

In Silico Screening of Natural Flavonoids against 3-Chymotrypsin-like Protease of SARS-CoV-2 Using Machine Learning and Molecular Modeling
 , , ,
, , , 
Abstract
:1. Introduction
2. Results and Discussion
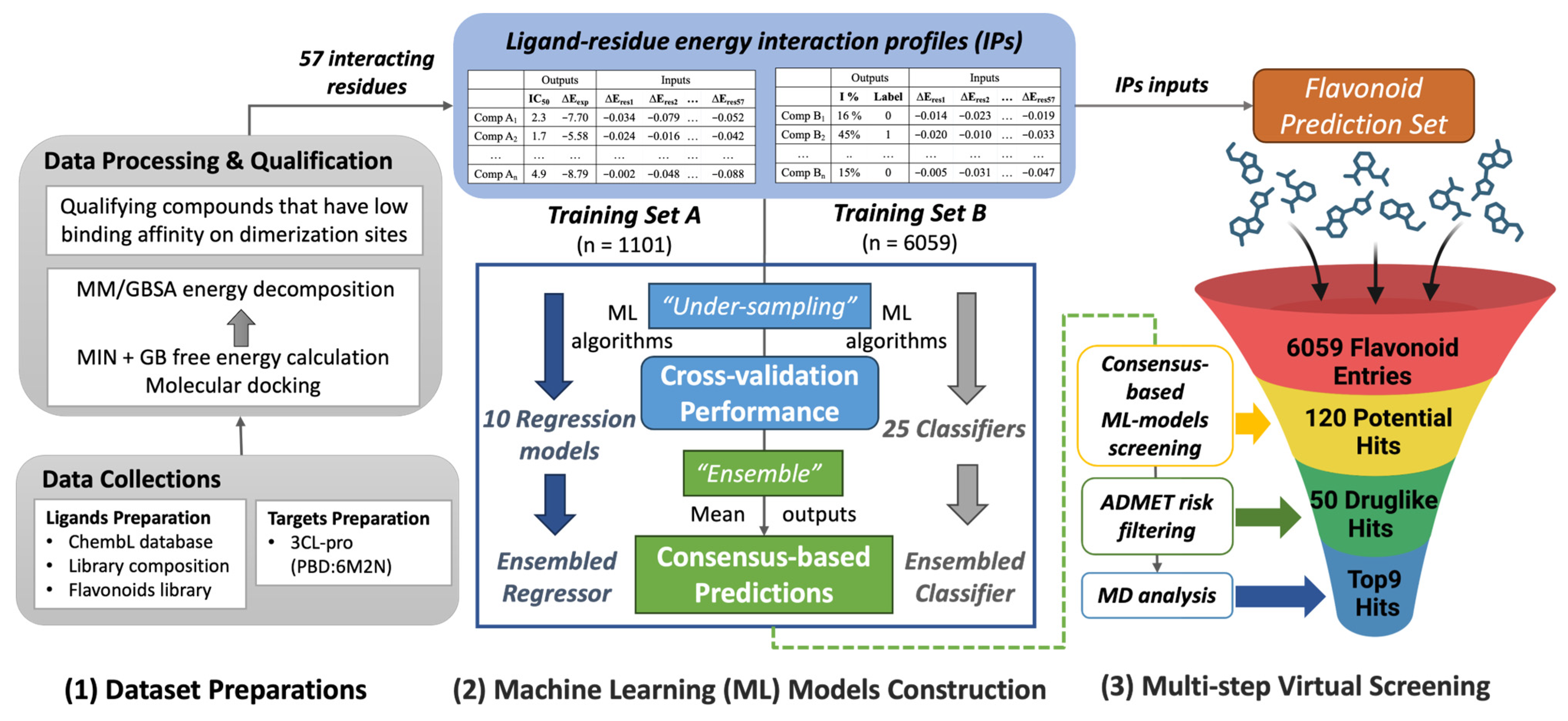
2.1. Dataset Preparation
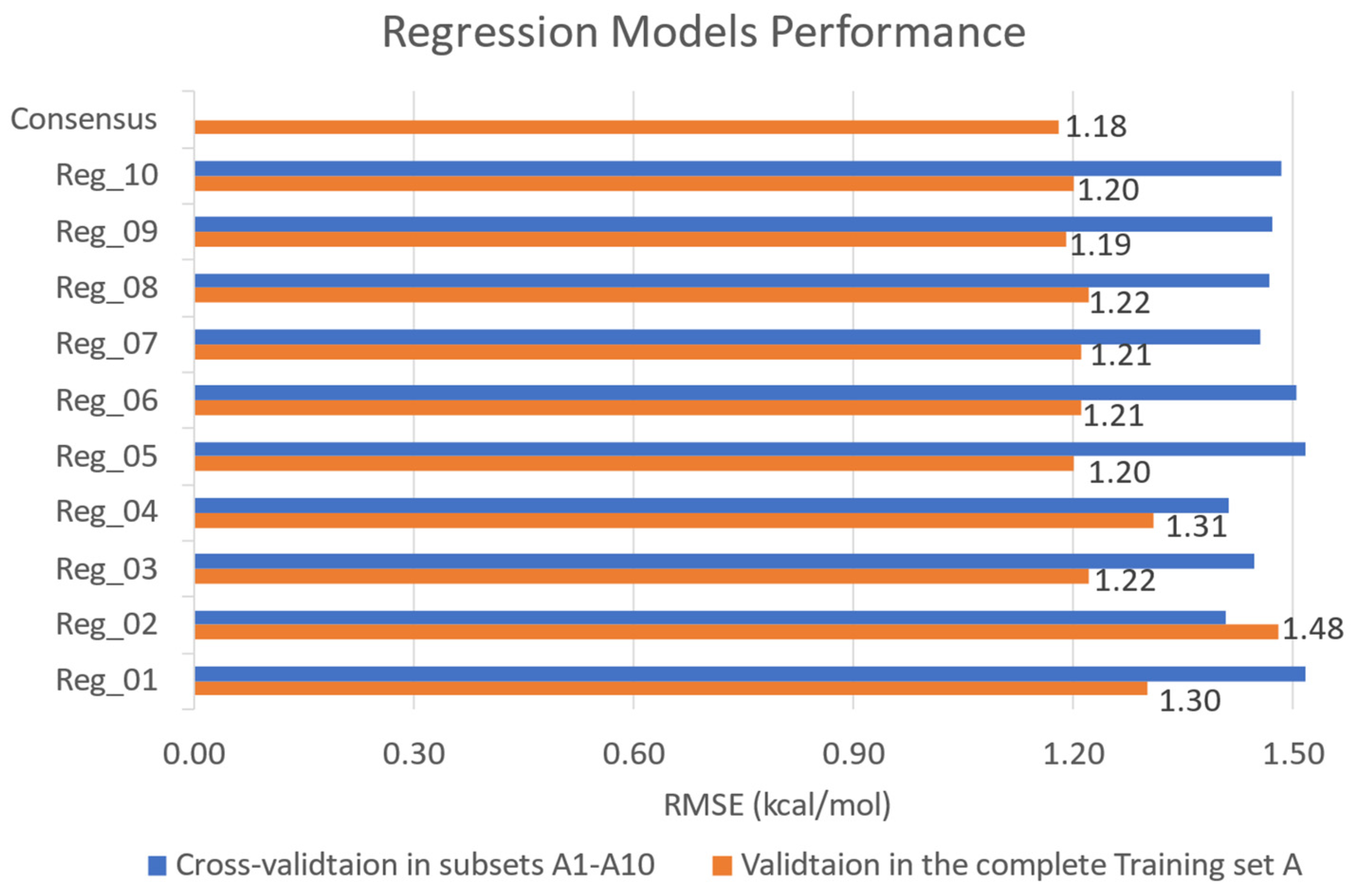
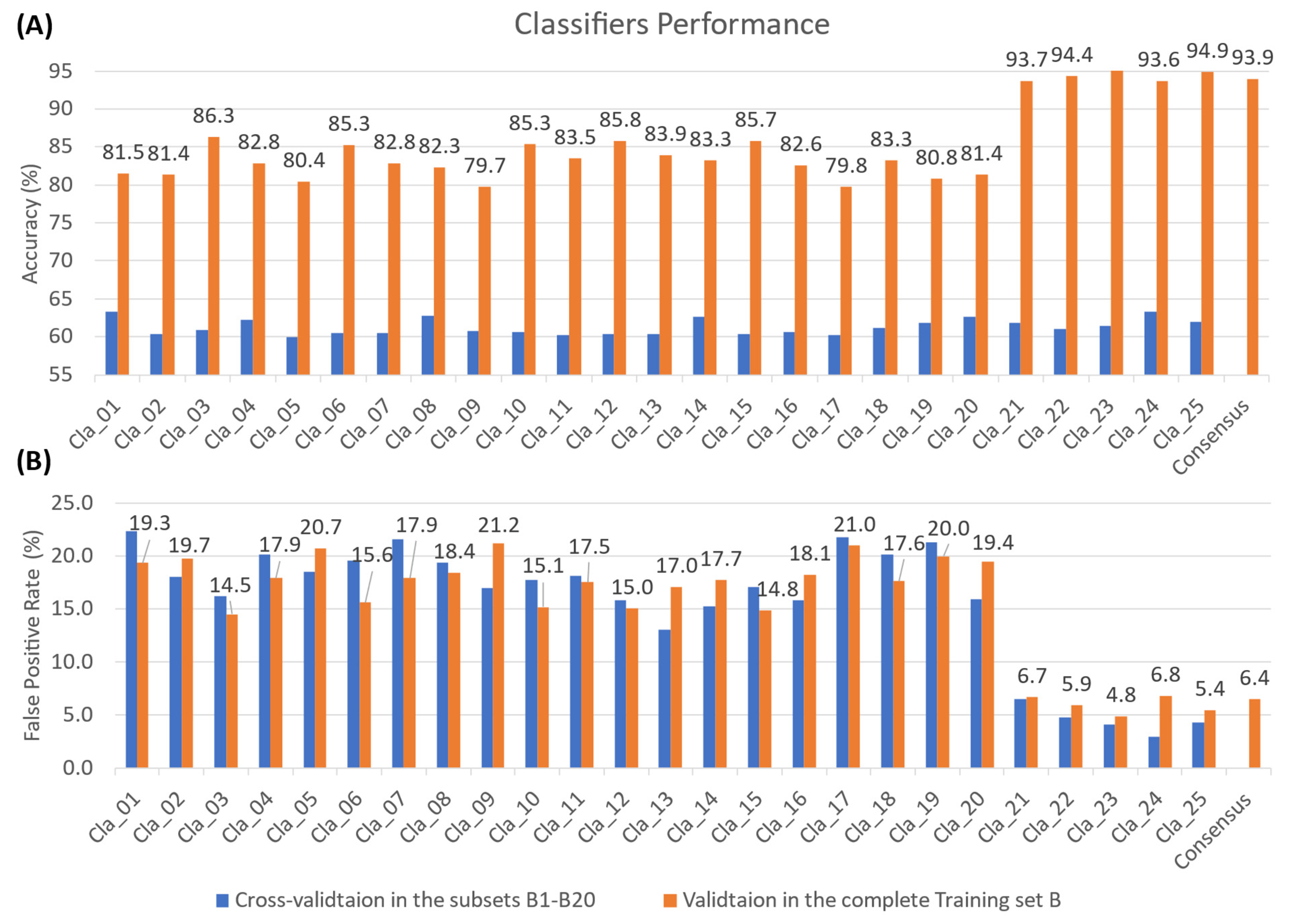
2.2. ML-Based Model Performance
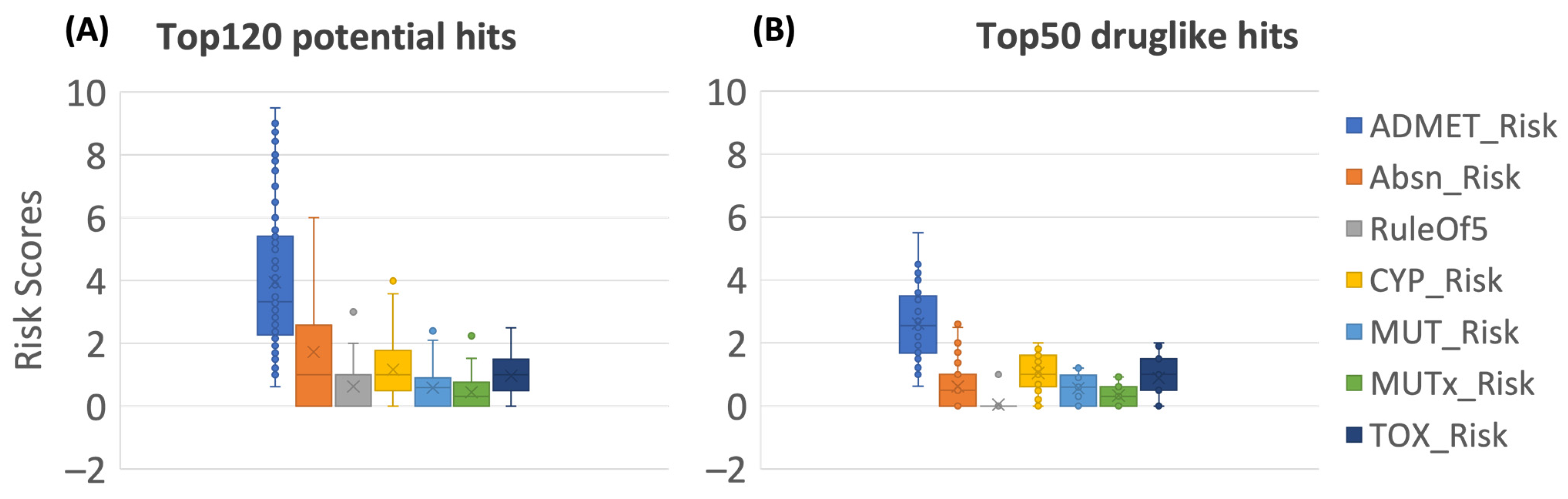
2.3. Flavonoid Hits Screening
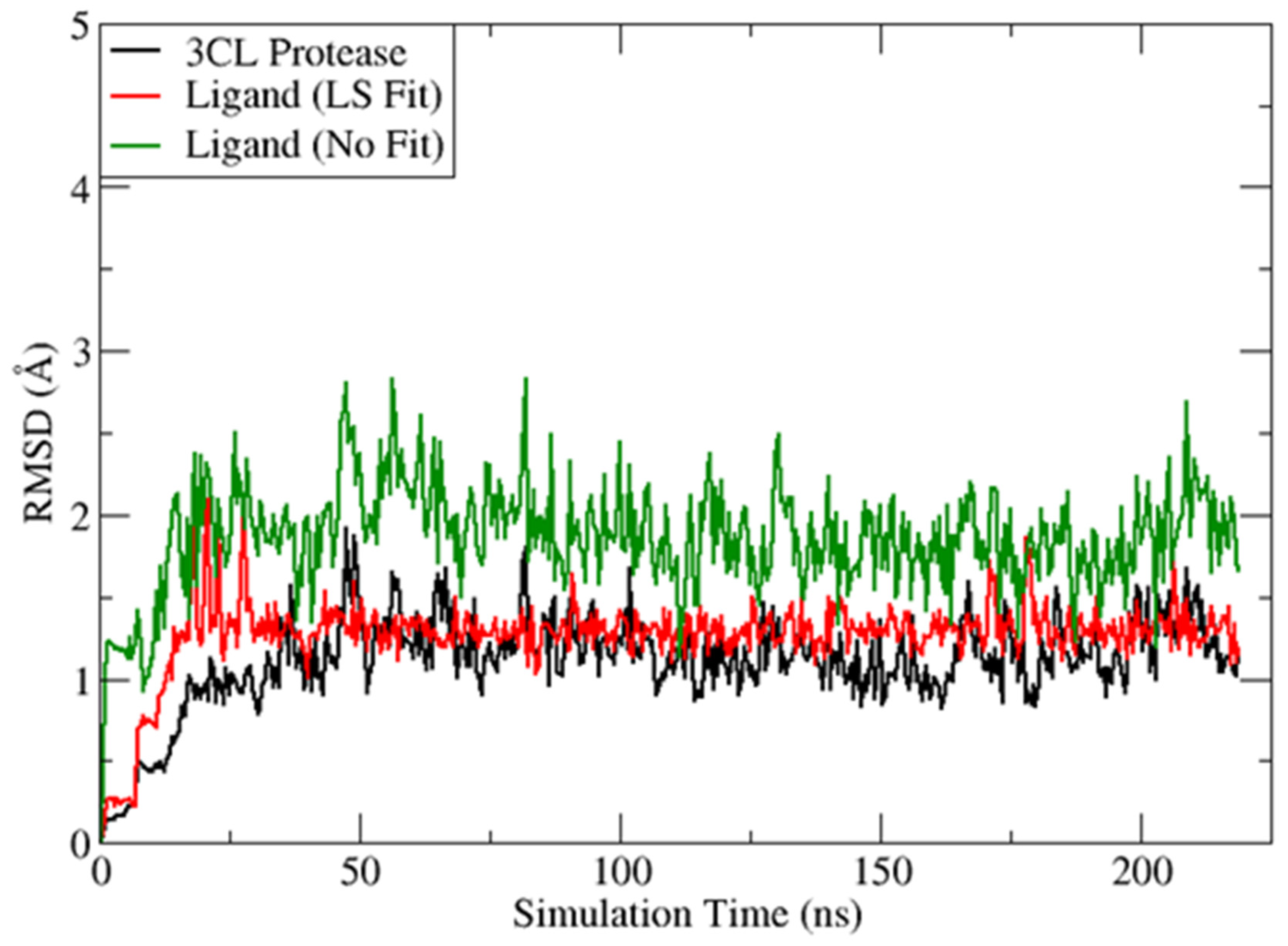
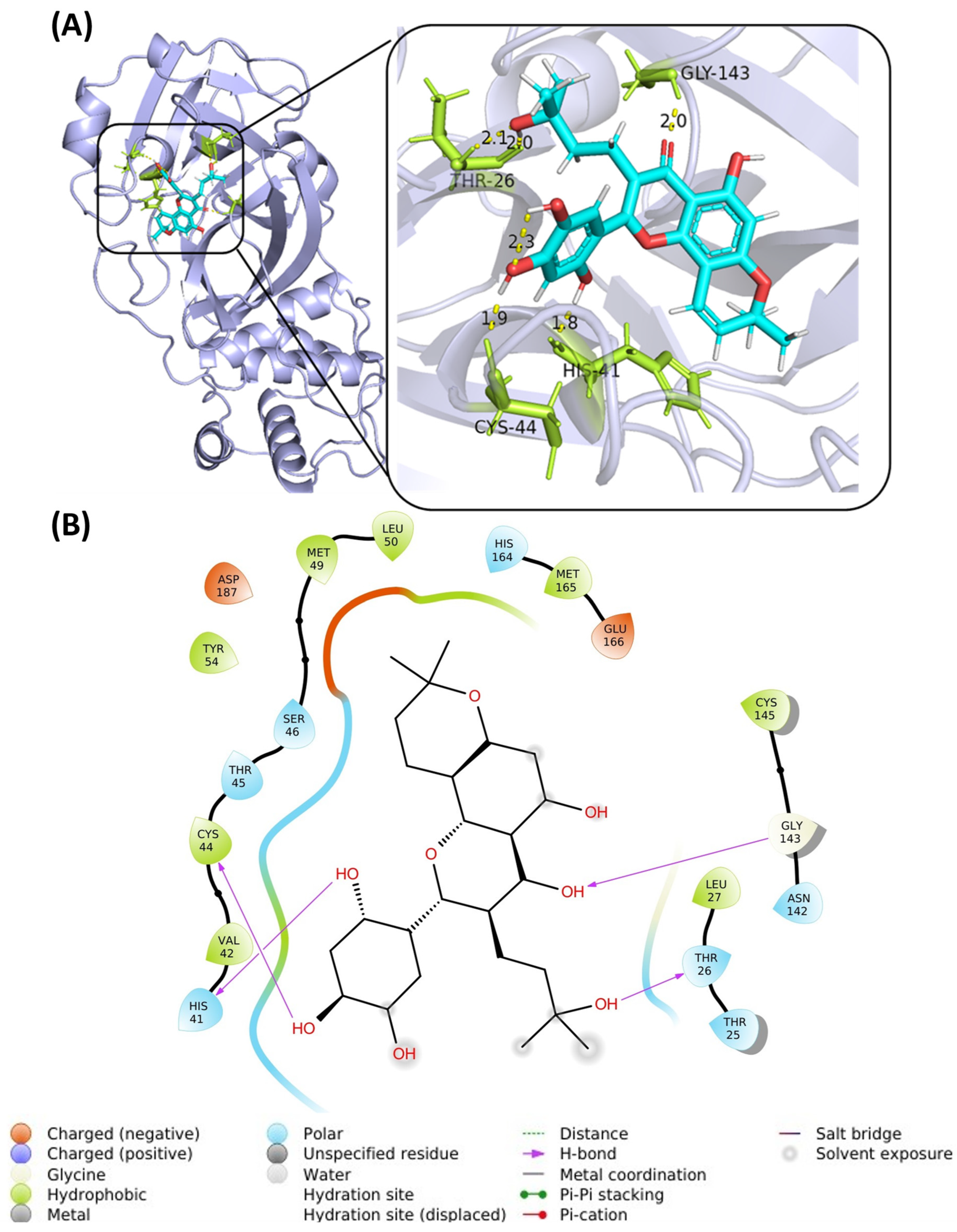
2.4. Molecular Dynamics Analysis
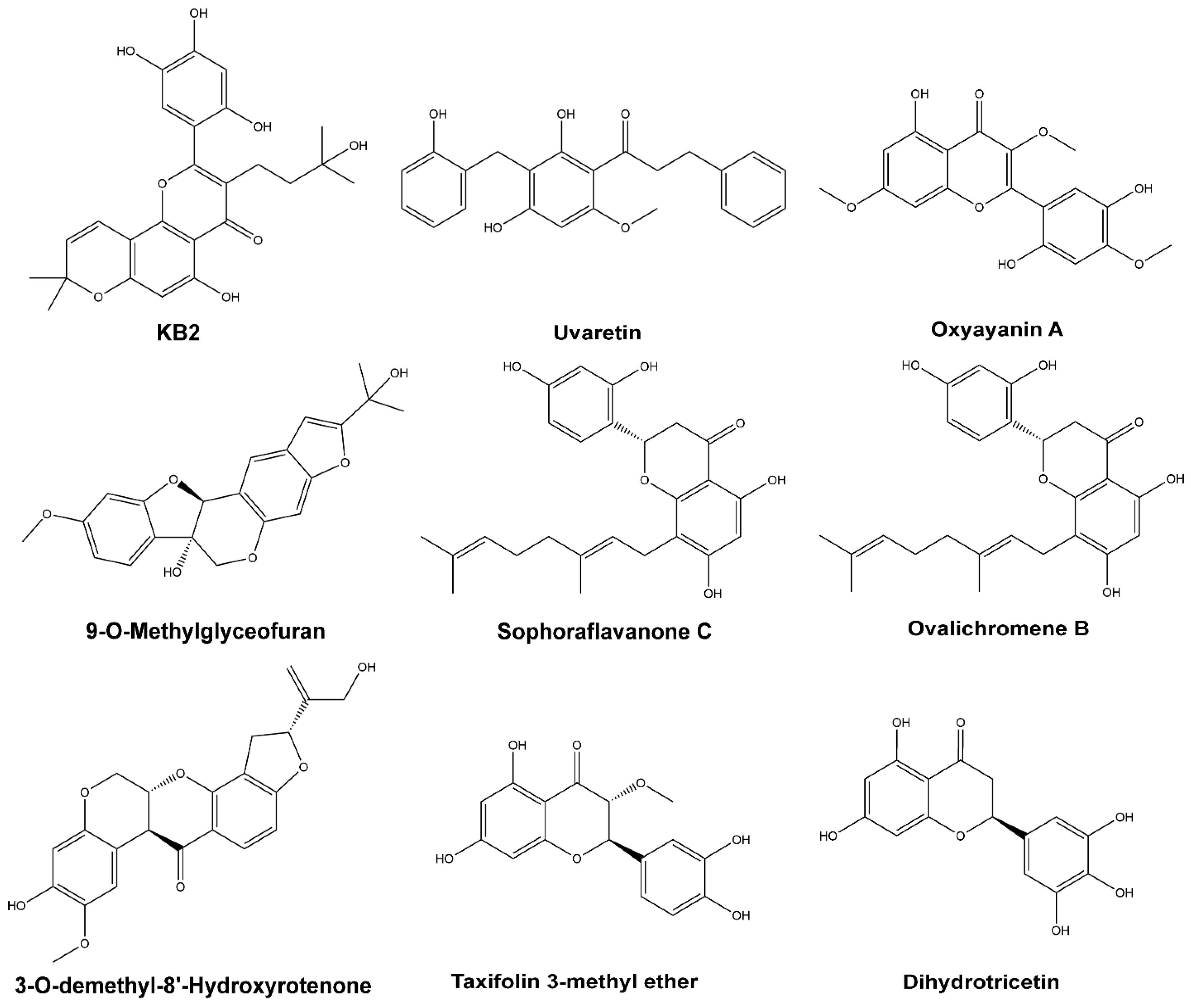
2.5. Top Flavonoids
3. Methods
3.1. Data Sources
3.2. Molecular Modeling Study and Interaction-Profile (IP) Calculation
3.2.1. Molecular Docking and System Setup
3.2.2. Data Processing and Qualification
3.3. Machine Learning Model Construction and Prediction
3.3.1. Model Training and Evaluation
3.3.2. Consensus-Based Model Prediction
3.4. ADMET Risk Filtering
3.5. Molecular Dynamics (MD) Simulation
3.6. MM-PBSA Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.-H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 2022, 602, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rocklov, J. The reproductive number of the Delta variant of SARS-CoV-2 is far higher compared to the ancestral SARS-CoV-2 virus. J. Travel Med. 2021, 28, taab124. [Google Scholar] [CrossRef] [PubMed]
- Nasserie, T.; Hittle, M.; Goodman, S.N. Assessment of the Frequency and Variety of Persistent Symptoms Among Patients With COVID-19. JAMA Netw. Open 2021, 4, e2111417. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, T.; Knight, M.; A’Court, C.; Buxton, M.; Husain, L. Management of post-acute covid-19 in primary care. BMJ 2020, 370, m3026. [Google Scholar] [CrossRef] [PubMed]
- Marshall, M. The lasting misery of coronavirus long-haulers. Nature 2020, 585, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Lutchmansingh, D.D.; Higuero Sevilla, J.P.; Possick, J.D.; Gulati, M. Long Haulers. Semin. Respir. Crit. Care Med. 2023, 44, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Catalano, A.; Iacopetta, D.; Ceramella, J.; Maio, A.C.D.; Basile, G.; Giuzio, F.; Bonomo, M.G.; Aquaro, S.; Walsh, T.J.; Sinicropi, M.S.; et al. Are Nutraceuticals Effective in COVID-19 and Post-COVID Prevention and Treatment? Foods 2022, 11, 2884. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef]
- Needle, D.; Lountos, G.T.; Waugh, D.S. Structures of the Middle East respiratory syndrome coronavirus 3C-like protease reveal insights into substrate specificity. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Choudhary, O.P.; Dhawan, M. Priyanka Omicron variant (B.1.1.529) of SARS-CoV-2: Threat assessment and plan of action. Int. J. Surg. 2022, 97, 106187. [Google Scholar] [CrossRef] [PubMed]
- Gouhar, S.A.; Elshahid, Z.A. Molecular docking and simulation studies of synthetic protease inhibitors against COVID-19: A computational study. J. Biomol. Struct. Dyn. 2021, 40, 13976–13996. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Yiu, C.B.; Wong, K.Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL (pro)) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Res 2020, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yang, L.; Zhang, X.; Zhang, Q.; Yang, Z.; Liu, Y.; Wei, S.; Liu, W. Discovery of Potential Flavonoid Inhibitors Against COVID-19 3CL Proteinase Based on Virtual Screening Strategy. Front. Mol. Biosci. 2020, 7, 556481. [Google Scholar] [CrossRef] [PubMed]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in Vitro Repurposing Screen. ACS Pharmacol. Transl. Sci. 2021, 4, 1096–1110. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Wanounou, M.; Caraco, Y.; Levy, R.H.; Bialer, M.; Perucca, E. Clinically Relevant Interactions Between Ritonavir-Boosted Nirmatrelvir and Concomitant Antiseizure Medications: Implications for the Management of COVID-19 in Patients with Epilepsy. Clin. Pharmacokinet. 2022, 61, 1219–1236. [Google Scholar] [CrossRef]
- Food and Drug Administration. Fact Sheet for Healthcare Providers: Emergency Use Authorization for Paxlovid; FDA: White Oak, MD, USA, 2022.
- EMA. EMA Issues Advice on Use of Paxlovid (PF-07321332 and Ritonavir) for the Treatment of COVID-19: Rolling Review Starts in Parallel. Available online: https://www.ema.europa.eu/en/news/ema-issues-advice-use-paxlovid-pf-07321332-ritonavir-treatment-covid-19-rolling-review-starts (accessed on 26 September 2023).
- Bardelčíková, A.; Miroššay, A.; Šoltýs, J.; Mojžiš, J. Therapeutic and prophylactic effect of flavonoids in post-COVID-19 therapy. Phytother. Res. 2022, 36, 2042–2060. [Google Scholar] [CrossRef]
- Alzaabi, M.M.; Hamdy, R.; Ashmawy, N.S.; Hamoda, A.M.; Alkhayat, F.; Khademi, N.N.; Al Joud, S.M.A.; El-Keblawy, A.A.; Soliman, S.S.M. Flavonoids are promising safe therapy against COVID-19. Phytochem. Rev. 2022, 21, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, F.R.; Howlader, S.; Raihan, T.; Hasan, M. Plants Metabolites: Possibility of Natural Therapeutics Against the COVID-19 Pandemic. Front. Med. 2020, 7, 444. [Google Scholar] [CrossRef]
- Jo, S.; Kim, S.; Kim, D.Y.; Kim, M.-S.; Shin, D.H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J. Enzym. Inhib. Med. Chem. 2020, 35, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Germano, C.; Messina, A.; Tavella, E.; Vitale, R.; Avellis, V.; Barboni, M.; Attini, R.; Revelli, A.; Zola, P.; Manzoni, P.; et al. Fetal Brain Damage during Maternal COVID-19: Emerging Hypothesis, Mechanism, and Possible Mitigation through Maternal-Targeted Nutritional Supplementation. Nutrients 2022, 14, 3303. [Google Scholar] [CrossRef] [PubMed]
- Pastor, N.; Collado, M.C.; Manzoni, P. Phytonutrient and Nutraceutical Action against COVID-19: Current Review of Characteristics and Benefits. Nutrients 2021, 13, 464. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Li, Y.; Su, M.; Liu, Z.; Li, J.; Liu, J.; Han, L.; Wang, R. Assessing protein–ligand interaction scoring functions with the CASF-2013 benchmark. Nat. Protoc. 2018, 13, 666–680. [Google Scholar] [CrossRef]
- Leach, A.R.; Shoichet, B.K.; Peishoff, C.E. Prediction of Protein−Ligand Interactions. Docking and Scoring: Successes and Gaps. J. Med. Chem. 2006, 49, 5851–5855. [Google Scholar] [CrossRef]
- Guedes, I.A.; Pereira, F.S.S.; Dardenne, L.E. Empirical Scoring Functions for Structure-Based Virtual Screening: Applications, Critical Aspects, and Challenges. Front. Pharmacol. 2018, 9, 1089. [Google Scholar] [CrossRef]
- Ji, B.; He, X.; Zhai, J.; Zhang, Y.; Man, V.H.; Wang, J. Machine learning on ligand-residue interaction profiles to significantly improve binding affinity prediction. Brief. Bioinform. 2021, 22, bbab054. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2018, 20, 1878–1912. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhang, C.; Bian, T.; Jiao, K.; Su, W.; Wu, K.-J.; Su, A. A Review on Artificial Intelligence Enabled Design, Synthesis, and Process Optimization of Chemical Products for Industry 4.0. Processes 2023, 11, 330. [Google Scholar] [CrossRef]
- Khamis, M.A.; Gomaa, W. Comparative assessment of machine-learning scoring functions on PDBbind 2013. Eng. Appl. Artif. Intell. 2015, 45, 136–151. [Google Scholar] [CrossRef]
- Crampon, K.; Giorkallos, A.; Deldossi, M.; Baud, S.; Steffenel, L.A. Machine-learning methods for ligand-protein molecular docking. Drug Discov. Today 2022, 27, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, E.A.; Zhang, Y. Protein–Ligand Docking in the Machine-Learning Era. Molecules 2022, 27, 4568. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2018, 47, D930–D940. [Google Scholar] [CrossRef]
- Arita, M.; Suwa, K. Search extension transforms Wiki into a relational system: A case for flavonoid metabolite database. BioData Min. 2008, 1, 7. [Google Scholar] [CrossRef]
- Haibo, H.; Garcia, E.A. Learning from Imbalanced Data. IEEE Trans. Knowl. Data Eng. 2009, 21, 1263–1284. [Google Scholar] [CrossRef]
- Weiss, G.M.; Provost, F. Learning When Training Data are Costly: The Effect of Class Distribution on Tree Induction. J. Artif. Intell. Res. 2003, 19, 315–354. [Google Scholar] [CrossRef]
- Mukherjee, A.; Su, A.; Rajan, K. Deep Learning Model for Identifying Critical Structural Motifs in Potential Endocrine Disruptors. J. Chem. Inf. Model 2021, 61, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Prentis, R.; Lis, Y.; Walker, S. Pharmaceutical innovation by the seven UK-owned pharmaceutical companies (1964–1985). Br. J. Clin. Pharmacol. 1988, 25, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T. Managing the drug discovery/development interface. Drug Discov. Today 1997, 2, 436–444. [Google Scholar] [CrossRef]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ma, C.; Wipf, P.; Liu, H.; Su, W.; Xie, X.Q. TargetHunter: An in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J. 2013, 15, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Singh, B. Flavonoids as lead compounds modulating the enzyme targets in Alzheimer’s disease. Med. Chem. Res. 2013, 22, 3061–3075. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, P.; Yu, H.; Li, J.; Wang, M.-W.; Zhao, W. Anti-Helicobacter pylori and Thrombin Inhibitory Components from Chinese Dragon’s Blood, Dracaena cochinchinensis. J. Nat. Prod. 2007, 70, 1570–1577. [Google Scholar] [CrossRef]
- Moro, S.; van Rhee, A.M.; Sanders, L.H.; Jacobson, K.A. Flavonoid Derivatives as Adenosine Receptor Antagonists: A Comparison of the Hypothetical Receptor Binding Site Based on a Comparative Molecular Field Analysis Model. J. Med. Chem. 1998, 41, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Subbarao, N. Deep learning model for virtual screening of novel 3C-like protease enzyme inhibitors against SARS coronavirus diseases. Comput. Biol. Med. 2021, 132, 104317. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cisneros, G.A.; Cruzeiro, V.W.D.; Darden, T.A.; et al. Amber 2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Das, S.K.; Chen, S.; Deasy, J.O.; Zhou, S.; Yin, F.-F.; Marks, L.B. Combining multiple models to generate consensus: Application to radiation-induced pneumonitis prediction. Med. Phys. 2008, 35, 5098–5109. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The Resp Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Rocchia, W.; Alexov, E.; Honig, B. Extending the applicability of the nonlinear Poisson-Boltzmann equation: Multiple dielectric constants and multivalent ions. J. Phys. Chem. B 2001, 105, 6507–6514. [Google Scholar] [CrossRef]
- Li, L.; Li, C.A.; Sarkar, S.; Zhang, J.; Witham, S.; Zhang, Z.; Wang, L.; Smith, N.; Petukh, M.; Alexov, E. DelPhi: A comprehensive suite for DelPhi software and associated resources. BMC Biophys. 2012, 5, 9. [Google Scholar] [CrossRef]
- Wang, E.C.; Sun, H.Y.; Wang, J.M.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T.J. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Wang, J.; Hou, T.; Xu, X. Recent Advances in Free Energy Calculations with a Combination of Molecular Mechanics and Continuum Models. Curr. Comput. Aided-Drug Des. 2006, 2, 287–306. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Training Set A | Training Set B | Prediction Set | |
|---|---|---|---|
| Sources | ChEMBL Database [38] | Library Composition [17] | Flavonoids Metabolites Database [39] |
| Usage | Regression Training | Classification Training | Prediction and Screening |
| Input | Ligand-residue energy interaction profiles (57 interacting residues) | ||
| Bioactivity Values | Half Maximal Inhibitory Concentration, IC50 (nM) | Normalized Inhibition, inhibition % | |
| Output (Transformed) | Continuous Free Energy, ΔEexp (kcal/mol) | Binary Bioactivity Label | |
| # Total Compounds | 1240 | 8702 | 6961 |
| # Processed Compounds | 1118 | 7860 | 6001 |
| # Qualified Compounds | 1101 | 6059 | |
| Ndecoys/Nactives | 1002/99 | 5729/330 | |
| # Balanced Subsets | 10 (A1–A10) | 20 (B1–B20) | |
| Compound Name | Structure Class | PubChem CID | PBSA Energy (kcal/mol) | RMSD Max (Å) | Full ADMET Scores |
|---|---|---|---|---|---|
| KB-2 | Flavones | 14630497 | −9.89 | 2.83 | 3.500 |
| 9-O-Methylglyceofuran | Isoflavonoids | 44257401 | −8.81 | 3.52 | 3.016 |
| 3-O-demethyl-8’-Hydroxyrotenone | Isoflavonoids | 44257401 | −7.73 | 4.65 | 4.220 |
| Uvaretin | Chalcones | 73447 | −7.41 | 4.93 | 4.010 |
| Sophoraflavanone C | Flavanones | 85403243 | −5.89 | 4.01 | 2.691 |
| Taxifolin 3-methyl ether | Dihydroflavonols | 14794885 | −5.66 | 3.97 | 4.163 |
| Oxyayanin A | Flavonols | 5281676 | −4.38 | 4.79 | 2.500 |
| Ovalichromene B | Flavanones | 10981007 | −4.35 | 4.75 | 5.503 |
| Dihydrotricetin | Flavanones | 5258991 | −4.34 | 4.70 | 1.188 |
| Risk Model | Thresholds (Range) | Criteria |
|---|---|---|
| Full ADMET Risk | 7.0 (0–22.0) | Exceeds 7 for 10% of a focused WDI subset when ALL component risks are included. |
| Absorption Risk (Absn Risk) | 4.0 (0–8.0) | Exceeds 4 for 9% of a focused WDI subset. |
| Lipinski’s Rule of 5 (Ro5) | 1.0 (0–5.0) | Exceeds 1 for 8% of a focused WDI subset. |
| Risk connected with P450 oxidation (CYP Risk) | 2.0 (0–6.0) | Exceeds 2.0 for 10% of a focused WDI subset. |
| Risk of mutagenicity (MUT Risk) | 1.2 (0–5.4) | Exceeds 1.2 for 12% of a focused WDI subset. |
| Enhanced risk of mutagenicity (MUT_x) | 1.0 (0–4.0) | Exceeds 1.0 for 12% of a focused WDI subset. |
| Risk connected with predicted toxicity (TOX Risk) | 2.0 (0–6.0) | Exceeds 2.0 for 9% of a focused WDI subset. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, L.; Han, F.; Ji, B.; He, X.; Wang, L.; Niu, T.; Zhai, J.; Wang, J. In Silico Screening of Natural Flavonoids against 3-Chymotrypsin-like Protease of SARS-CoV-2 Using Machine Learning and Molecular Modeling. Molecules 2023, 28, 8034. https://doi.org/10.3390/molecules28248034
Cai L, Han F, Ji B, He X, Wang L, Niu T, Zhai J, Wang J. In Silico Screening of Natural Flavonoids against 3-Chymotrypsin-like Protease of SARS-CoV-2 Using Machine Learning and Molecular Modeling. Molecules. 2023; 28(24):8034. https://doi.org/10.3390/molecules28248034
Chicago/Turabian StyleCai, Lianjin, Fengyang Han, Beihong Ji, Xibing He, Luxuan Wang, Taoyu Niu, Jingchen Zhai, and Junmei Wang. 2023. "In Silico Screening of Natural Flavonoids against 3-Chymotrypsin-like Protease of SARS-CoV-2 Using Machine Learning and Molecular Modeling" Molecules 28, no. 24: 8034. https://doi.org/10.3390/molecules28248034
APA StyleCai, L., Han, F., Ji, B., He, X., Wang, L., Niu, T., Zhai, J., & Wang, J. (2023). In Silico Screening of Natural Flavonoids against 3-Chymotrypsin-like Protease of SARS-CoV-2 Using Machine Learning and Molecular Modeling. Molecules, 28(24), 8034. https://doi.org/10.3390/molecules28248034