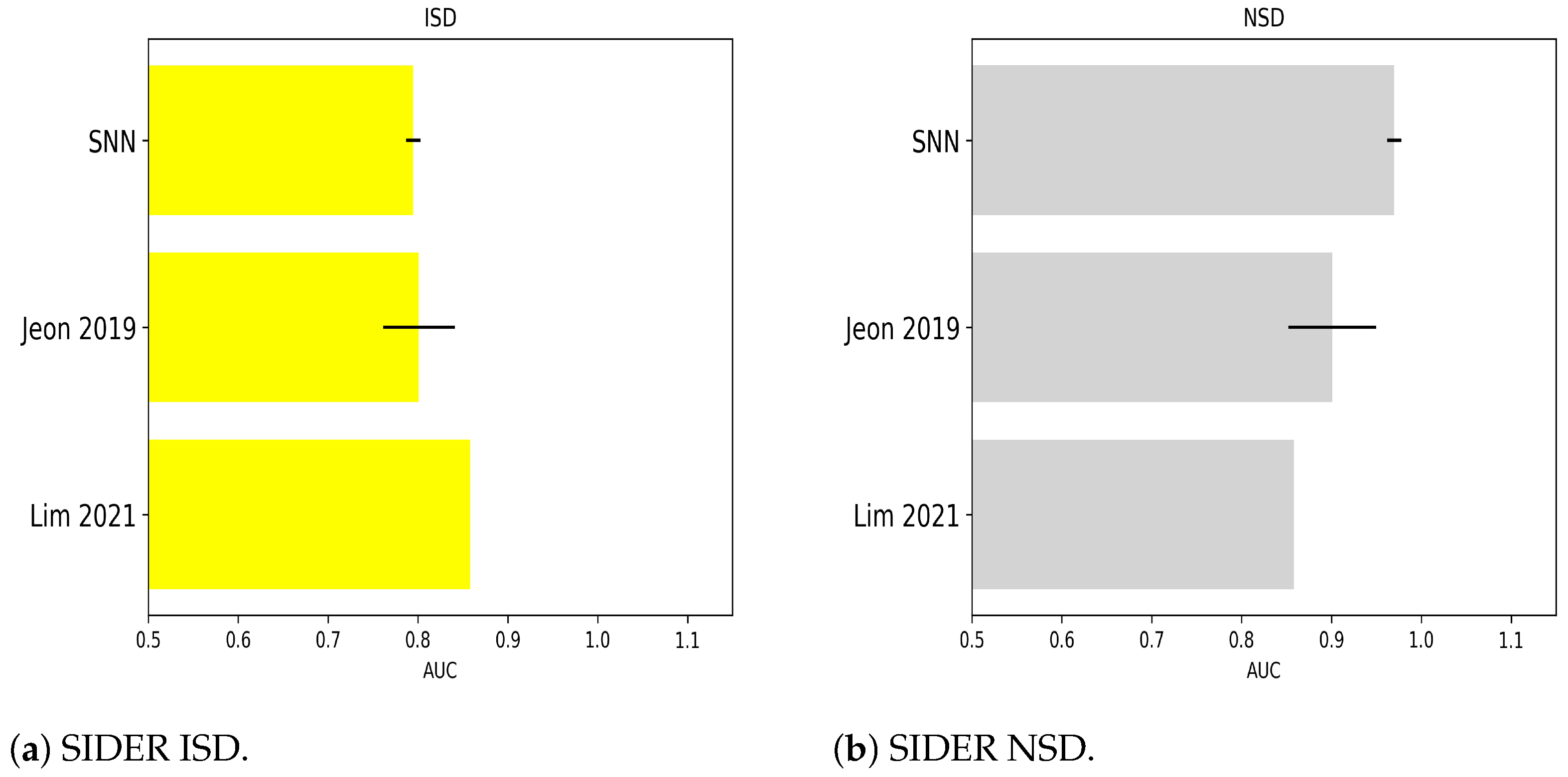
2.1.7. Numerical Experiments on SIDER ISD
2.1.8. Numerical Experiments on SIDER NSD
2.2. Hyperparameters Evaluation
2.3. Meta-Analysis
2.3.1. Previous Literature on Virtual Screenings from MFs
2.3.2. Previous Literature Applying More Sophisticated Data Representations
2.3.3. Positioning of SNN Results in the Current Body of Knowledge
2.4. Scalability of SNNs with Longer MF
3. Discussion
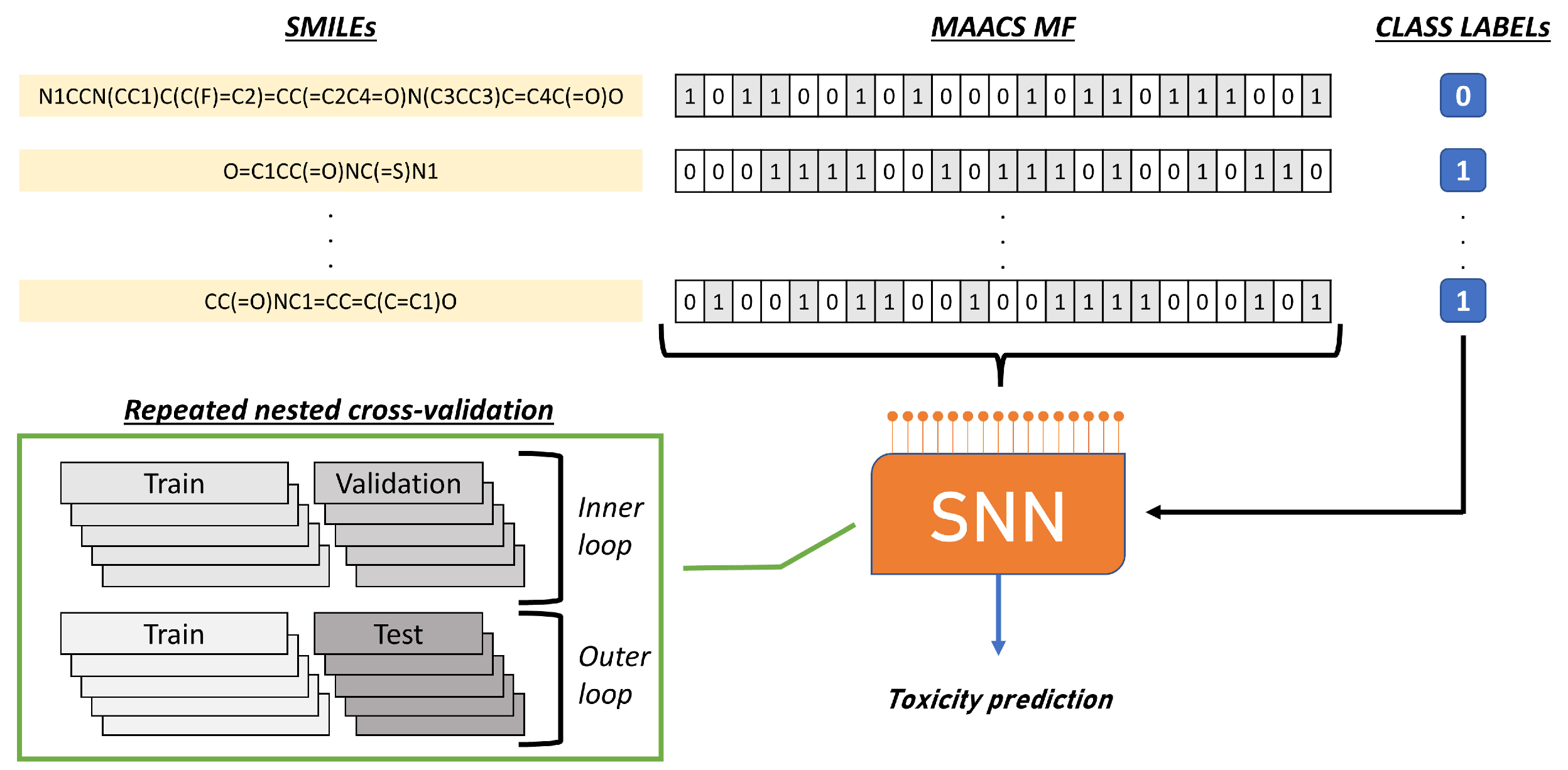
4. Materials and Methods
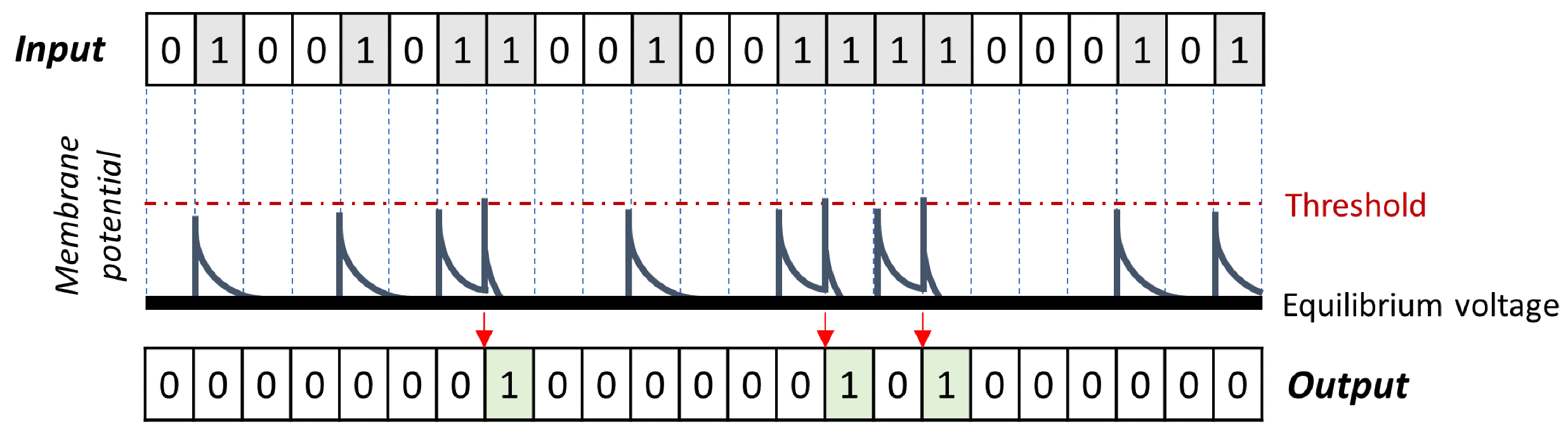
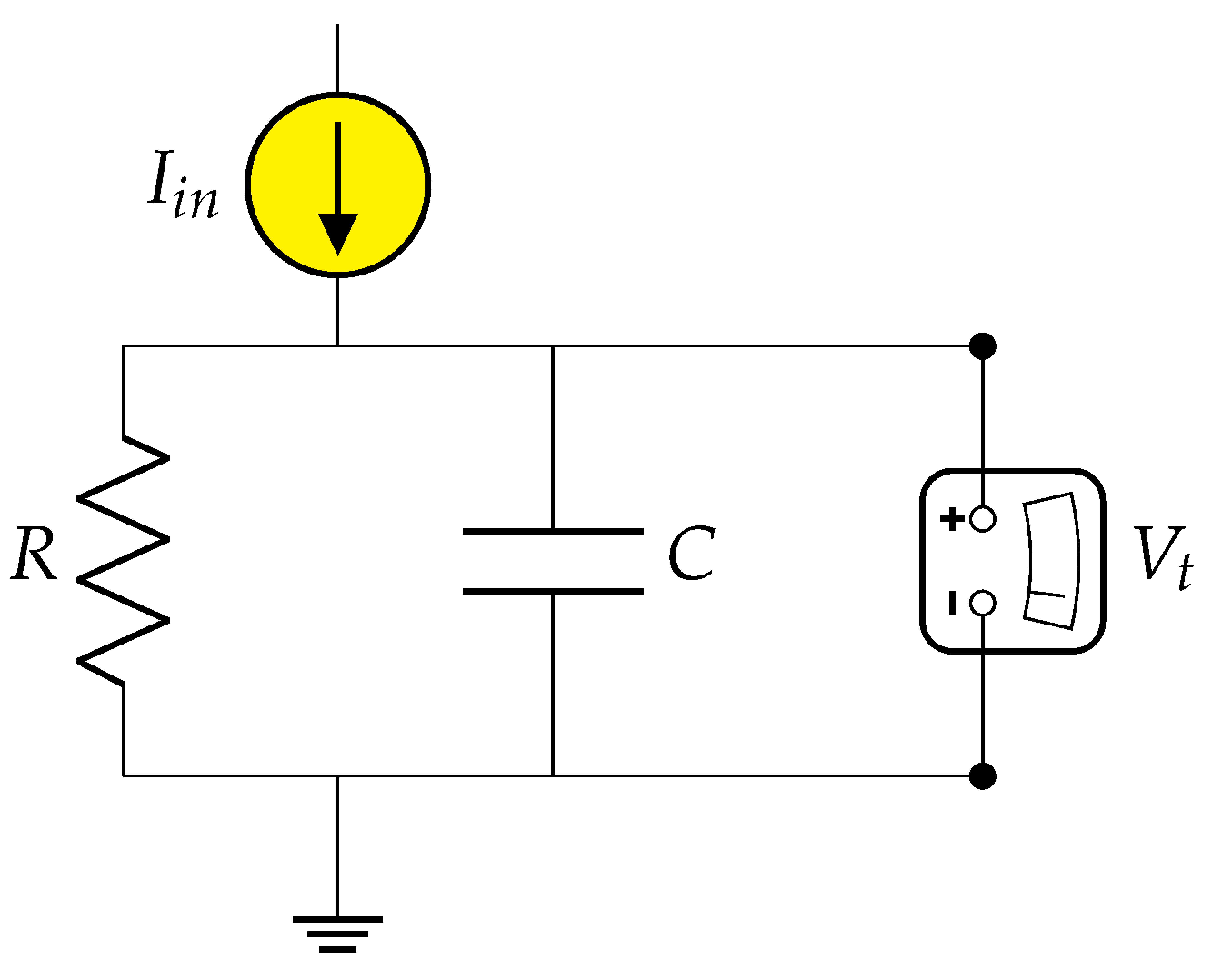
4.1. Neuron Model
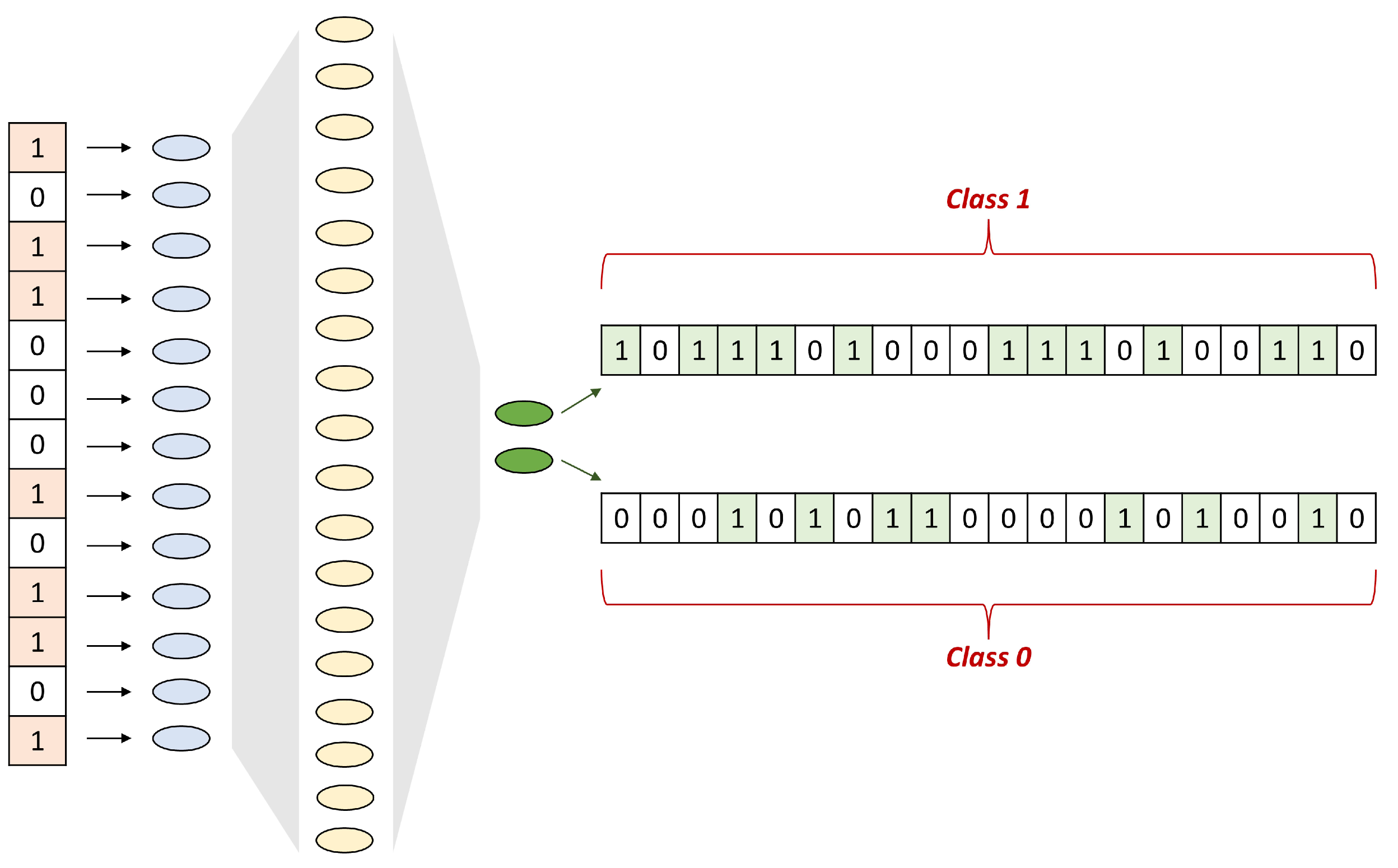
4.2. SNN Architecture
4.3. Model Evaluation and Hyperparameters Tuning
4.4. Benchmark Datasets Employed in the Study
- TOXCAST [83], containing results of in vitro toxicological experiments. In particular, the outcomes for “Tox21-TR-LUC-GH3-Antagonist” were considered due to the best sample ratio;
- Tox21 [84], predicting the toxicity on biological targets, including nuclear receptors or stress response pathways. Activities selected were “SR-ATAD5”, “NR-EL-LBD”, and “NR-AR” for the relatively low number of missing entries compared to the others inside the dataset;
- BBBP [85] assessing drug’s blood–brain barrier penetration;
- SIDER [86], employed for predicting drug’s side effects on the immune and nervous systems;
- Clintox [87], containing drugs that failed or passed clinical trials for toxicity.
4.5. Fingerprints Characteristics
4.6. Meta-Analysis Criteria
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| AUC | Area Under the ROC Curve |
| BA | Balanced Accuracy |
| CV | Cross-Validation |
| ECFPs | Extended-Connectivity Fingerprints |
| GC | Gradient Clipping |
| HPs | Hyperparameters |
| LIF | Leaky Integrate-and-Fire (neuron model) |
| LR | Learning Rate |
| MFs | Molecular Fingerprints |
| ML | Machine Learning |
| NN | Neural Network (other than SNN) |
| QSAR | Quantitative Structure–Activity Relationship |
| QSPR | Quantitative Structure–Property Relationship |
| RF | Random Forest |
| ROC | Receiver Operating Characteristic |
| SGO | Stochastic Gradient Optimizer |
| SMILES | Simplified Molecular-Input Line-Entry System |
| SNN | Spiking Neural Network |
| WD | Weight Decay |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Validation Set | Test Set |
|---|---|---|
| Clintox | 96.6 ± 0.5 | 97.8 ± 0.7 |
| NR-AR | 98.8 ± 0.4 | 98.3 ± 0.5 |
| NR-ER-LBD | 97.8 ± 0.4 | 98.5 ± 0.4 |
| SR-ATAD5 | 95.0 ± 0.6 | 99.0 ± 0.2 |
| BBBP | 92.7 ± 1.3 | 94.5 ± 1.1 |
| TOXCAST | 89.0 ± 1.0 | 91.4 ± 0.9 |
| NSD | 95.8 ± 1.1 | 97.0 ± 0.8 |
| ISD | 78.5 ± 2.3 | 81.8 ± 2.0 |
Appendix B
| Benchmark | Single | Multiple |
|---|---|---|
| ISD | 0.670 ± 0.038 | 0.801 ± 0.040 |
| NSD | 0.820 ± 0.083 | 0.901 ± 0.049 |
| NR-AR | 0.810 ± 0.027 | 0.851 ± 0.043 |
| NR-ER-LBD | 0.898 ± 0.040 | 0.952 ± 0.041 |
| SR-ATAD5 | 0.844 ± 0.033 | 0.905 ± 0.038 |
| BBBP | 0.713 ± 0.006 |
| Method | Ref. | Clintox |
|---|---|---|
| TOP | [49] | 0.946 ± 0.003 |
| CheMixNet | [53] | 0.944 ± 0.004 |
| DeepAOT | [54] | 0.894 ± 0.003 |
| DeepTox | [46] | 0.843 ± 0.003 |
| Chemception | [55] | 0.745 ± 0.006 |
| SMILES2Vec | [56] | 0.693 ± 0.004 |
| RF | 0.769 ± 0.002 | |
| SVM | 0.751 ± 0.002 | |
| KNN 1 | 0.698 ± 0.003 |
| Benchmark | D-MPNN 1 | D-MPNN (Ensemble) |
|---|---|---|
| BBBP | 0.913 ± 0.026 | 0.925 ± 0.036 |
| Tox21 | 0.845 ± 0.015 | 0.861 ± 0.012 |
| SIDER | 0.646 ± 0.016 | 0.664 ± 0.021 |
| Clintox | 0.894 ± 0.027 | 0.906 ± 0.043 |
| MF Type | Features | BBBP | Clintox |
|---|---|---|---|
| SSLP-FPs | C [51] | 0.949 ± 0.016 | 0.963 ± 0.044 |
| SSLP-FPs | CP [51] | 0.953 ± 0.009 | 0.939 ± 0.047 |
| SSLP-FPs | CPZ [51] | 0.946 ± 0.015 | 0.941 ± 0.035 |
| Auto-FPs | 0.969 ± 0.01 | 0.95 ± 0.037 | |
| ECFC2 | 1024 bits | 0.92 ± 0.015 | 0.821 ± 0.058 |
| ECFC2 | 2048 bits | 0.919 ± 0.021 | 0.833 ± 0.053 |
| ECFC2 | 512 bits | 0.913 ± 0.019 | 0.831 ± 0.056 |
| ECFC4 | 1024 bits | 0.914 ± 0.024 | 0.782 ± 0.052 |
| ECFC4 | 2048 bits | 0.916 ± 0.021 | 0.784 ± 0.053 |
| ECFC4 | 512 bits | 0.908 ± 0.025 | 0.801 ± 0.049 |
| ECFC6 | 1024 bits | 0.907 ± 0.029 | 0.77 ± 0.054 |
| ECFC6 | 2048 bits | 0.911 ± 0.026 | 0.75 ± 0.059 |
| ECFC6 | 512 bits | 0.9 ± 0.032 | 0.77 ± 0.048 |
Appendix C
| Dataset | Computational Time (s) | Training Epochs |
|---|---|---|
| Clintox | 4109.55 | 300 |
| BBBP | 1522.74 | 100 |
References
- Mitchell, J.B. Machine learning methods in chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Front. Pharmacol. 2018, 9, 1275. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Polishchuk, P.G.; Muratov, E.N.; Artemenko, A.G.; Kolumbin, O.G.; Muratov, N.N.; Kuz’min, V.E. Application of random forest approach to QSAR prediction of aquatic toxicity. J. Chem. Inf. Model. 2009, 49, 2481–2488. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Singh, S.; Singla, D.; Agarwal, S.M.; Raghava, G.P. QSAR based model for discriminating EGFR inhibitors and non-inhibitors using Random forest. Biol. Direct 2015, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Motamedi, F.; Pérez-Sánchez, H.; Mehridehnavi, A.; Fassihi, A.; Ghasemi, F. Accelerating big data analysis through LASSO-random forest algorithm in QSAR studies. Bioinformatics 2022, 38, 469–475. [Google Scholar] [CrossRef]
- Ancuceanu, R.; Dinu, M.; Neaga, I.; Laszlo, F.G.; Boda, D. Development of QSAR machine learning-based models to forecast the effect of substances on malignant melanoma cells. Oncol. Lett. 2019, 17, 4188–4196. [Google Scholar] [CrossRef]
- Doucet, J.P.; Barbault, F.; Xia, H.; Panaye, A.; Fan, B. Nonlinear SVM approaches to QSPR/QSAR studies and drug design. Curr. Comput. -Aided Drug Des. 2007, 3, 263–289. [Google Scholar] [CrossRef]
- Czermiński, R.; Yasri, A.; Hartsough, D. Use of support vector machine in pattern classification: Application to QSAR studies. Quant. Struct. -Act. Relatsh. 2001, 20, 227–240. [Google Scholar] [CrossRef]
- Chavan, S.; Nicholls, I.A.; Karlsson, B.C.; Rosengren, A.M.; Ballabio, D.; Consonni, V.; Todeschini, R. Towards global QSAR model building for acute toxicity: Munro database case study. Int. J. Mol. Sci. 2014, 15, 18162–18174. [Google Scholar] [CrossRef] [PubMed]
- Konovalov, D.A.; Coomans, D.; Deconinck, E.; Vander Heyden, Y. Benchmarking of QSAR models for blood–brain barrier permeation. J. Chem. Inf. Model. 2007, 47, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.B.; Hodas, N.O.; Vishnu, A. Deep learning for computational chemistry. J. Comput. Chem. 2017, 38, 1291–1307. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, F. The perceptron: A probabilistic model for information storage and organization in the brain. Psychol. Rev. 1958, 65, 386. [Google Scholar] [CrossRef]
- Maass, W.; Bishop, C.M. Pulsed Neural Networks; MIT Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Marković, D.; Mizrahi, A.; Querlioz, D.; Grollier, J. Physics for neuromorphic computing. Nat. Rev. Phys. 2020, 2, 499–510. [Google Scholar] [CrossRef]
- Roy, K.; Jaiswal, A.; Panda, P. Towards spike-based machine intelligence with neuromorphic computing. Nature 2019, 575, 607–617. [Google Scholar] [CrossRef]
- Gerum, R.C.; Schilling, A. Integration of leaky-integrate-and-fire neurons in standard machine learning architectures to generate hybrid networks: A surrogate gradient approach. Neural Comput. 2021, 33, 2827–2852. [Google Scholar] [CrossRef] [PubMed]
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R. Dragon software: An easy approach to molecular descriptor calculations. Match 2006, 56, 237–248. [Google Scholar]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminformatics 2018, 10, 1–14. [Google Scholar] [CrossRef]
- Karpov, P.; Godin, G.; Tetko, I.V. Transformer-CNN: Swiss knife for QSAR modeling and interpretation. J. Cheminformatics 2020, 12, 1–12. [Google Scholar] [CrossRef]
- Hu, S.; Chen, P.; Gu, P.; Wang, B. A Deep Learning-Based Chemical System for QSAR Prediction. IEEE J. Biomed. Health Inform. 2020, 24, 3020–3028. [Google Scholar] [CrossRef] [PubMed]
- Atz, K.; Grisoni, F.; Schneider, G. Geometric deep learning on molecular representations. Nat. Mach. Intell. 2021, 3, 1023–1032. [Google Scholar] [CrossRef]
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. In Proceedings of the International Conference on Machine Learning, PMLR, Stockholm, Sweden, 10–15 July 2018; pp. 2323–2332. [Google Scholar]
- Xu, K.; Hu, W.; Leskovec, J.; Jegelka, S. How powerful are graph neural networks? arXiv 2018, arXiv:1810.00826. [Google Scholar]
- Fung, V.; Zhang, J.; Juarez, E.; Sumpter, B.G. Benchmarking graph neural networks for materials chemistry. Npj Comput. Mater. 2021, 7, 1–8. [Google Scholar] [CrossRef]
- Chen, D.; Lin, Y.; Li, W.; Li, P.; Zhou, J.; Sun, X. Measuring and relieving the over-smoothing problem for graph neural networks from the topological view. In Proceedings of the AAAI Conference on Artificial Intelligence, New York, NY, USA, 7–12 February 2020; AAAI Press: Palo Alto, CA, USA, 2020; Volume 34, pp. 3438–3445. [Google Scholar]
- Oono, K.; Suzuki, T. Graph neural networks exponentially lose expressive power for node classification. arXiv 2019, arXiv:1905.10947. [Google Scholar]
- Ying, C.; Cai, T.; Luo, S.; Zheng, S.; Ke, G.; He, D.; Shen, Y.; Liu, T.Y. Do transformers really perform badly for graph representation? Adv. Neural Inf. Process. Syst. 2021, 34, 28877–28888. [Google Scholar]
- Wu, Z.; Zhu, M.; Kang, Y.; Leung, E.L.H.; Lei, T.; Shen, C.; Jiang, D.; Wang, Z.; Cao, D.; Hou, T. Do we need different machine learning algorithms for QSAR modeling? A comprehensive assessment of 16 machine learning algorithms on 14 QSAR data sets. Brief. Bioinform. 2021, 22, bbaa321. [Google Scholar] [CrossRef]
- Triolascarya, K.; Septiawan, R.R.; Kurniawan, I. QSAR Study of Larvicidal Phytocompounds as Anti-Aedes Aegypti by using GA-SVM Method. J. RESTI (Rekayasa Sist. Dan Teknol. Inf.) 2022, 6, 632–638. [Google Scholar] [CrossRef]
- Rahmani, N.; Abbasi-Radmoghaddam, Z.; Riahi, S.; Mohammadi-Khanaposhtanai, M. Predictive QSAR models for the anti-cancer activity of topoisomerase IIα catalytic inhibitors against breast cancer cell line HCT15: GA-MLR and LS-SVM modeling. Struct. Chem. 2020, 31, 2129–2145. [Google Scholar] [CrossRef]
- Wigh, D.S.; Goodman, J.M.; Lapkin, A.A. A review of molecular representation in the age of machine learning. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1603. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef]
- Daylight Toolkit. SMARTS-A Language for Describing Molecular Patterns; Daylight Chemical Information Systems Inc.: Aliso Viejo, CA, USA, 2007. [Google Scholar]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Dastidar, S.; Adeli, H. Spiking neural networks. Int. J. Neural Syst. 2009, 19, 295–308. [Google Scholar] [CrossRef]
- Carlson, K.D.; Nageswaran, J.M.; Dutt, N.; Krichmar, J.L. An efficient automated parameter tuning framework for spiking neural networks. Front. Neurosci. 2014, 8, 10. [Google Scholar] [CrossRef]
- Hassabis, D.; Kumaran, D.; Summerfield, C.; Botvinick, M. Neuroscience-inspired artificial intelligence. Neuron 2017, 95, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing learned molecular representations for property prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, Y.; Cheng, J.; Li, Y.; Liu, S.; Duan, Y.; Zhang, L.; Luo, S. An Ensemble Structure and Physiochemical (SPOC) Descriptor for Machine-Learning Prediction of Chemical Reaction and Molecular Properties. ChemPhysChem 2022, 23, e202200255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mucs, D.; Norinder, U.; Svensson, F. LightGBM: An effective and scalable algorithm for prediction of chemical toxicity–application to the Tox21 and mutagenicity data sets. J. Chem. Inf. Model. 2019, 59, 4150–4158. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, R.; Wei, G.W. GGL-Tox: Geometric graph learning for toxicity prediction. J. Chem. Inf. Model. 2021, 61, 1691–1700. [Google Scholar] [CrossRef]
- Banerjee, P.; Siramshetty, V.B.; Drwal, M.N.; Preissner, R. Computational methods for prediction of in vitro effects of new chemical structures. J. Cheminformatics 2016, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jeon, W.; Kim, D. FP2VEC: A new molecular featurizer for learning molecular properties. Bioinformatics 2019, 35, 4979–4985. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity prediction using deep learning. Front. Environ. Sci. 2016, 3, 80. [Google Scholar] [CrossRef]
- Drwal, M.N.; Siramshetty, V.B.; Banerjee, P.; Goede, A.; Preissner, R.; Dunkel, M. Molecular similarity-based predictions of the Tox21 screening outcome. Front. Environ. Sci. 2015, 3, 54. [Google Scholar] [CrossRef]
- Idakwo, G.; Thangapandian, S.; Luttrell, J.; Li, Y.; Wang, N.; Zhou, Z.; Hong, H.; Yang, B.; Zhang, C.; Gong, P. Structure–activity relationship-based chemical classification of highly imbalanced Tox21 datasets. J. Cheminformatics 2020, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zhang, Z.; Jiang, Q.; Guan, J.; Zhou, S. TOP: A deep mixture representation learning method for boosting molecular toxicity prediction. Methods 2020, 179, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Dai, B.; Song, L. Discriminative embeddings of latent variable models for structured data. In Proceedings of the International Conference on Machine Learning, PMLR, New York, NY, USA, 19–24 June 2016; pp. 2702–2711. [Google Scholar]
- Chen, D.; Zheng, J.; Wei, G.W.; Pan, F. Extracting Predictive Representations from Hundreds of Millions of Molecules. J. Phys. Chem. Lett. 2021, 12, 10793–10801. [Google Scholar] [CrossRef]
- Lim, S.; Lee, Y.O. Predicting chemical properties using self-attention multi-task learning based on SMILES representation. In Proceedings of the IEEE 2020 25th International Conference on Pattern Recognition (ICPR), Milan, Italy, 10–15 January 2021; pp. 3146–3153. [Google Scholar]
- Paul, A.; Jha, D.; Al-Bahrani, R.; Liao, W.k.; Choudhary, A.; Agrawal, A. Chemixnet: Mixed dnn architectures for predicting chemical properties using multiple molecular representations. arXiv 2018, arXiv:1811.08283. [Google Scholar]
- Xu, Y.; Pei, J.; Lai, L. Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J. Chem. Inf. Model. 2017, 57, 2672–2685. [Google Scholar] [CrossRef]
- Goh, G.B.; Siegel, C.; Vishnu, A.; Hodas, N.O.; Baker, N. Chemception: A deep neural network with minimal chemistry knowledge matches the performance of expert-developed QSAR/QSPR models. arXiv 2017, arXiv:1706.06689. [Google Scholar]
- Goh, G.B.; Siegel, C.; Vishnu, A.; Hodas, N. Using rule-based labels for weak supervised learning: A ChemNet for transferable chemical property prediction. In Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, London, UK, 19–23 August 2018; pp. 302–310. [Google Scholar]
- Zou, X.; Xu, S.; Chen, X.; Yan, L.; Han, Y. Breaking the von Neumann bottleneck: Architecture-level processing-in-memory technology. Sci. China Inf. Sci. 2021, 64, 1–10. [Google Scholar] [CrossRef]
- Young, A.R.; Dean, M.E.; Plank, J.S.; Rose, G.S. A review of spiking neuromorphic hardware communication systems. IEEE Access 2019, 7, 135606–135620. [Google Scholar] [CrossRef]
- Kaiser, F.; Feldbusch, F. Building a bridge between spiking and artificial neural networks. In Proceedings of the International Conference on Artificial Neural Networks, Porto, Portugal, 9–13 September 2007; Springer: Berlin/Heidelberg, Germany, 2007; pp. 380–389. [Google Scholar]
- Rao, V.R.; Finkbeiner, S. NMDA and AMPA receptors: Old channels, new tricks. Trends Neurosci. 2007, 30, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bote, R.; Parga, N. Simple model neurons with AMPA and NMDA filters: Role of synaptic time scales. Neurocomputing 2005, 65, 441–448. [Google Scholar] [CrossRef]
- Rao, A.; Plank, P.; Wild, A.; Maass, W. A Long Short-Term Memory for AI Applications in Spike-based Neuromorphic Hardware. Nat. Mach. Intell. 2022, 4, 467–479. [Google Scholar] [CrossRef]
- Ruiz Puentes, P.; Valderrama, N.; González, C.; Daza, L.; Muñoz-Camargo, C.; Cruz, J.C.; Arbeláez, P. PharmaNet: Pharmaceutical discovery with deep recurrent neural networks. PLoS ONE 2021, 16, e0241728. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, A.N. A review of the integrate-and-fire neuron model: I. Homogeneous synaptic input. Biol. Cybern. 2006, 95, 1–19. [Google Scholar] [CrossRef]
- Fukutome, H.; Tamura, H.; Sugata, K. An electric analogue of the neuron. Kybernetik 1963, 2, 28–32. [Google Scholar] [CrossRef]
- Dayan, P.; Abbott, L.F. Theoretical Neuroscience: Computational and Mathematical Modeling of Neural Systems; MIT Press: Cambridge, MA, USA, 2005. [Google Scholar]
- Brunel, N.; Van Rossum, M.C. Lapicque’s 1907 paper: From frogs to integrate-and-fire. Biol. Cybern. 2007, 97, 337–339. [Google Scholar] [CrossRef]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. Pytorch: An imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. 2019, 32. [Google Scholar] [CrossRef]
- Eshraghian, J.K.; Ward, M.; Neftci, E.; Wang, X.; Lenz, G.; Dwivedi, G.; Bennamoun, M.; Jeong, D.S.; Lu, W.D. Training spiking neural networks using lessons from deep learning. arXiv 2021, arXiv:2109.12894. [Google Scholar]
- Oyedotun, O.K.; Papadopoulos, K.; Aouada, D. A new perspective for understanding generalization gap of deep neural networks trained with large batch sizes. Appl. Intell. 2022, 1–17. [Google Scholar] [CrossRef]
- Neftci, E.O.; Mostafa, H.; Zenke, F. Surrogate gradient learning in spiking neural networks: Bringing the power of gradient-based optimization to spiking neural networks. IEEE Signal Process. Mag. 2019, 36, 51–63. [Google Scholar] [CrossRef]
- Russell, A.; Orchard, G.; Dong, Y.; Mihalaş, Ş.; Niebur, E.; Tapson, J.; Etienne-Cummings, R. Optimization methods for spiking neurons and networks. IEEE Trans. Neural Netw. 2010, 21, 1950–1962. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Yantır, H.E.; Fouda, M.E.; Eltawil, A.M.; Salama, K.N. Toward the optimal design and FPGA implementation of spiking neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2021, 33, 3988–4002. [Google Scholar] [CrossRef]
- Manna, D.L.; Vicente-Sola, A.; Kirkland, P.; Bihl, T.; Di Caterina, G. Simple and complex spiking neurons: Perspectives and analysis in a simple STDP scenario. Neuromorphic Comput. Eng. 2022, 2, 044009. [Google Scholar] [CrossRef]
- Zhang, J.; He, T.; Sra, S.; Jadbabaie, A. Why gradient clipping accelerates training: A theoretical justification for adaptivity. arXiv 2019, arXiv:1905.11881. [Google Scholar]
- Krogh, A.; Hertz, J. A simple weight decay can improve generalization. Adv. Neural Inf. Process. Syst. 1991, 4, 950–957. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Sutskever, I.; Martens, J.; Dahl, G.; Hinton, G. On the importance of initialization and momentum in deep learning. In Proceedings of the International Conference on Machine Learning, PMLR, Atlanta, GA, USA, 16–21 June 2013; pp. 1139–1147. [Google Scholar]
- Duchi, J.; Hazan, E.; Singer, Y. Adaptive subgradient methods for online learning and stochastic optimization. J. Mach. Learn. Res. 2011, 12, 2121–2159. [Google Scholar]
- Zeiler, M.D. Adadelta: An adaptive learning rate method. arXiv 2012, arXiv:1212.5701. [Google Scholar]
- Loshchilov, I.; Hutter, F. Decoupled weight decay regularization. arXiv 2017, arXiv:1711.05101. [Google Scholar]
- Hinton, G.; Srivastava, N.; Swersky, K. Neural networks for machine learning lecture 6a overview of mini-batch gradient descent. Cited 2012, 14, 2. [Google Scholar]
- Judson, R.S.; Houck, K.A.; Kavlock, R.J.; Knudsen, T.B.; Martin, M.T.; Mortensen, H.M.; Reif, D.M.; Rotroff, D.M.; Shah, I.; Richard, A.M.; et al. In vitro screening of environmental chemicals for targeted testing prioritization: The ToxCast project. Environ. Health Perspect. 2010, 118, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.M.; Huang, R.; Waidyanatha, S.; Shinn, P.; Collins, B.J.; Thillainadarajah, I.; Grulke, C.M.; Williams, A.J.; Lougee, R.R.; Judson, R.S.; et al. The Tox21 10K compound library: Collaborative chemistry advancing toxicology. Chem. Res. Toxicol. 2020, 34, 189–216. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef]
- Gayvert, K.M.; Madhukar, N.S.; Elemento, O. A data-driven approach to predicting successes and failures of clinical trials. Cell Chem. Biol. 2016, 23, 1294–1301. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. Greg Landrum 2013, 8. [Google Scholar] [CrossRef]
- Swain, M. MolVS: Molecule Validation and Standardization. Available online: https://molvs.readthedocs.io/en/latest/ (accessed on 15 August 2022).
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminformatics 2015, 7, 1–13. [Google Scholar] [CrossRef]







| Hidden Neur. | Grad. Slope | Opt. | LR | WD | GC | Mean BA | Std BA | |
|---|---|---|---|---|---|---|---|---|
| 1000 | 0.8 | 75 | Adam | 0.0001 | 0 | True | 97.845 | 0.661 |
| 1000 | 0.65 | 50 | Adam | 1 × 10−5 | 0 | True | 97.663 | 0.605 |
| 1000 | 0.6 | 25 | Adam | 0.0001 | 0 | True | 97.669 | 0.317 |
| 1000 | 0.95 | 50 | Adam | 1 × 10−5 | 0 | True | 97.530 | 0.774 |
| 1000 | 0.8 | 25 | Adam | 0.0001 | 0 | False | 97.339 | 0.556 |
| Hidden Neur. | Grad. Slope | Opt. | LR | WD | GC | Mean BA | Std BA | |
|---|---|---|---|---|---|---|---|---|
| 1000 | 0.95 | 50 | Adagrad | 0.01 | 0 | False | 98.840 | 0.368 |
| 1000 | 0.95 | 50 | AdamW | 0.0001 | 0 | False | 98.829 | 0.232 |
| 1000 | 0.95 | 50 | Adamax | 0.002 | 0 | False | 98.823 | 0.369 |
| 1500 | 0.95 | 50 | Adam | 5 × 10−5 | 0 | False | 98.810 | 0.215 |
| 800 | 0.95 | 50 | Adam | 5 × 10−5 | 0 | False | 98.778 | 0.241 |
| Hidden Neur. | Grad. Slope | Opt. | LR | WD | GC | Mean BA | Std BA | |
|---|---|---|---|---|---|---|---|---|
| 1000 | 0.95 | 50 | Adamax | 0.002 | 0 | False | 98.491 | 0.399 |
| 1200 | 0.85 | 75 | Adam | 1 × 10−5 | 0 | False | 98.457 | 0.270 |
| 1000 | 0.95 | 50 | Adagrad | 0.01 | 0 | False | 98.456 | 0.268 |
| 1000 | 0.95 | 50 | Adam | 0.0001 | 0.001 | False | 98.354 | 0.373 |
| 1000 | 0.95 | 50 | SGO | 0.001 | 0 | False | 98.283 | 0.362 |
| Hidden Neur. | Grad. Slope | Opt. | LR | WD | GC | Mean BA | Std BA | |
|---|---|---|---|---|---|---|---|---|
| 1200 | 0.85 | 75 | Adam | 1 × 10−5 | 0 | False | 99.055 | 0.232 |
| 1000 | 0.95 | 50 | Adam | 0.0001 | 0.001 | False | 98.837 | 0.257 |
| 1000 | 0.95 | 50 | Adagrad | 0.01 | 0 | False | 98.818 | 0.182 |
| 1500 | 0.95 | 50 | SGO | 0.005 | 0.001 | False | 98.798 | 0.221 |
| 1000 | 0.95 | 50 | Adam | 0.0001 | 0 | False | 98.795 | 0.251 |
| Hidden Neur. | Grad. Slope | Opt. | LR | WD | GC | Mean BA | Std BA | |
|---|---|---|---|---|---|---|---|---|
| 1000 | 0.95 | 50 | Adamax | 0.002 | 0 | False | 91.379 | 0.819 |
| 1000 | 0.95 | 50 | Adamax | 0.002 | 0.0001 | False | 91.358 | 0.417 |
| 1200 | 0.95 | 50 | Adamax | 0.002 | 0 | False | 91.190 | 0.877 |
| 1000 | 0.95 | 50 | Adagrad | 0.01 | 0.001 | False | 91.166 | 0.391 |
| 1200 | 0.95 | 50 | Adagrad | 0.01 | 0 | False | 91.138 | 0.699 |
| Hidden Neur. | Grad. Slope | Opt. | LR | WD | GC | Mean BA | Std BA | |
|---|---|---|---|---|---|---|---|---|
| 1000 | 0.95 | 50 | Adamax | 0.002 | 0 | False | 94.481 | 1.068 |
| 1000 | 0.95 | 50 | SGO | 0.005 | 0 | False | 94.230 | 1.342 |
| 1000 | 0.95 | 50 | RMSProp | 0.001 | 0 | False | 93.883 | 1.434 |
| 1000 | 0.8 | 25 | Adam | 0.0001 | 0 | False | 93.838 | 0.696 |
| 1000 | 0.95 | 50 | Adam | 0.0001 | 0 | False | 93.651 | 1.005 |
| Hidden Neur. | Grad. Slope | Opt. | LR | WD | GC | Mean BA | Std BA | |
|---|---|---|---|---|---|---|---|---|
| 1000 | 0.95 | 50 | Adagrad | 0.01 | 0 | False | 81.750 | 2.027 |
| 1500 | 0.95 | 50 | SGO | 0.005 | 0.001 | False | 81.376 | 1.998 |
| 1000 | 0.95 | 50 | SGO | 0.001 | 0 | False | 81.254 | 3.243 |
| 2000 | 0.95 | 50 | Adagrad | 0.01 | 0 | False | 80.981 | 1.905 |
| 1000 | 0.8 | 25 | Adagrad | 0.01 | 0 | False | 80.937 | 1.721 |
| Hidden Neur. | Grad. Slope | Opt. | LR | WD | GC | Mean BA | Std BA | |
|---|---|---|---|---|---|---|---|---|
| 1000 | 0.95 | 50 | SGO | 0.001 | 0.001 | False | 96.978 | 0.740 |
| 1000 | 0.95 | 50 | Adadelta | 1.0 | 0 | False | 96.520 | 1.197 |
| 1000 | 0.95 | 50 | Adagrad | 0.01 | 0 | False | 96.515 | 1.211 |
| 1000 | 0.95 | 50 | AdamW | 1 × 10−5 | 0 | True | 96.450 | 0.840 |
| 1000 | 0.95 | 50 | Adamax | 0.002 | 0 | False | 96.374 | 0.697 |
| Dataset | Hidden Neur. | Grad. Slope | Mean BA | Std BA | Mean AUC | Std AUC | |
|---|---|---|---|---|---|---|---|
| BBBP | 1000 | 0.95 | 50 | 94.481 | 1.068 | 0.946 | 0.008 |
| Clintox | 1000 | 0.8 | 75 | 97.845 | 0.661 | 0.974 | 0.01 |
| ISD | 1000 | 0.95 | 50 | 81.750 | 2.027 | 0.795 | 0.008 |
| NR-AR | 1000 | 0.95 | 50 | 98.840 | 0.368 | 0.988 | 0.002 |
| NR-ER-LBD | 1000 | 0.95 | 50 | 98.491 | 0.399 | 0.986 | 0.003 |
| NSD | 1000 | 0.95 | 50 | 96.977 | 0.740 | 0.97 | 0.008 |
| SR-ATAD5 | 1200 | 0.85 | 75 | 99.055 | 0.232 | 0.991 | 0.002 |
| TOXCAST | 1000 | 0.95 | 50 | 91.379 | 0.819 | 0.912 | 0.007 |
| MF Type | BBBP | Clintox |
|---|---|---|
| ECFP | 93.451 ± 0.684 | 97.772 ± 0.522 |
| MAACS | 94.481 ± 1.068 | 97.845 ± 0.661 |
| Hyperparameter | Lower Limit | Upper Limit | Levels |
|---|---|---|---|
| Hidden neurons | 500 | 2000 | 5 |
| 0.6 | 0.95 | 6 | |
| Grad. slope | 25 | 75 | 3 |
| LR | 1 × 10−5 | 0.5 | 15 |
| WD | 0.001 | 0.05 | 4 |
| Dataset | Activity Studied | Acronym | Instances |
|---|---|---|---|
| Clintox | Toxicity in clinical trials | Clintox | 1366 (1366)–112 (1366) |
| Tox21 | Androgen receptor nipple retention | NR-AR | 6956 (6956)–309 (6956) |
| Tox21 | Hepatotoxicity | NR-ER-LBD | 6605 (6605)–350 (6605) |
| Tox21 | DNA damage | SR-ATAD5 | 6808 (6808)–264 (6808) |
| TOXCAST | Thyroid homeostasis disruption | TR-LUC-GH3-Ant 1 | 6170 (6170)–1761 (6170) |
| BBBP | Blood–brain barrier permeability | BBBP | 479 (1560)–1560 (1560) |
| SIDER | Immune system disorders (iatrogenic toxicity) | ISD | 403 (1024)–1024 (1024) |
| SIDER | Nervous system disorders (iatrogenic toxicity) | NSD | 123 (1304)–1304 (1304) |
| Dataset | Tanimoto Index |
|---|---|
| Clintox | 0.315 |
| NR-AR | 0.21 |
| NR-ER-LBD | 0.209 |
| SR-ATAD5 | 0.208 |
| TR-LUC-GH3-Ant | 0.211 |
| BBBP | 0.335 |
| ISD | 0.308 |
| NSD | 0.308 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nascimben, M.; Rimondini, L. Molecular Toxicity Virtual Screening Applying a Quantized Computational SNN-Based Framework. Molecules 2023, 28, 1342. https://doi.org/10.3390/molecules28031342
Nascimben M, Rimondini L. Molecular Toxicity Virtual Screening Applying a Quantized Computational SNN-Based Framework. Molecules. 2023; 28(3):1342. https://doi.org/10.3390/molecules28031342
Chicago/Turabian StyleNascimben, Mauro, and Lia Rimondini. 2023. "Molecular Toxicity Virtual Screening Applying a Quantized Computational SNN-Based Framework" Molecules 28, no. 3: 1342. https://doi.org/10.3390/molecules28031342
APA StyleNascimben, M., & Rimondini, L. (2023). Molecular Toxicity Virtual Screening Applying a Quantized Computational SNN-Based Framework. Molecules, 28(3), 1342. https://doi.org/10.3390/molecules28031342