3. Materials and Methods
3.1. General Information
All reagents and solvents were purchased from commercial suppliers and used without purification unless otherwise specialized. All reactions were carried out under Ar atmosphere. PAGE images were recorded on Chemi-Doc (Bio-Rad, Hercules, CA, USA) and analyzed by Image LabTM (Bio-Rad). 1H, 13C and 31P NMR spectra were recorded on an AVHD 400 NB (Bruker Daltonics, Billerica, MA, USA) using CDCl3, DMSO-d6, and D2O as the solvents. Mass spectra of all new compounds were measured on a JMS-700 instrument (JEOL, Tokyo, Japan) (for fast atom bombardment, FAB). For HPLC, Shimadzu SLC-20A3R, LC-20AD, CTO-20AC, SPD-20A, and FRC-10A were utilized.
3.2. Synthesis of N3-Benzoyl-2′-O-ethyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-5-methyluridine (2a)
Compound 1 (1.5 g, 2.5 mmol) was dissolved in dry toluene (7.5 mL). Ag2O (1460 mg, 6.3 mmol) and iodoethane (5.0 mL, 63 mmol) were added and the reaction mixture was stirred at 60 °C for 13 h. The resulting reaction mixture was filtered by celite pad and the filtrate was diluted with CHCl3. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 85/15) to afford compound 2a (456 mg, 0.72 mmol, 29%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.93 (dd, J = 1.2 Hz, 8.4 Hz, 2H), 7.73 (d, J = 1.2 Hz, 1H), 7.67–7.63 (m, 1H), 7.49 (t, J = 7.5 Hz, 2H), 5.70 (s, 1H), 4.29–4.21 (m, 2 H), 4.12 (dd, J = 2.0 Hz, 9.6 Hz, 1H), 3.87–3.77 (d, 3H), 1.95 (d, J = 1.1 Hz, 3H), 1.19 (t, J = 7.0 Hz, 3H), 1.21–1.05 (m, 28H). 13C NMR (100 MHz, CDCl3): δ 169.12, 162.95, 149.01, 135.11, 134.85, 131.52, 130.55, 129.17, 110.10, 89.79, 82.00, 81.88, 68.18, 66.97, 59.38, 17.48, 17.43, 17.31, 17.27, 17.21, 17.09, 17.04, 16.92, 15.34, 13.57, 12.91, 12.81, 12.77, 12.60. HRMS (MALDI): calcd for C31H48N2O8NaSi2 [M + Na]+ 655.2841, found 655.2844.
3.3. Synthesis of N3-Benzoyl-2′-O-ethyl-5-methyluridine (3a)
Compound 2a (426 mg, 0.67 mmol) was dissolved in dry THF (6.7 mL). Then, 1 M TBAF in THF (2.0 mL, 2.0 mmol) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 99/1 to 97/3) to afford compound 3a (238 mg, 0.61 mmol, 91%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.92 (dd, J = 1.2 Hz, 8.4 Hz, 2H), 7.75 (s, 1H), 7.68–7.63 (m, 1H), 7.49 (t, J = 7.6 Hz, 2H), 5.78 (d, J = 2.8 Hz, 1H), 4.32 (d, J = 5.3 Hz, 1H), 4.09 (dd, J = 3.0 Hz, 5.2 Hz, 1H), 4.03 (dd, J = 2.2 Hz, 8.3 Hz, 2H), 3.90–3.83 (m, 2H), 3.67–3.59 (m, 1H), 2.73 (d, J = 6.2 Hz, 1H), 2.50 (br, 1H), 1.95 (d, J = 1.2 Hz, 3H), 1.23 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 168.77, 162.79, 149.31, 136.74, 135.18, 131.43, 130.49, 129.20, 110.88, 89.85, 84.80, 81.23, 68.35, 66.79, 61.10, 15.24, 12.61. HRMS (MALDI): calcd for C19H22N2O7Na [M + Na]+ 413.1319, found 413.1316.
3.4. Synthesis of 3′-O-Acetyl-N3-benzoyl-5′-O-dimethoxytrityl-2′-O-ethyl-5-methyluridine (4a)
Compound 3a (220 mg, 0.56 mmol) was dissolved in dry pyridine (5.6 mL). DMTrCl (209 mg, 0.62 mmol) was added and the reaction mixture was stirred at room temperature for 6 h. DMAP (7 mg, 0.06 mmol) and Ac2O (262 µL, 2.8 mmol) were added and the reaction mixture was stirred at room temperature for 18 h. The resulting reaction mixture was concentrated in vacuo and diluted with ethyl acetate. The combined organic layers were washed with saturated NaHCO3 aq. and brine, dried over anhydrous Na2SO4, andconcentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 3/1 to 2/1) to afford compound 4a (291 mg, 0.40 mmol, 71%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.94 (d, J = 7.2 Hz, 2H), 7.72 (d, J = 1.0 Hz, 1H), 7.64 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.8 Hz, 2H), 7.41 (d, J = 7.2 Hz, 2H), 7.34–7.26 (m, 7H), 6.86 (dd, J = 1.9 Hz, 8.9 Hz, 4H), 6.03 (d, J = 4.4 Hz, 1H), 5.36 (t, J = 5.3 Hz, 1H), 4.32–4.28 (m, 2H), 3.81 (s, 6H), 3.76–3.69 (m, 1H), 3.63–3.56 (m, 2H), 3.39 (dd, J = 2.3 Hz, 11.0 Hz, 1H), 2.10 (s, 3H), 1.40 (s, 3H), 1.16 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 170.13, 168.85, 162.76, 158.85, 158.83, 149.41, 144.05, 135.12, 135.00, 131.59, 130.52, 130.14, 129.11, 128.18, 128.09, 127.31, 113.36, 113.35, 114.41, 87.59, 87.24, 81.16, 80.18, 70.58, 66.93, 62.09, 55.29, 20.68, 15.16, 11.82. HRMS (MALDI): calcd for C42H42N2O10Na [M + Na]+ 757.2732, found 757.2738.
3.5. Synthesis of 3′-O-Acetyl-N3-benzoyl-2′-O-ethyl-5-methyluridine (5a)
Compound 4a (276 mg, 0.38 mmol) was dissolved in CH2Cl2 (3.8 mL). TFA (1.0 mL) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting reaction mixture was concentrated in vacuo and diluted with CHCl3. The combined organic layers were washed with saturated NaHCO3 aq. and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 1/1 to 1/2) to afford compound 5a (125 mg, 0.29 mmol, 76%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.92 (dd, J = 1.1 Hz, 8.3 Hz, 2H), 7.66 (t, J = 7.4 Hz, 1H), 7.62 (br, 1H), 7.50 (t, J = 7.9 Hz, 2H), 5.69 (d, J = 4.8 Hz, 1H), 5.27 (t, J = 5.0 Hz, 1H), 4.37 (t, J = 5.0 Hz, 1H), 4.23–4.21 (m, 1H), 3.97 (dd, J = 1.8 Hz, 12.6 Hz, 1H), 3.76 (dd, J = 2.0 Hz, 12.6 Hz, 1H), 3.70–3.53 (m, 2H), 2.15 (s, 3H), 1.96 (d, J = 1.0 Hz, 3H), 1.15 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 170.50, 168.66, 162.72, 149.43, 137.33, 135.20, 131.41, 130.53, 129.21, 111.19, 91.09, 83.01, 79.14, 70.43, 66.94, 61.55, 20.83, 15.22, 12.66. HRMS (MALDI): calcd for C21H24N2O8Na [M + Na]+ 455.1425, found 455.1427.
3.6. Synthesis of 2′-O-Ethyl-5-methyluridine-5′-triphospahte (6a)
Compound 5a was dissolved in dry pyridine and the solution was evaporated to dryness in vacuo. Compound 5a (23 mg, 0.065 mmol) was dissolved in dry pyridine (0.25 mL) and 1,4-dioxane (0.25 mL). Then, 2-Chloro-4H-1,3,2-benzodioxaphosphorin-4-one (15 mg, 0.072 mmol) was added and the reaction mixture was stirred for 30 min at room temperature. Tris(tetrabutylammonium) hydrogen pyrophosphate (43 mg, 0.078 mmol) in DMF (0.5 mL) and tributylamine (52 µL, 0.22 mmol) were added and the reaction mixture was stirred for 1 h at room temperature. Next, 0.1 M I2 in pyridine/H2O (98:2, v/v) was added and the reaction mixture was stirred for 30 min at 0 °C. Then, 10% Na2S2O3 aq. was added and the reaction mixture was stirred for 10 min at room temperature. The reaction mixture was purified by ethanol precipitation. The precipitate was dissolved in 7% NH3 aq. and the reaction mixture was stirred for 2 h at room temperature. The solvent was removed in vacuo and the residue was purified by HPLC to give compound 6a (14 µmol, 22%). 1H NMR (400 MHz, D2O): δ7.78 (s, 1H), 6.02 (d, J = 5.0 Hz, 1H), 4.53 (t, J = 4.5 Hz, 1H), 4.25 (d, J = 4.9 Hz, 3H), 4.19 (t, J = 5.2 Hz, 1H), 3.71 (dd, J = 7.0 Hz, 14.1 Hz, 2H), 1.92 (s, 3H), 1.18 (t, J = 7.0 Hz, 3H). 31P NMR (120 MHz, D2O): δ −9.60 (d, J = 14.5 Hz), −12.41 (d, J = 14.5 Hz), −23.38 (t, J = 14.7 Hz). HRMS (MALDI): calcd for C12H20N2O15P3 [M – H]− 525.0082, found 525.0089.
3.7. Synthesis of N3-Benzoyl-2′-O-propyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-5-methyluridine (2b)
Compound 1 (1.5 g, 2.5 mmol) was dissolved in dry toluene (7.5 mL). Ag2O (1460 mg 6.3 mmol) and 1-iodopropane (6.0 mL, 63 mmol) were added and the reaction mixture was stirred at 80 °C for 15 h. The resulting reaction mixture was filtered by celite pad and the filtrate was diluted with CHCl3. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 85/15) to afford compound 2b (369 mg, 0.57 mmol, 23%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.93 (dd, J = 1.2 Hz, 8.4 Hz, 2H), 7.73 (d, J = 1.2 Hz, 1H), 7.67–7.63 (m, 1H), 7.49 (t, J = 8.0 Hz, 2H), 5.71 (s, 1H), 4.29–4.21 (m, 2 H), 4.16 (dd, J = 2.1 Hz, 9.6 Hz, 1H), 3.84 (d, J = 4.3 Hz, 1H), 3.71 (t, J = 6.4 Hz, 2H), 1.95 (d, J = 1.1 Hz, 3H), 1.62–1.53 (m, 2H), 1.13–1.04 (m, 28H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 169.14, 162.98, 148.98, 135.11, 134.87, 131.53, 130.56, 129.17, 110.06, 89.65, 82.16, 81.90, 73.08, 68.24, 59.39, 23.01, 17.49, 17.43, 17.31, 17.28, 17.21, 17.09, 17.03, 16.93, 13.58, 12.91, 12.81, 12.77, 12.65. HRMS (MALDI): calcd for C32H50N2O8NaSi2 [M + Na]+ 699.2998, found 699.2986.
3.8. Synthesis of N3-Benzoyl-2′-O-propyl-5-methyluridine (3b)
Compound 2b (185 mg, 0.29 mmol) was dissolved in dry THF (3.0 mL). 1 M TBAF in THF (0.86 mL, 0.86 mmol) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 100/0 to 97/3) to afford compound 3b (90 mg, 0.22 mmol, 77%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.92 (dd, J = 1.2 Hz, 8.4 Hz, 2H), 7.73 (s, 1H), 7.68–7.63 (m, 1H), 7.50 (t, J = 8.0 Hz, 2H), 5.78 (d, J = 2.9 Hz, 1H), 4.33 (dd, J = 5.8 Hz, 12.9 Hz, 1H), 4.10 (dd, J = 3.0 Hz, 5.2 Hz, 1H), 4.04 (dd, J = 2.3 Hz, 8.6 Hz, 2H), 3.90–3.84 (m, 1H), 3.79–3.73 (m, 1H), 3.57–3.51 (m, 1H), 2.70 (d, J = 7.3 Hz, 1H), 2.40 (br, 1H), 1.95 (d, J = 1.1 Hz, 3H), 1.66–1.57 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 168.75, 162.77, 149.30, 136.74, 135.16, 131.45, 130.50, 129.19, 110.91, 89.91, 84.82, 81.27, 72.82, 68.46, 61.18, 22.91, 12.62, 10.42. HRMS (MALDI): calcd for C20H24N2O7Na [M + Na]+ 427.1476, found 427.1473.
3.9. Synthesis of 3′-O-Acetyl-N3-benzoyl-5′-O-dimethoxytrityl-2′-O-propyl-5-methyluridine (4b)
Compound 3b (80 mg, 0.20 mmol) was dissolved in dry pyridine (2.0 mL). DMTrCl (74 mg, 0.22 mmol) was added and the reaction mixture was stirred at room temperature for 4.5 h. DMAP (2 mg, 0.02 mmol) and Ac2O (93 µL, 1.0 mmol) were added and the reaction mixture was stirred at room temperature for 13 h. The resulting reaction mixture was concentrated in vacuo and diluted with ethyl acetate. The combined organic layers were washed with saturated NaHCO3 aq. and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 3/1 to 2/1) to afford compound 4b (104 mg, 0.14 mmol, 69%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.94 (dd, J = 1.1 Hz, 8.4 Hz, 2H), 7.72 (d, J = 1.2 Hz, 1H), 7.66–7.62 (m, 1H), 7.49 (t, J = 7.8 Hz, 2H), 7.41 (d, J = 7.1 Hz, 2H), 7.34–7.26 (m, 7H), 6.86 (dd, J = 2.0 Hz, 8.9 Hz, 4H), 6.04 (d, J = 4.4 Hz, 1H), 5.36 (t, J = 5.3 Hz, 1H), 4.31–4.28 (m, 2H), 3.81 (s, 6H), 3.65–3.59 (m, 2H), 3.51–3.46 (m, 1H), 3.39 (dd, J = 2.4 Hz, 11.0 Hz, 1H), 2.10 (s, 3H), 1.59–1.50 (m, 2H), 1.40 (d, J = 0.9 Hz, 3H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 170.13, 168.85, 162.78, 158.86, 158.84, 149.40, 144.07, 135.13, 135.00, 131.60, 130.54, 130.15, 129.11, 128.20, 128.10, 127.32, 113.37, 113.36, 111.40, 87.51, 87.24, 81.21, 80.37, 73.01, 70.62, 62.14, 55.29, 22.86, 20.70, 11.82, 10.41. HRMS (MALDI): calcd for C43H44N2O10Na [M + Na]+ 771.2888, found 771.2887.
3.10. Synthesis of 3′-O-Acetyl-N3-benzoyl-2′-O-propyl-5-methyluridine (5b)
Compound 4b (80 mg, 0.11 mmol) was dissolved in CH2Cl2 (1.1 mL). TFA (0.3 mL) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting reaction mixture was concentrated in vacuo and diluted with CHCl3. The combined organic layers were washed with saturated NaHCO3 aq. and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 1/1 to 1/2) to afford compound 5b (35 mg, 0.08 mmol, 72%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.92 (dd, J = 1.1 Hz, 8.4 Hz, 2H), 7.68–7.63 (m, 2H), 7.49 (t, J = 7.8 Hz, 2H), 5.71 (d, J = 4.8 Hz, 1H), 5.27 (t, J = 5.0 Hz, 1H), 4.35 (t, J = 5.0 Hz, 1H), 4.24–4.21 (m, 1H), 3.97 (dd, J = 1.8 Hz, 12.5 Hz, 1H), 3.76 (dd, J = 2.0 Hz, 12.5 Hz, 1H), 3.58–3.53 (m, 1H), 3.49–3.43 (m, 1H), 2.14 (s, 3H), 1.96 (d, J = 1.0 Hz, 3H), 1.58–1.49 (m, 2H), 0.88 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 170.49, 168.67, 162.74, 149.40, 137.24, 135.19, 131.42, 130.53, 129.20, 111.16, 90.89, 83.01, 79.36, 73.01, 70.43, 61.53, 22.88, 20.83, 12.66, 10.40. HRMS (MALDI): calcd for C22H26N2O8Na [M + Na]+ 469.1581, found 469.1580.
3.11. Synthesis of 2′-O-Propyl-5-methyluridine-5′-triphospahte (6b)
Compound 5b was dissolved in dry pyridine and the solution was evaporated to dryness in vacuo. Compound 5b (47 mg, 0.10 mmol) was dissolved in dry pyridine (0.5 mL) and 1,4-dioxane (0.5 mL). Then, 2-Chloro-4H-1,3,2-benzodioxaphosphorin-4-one (24 mg, 0.12 mmol) was added and the reaction mixture was stirred for 30 min at room temperature. Tris(tetrabutylammonium) hydrogen pyrophosphate (66 mg, 0.12 mmol) in DMF (1.0 mL) and tributylamine (86 µL, 0.36 mmol) were added and the reaction mixture was stirred for 1 h at room temperature. Next, 0.1 M I2 in pyridine/H2O (98:2, v/v) was added and the reaction mixture was stirred for 30 min at room temperature. Then, 10% Na2S2O3 aq. was added and the reaction mixture was stirred for 10 min at room temperature. The reaction mixture was purified by ethanol precipitation. The precipitate was dissolved in 7% NH3 aq. and the reaction mixture was stirred for 2 h at room temperature. The solvent was removed in vacuo and the residue was purified by HPLC to give compound 6b (46 µmol, 46%). 1H NMR (400 MHz, D2O): δ7.77 (s, 1H), 6.01 (d, J = 5.2 Hz, 1H), 4.53–4.50 (m, 1H), 4.24 (d, J = 4.2 Hz, 3H), 4.17 (t, J = 5.3 Hz, 1H), 3.66–3.55 (m, 2H), 1.91 (s, 3H), 1.59–1.50 (m, 2H), 0.84 (t, J = 7.4 Hz, 3H). 31P NMR (120 MHz, D2O): δ −9.57 (d, J = 13.4 Hz), −12.66 (d, J = 14.6 Hz), −23.64 (t, J = 14.7 Hz). HRMS (MALDI): calcd for C13H22N2O15P3 [M – H]− 539.0239, found 539.0243.
3.12. Synthesis of N3-Benzoyl-2′-O-butyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-5-methyluridine (2c)
Compound 1 (1.5 g, 2.5 mmol) was dissolved in dry toluene (7.5 mL). Ag2O (1460 mg, 6.3 mmol) and 1-iodobutane (7.1 mL, 63 mmol) were added and the reaction mixture was stirred at 80 °C for 15 h. The resulting reaction mixture was filtered by celite pad and the filtrate was diluted with CHCl3. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 85/15) to afford compound 2c (303 mg, 0.46 mmol, 18%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.93 (dd, J = 1.2 Hz, 8.4 Hz, 2H), 7.73 (d, J = 1.2 Hz, 1H), 7.67–7.62 (m, 1H), 7.49 (t, J = 8.0 Hz, 2H), 5.70 (s, 1H), 4.29–4.21 (m, 2 H), 4.15 (dd, J = 2.1 Hz, 9.6 Hz, 1H), 4.00 (dd, J = 2.5 Hz, 13.6 Hz, 1H), 3.84 (d, J = 4.3 Hz, 1H), 3.75 (t, J = 6.3 Hz, 2H), 1.95 (d, J = 1.1 Hz, 3H), 1.58–1.50 (m, 2H), 1.42–1.33 (m, 2H), 1.13–1.03 (m, 28H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 169.14, 162.97, 148.96, 135.09, 134.87, 131.53, 130.55, 129.16, 110.05, 89.64, 82.19, 81.88, 71.24, 68.25, 59.38, 31.84, 19.15, 17.49, 17.42, 17.30, 17.28, 17.21, 17.09, 17.01, 16.92, 13.83, 13.58, 12.91, 12.81, 12.76, 12.64. HRMS (MALDI): calcd for C33H52N2O8NaSi2 [M + Na]+ 683.3154, found 683.3158.
3.13. Synthesis of N3-Benzoyl-2′-O-butyl-5-methyluridine (3c)
Compound 2c (192 mg, 0.29 mmol) was dissolved in dry THF (3.0 mL). 1 M TBAF in THF (0.87 mL, 0.87 mmol) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 100/0 to 97/3) to afford compound 3c (95 mg, 0.23 mmol, 78%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.92 (dd, J = 1.2 Hz, 8.4 Hz, 2H), 7.73 (s, 1H), 7.68–7.63 (m, 1H), 7.50 (t, J = 7.8 Hz, 2H), 5.78 (d, J = 2.8 Hz, 1H), 4.33 (dd, J = 5.8 Hz, 12.8 Hz, 1H), 4.09 (dd, J = 3.0 Hz, 5.1 Hz, 1H), 4.04–4.02 (m, 2H), 3.89–3.84 (m, 1H), 3.83–3.77 (m, 1H), 3.60–3.55 (m, 1H), 2.69 (d, J = 7.3 Hz, 1H), 2.43 (br, 1H), 1.95 (d, J = 1.1 Hz, 3H), 1.61–1.54 (m, 2H), 1.41–1.32 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 168.74, 162.77, 149.27, 136.73, 135.15, 131.43, 130.49, 129.17, 110.87, 89.81, 84.80, 81.31, 71.02, 68.43, 61.14, 31.69, 19.16, 13.81, 12.60. HRMS (MALDI): calcd for C21H26N2O7Na [M + Na]+ 441.1632, found 441.1633.
3.14. Synthesis of 3′-O-Acetyl-N3-benzoyl-2′-O-butyl-5′-O-dimethoxytrityl-5-methyluridine (4c)
Compound 3c (85 mg, 0.20 mmol) was dissolved in dry pyridine (2.0 mL). DMTrCl (74 mg, 0.22 mmol) was added and the reaction mixture was stirred at room temperature for 4.5 h. DMAP (2 mg, 0.02 mmol) and Ac2O (93 µL, 1.0 mmol) were added and the reaction mixture was stirred at room temperature for 13 h. The resulting reaction mixture was concentrated in vacuo and diluted with ethyl acetate. The combined organic layers were washed with saturated NaHCO3 aq. and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 3/1 to 2/1) to afford compound 4c (113 mg, 0.15 mmol, 75%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.94 (d, J = 7.2 Hz, 2H), 7.71 (d, J = 1.0 Hz, 1H), 7.64 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.8 Hz, 2H), 7.41 (d, J = 7.2 Hz, 2H), 7.32–7.26 (m, 7H), 6.86 (dd, J = 2.0 Hz, 8.8 Hz, 4H), 6.04 (d, J = 4.5 Hz, 1H), 5.36 (t, J = 5.2 Hz, 1H), 4.30–4.27 (m, 2H), 3.80 (s, 6H), 3.69–3.60 (m, 2H), 3.56–3.47 (m, 1H), 3.39 (dd, J = 2.2 Hz, 11.0 Hz, 1H), 2.09 (s, 3H), 1.57–1.49 (m, 2H), 1.40 (s, 3H), 1.38–1.31 (m, 2H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 170.13, 168.86, 162.79, 158.86, 158.85, 149.40, 144.08, 135.14, 135.03, 135.01, 131.63, 130.54, 130.16, 129.12, 128.20, 128.10, 127.33, 113.38, 113.37, 114.40, 87.46, 87.25, 81.22, 80.35, 73.02, 71.15, 70.63, 62.16, 55.30, 31.65, 22.87, 20.70, 19.10, 13.83, 11.82, 10.42. HRMS (MALDI): calcd for C44H46N2O10Na [M + Na]+ 785.3045, found 785.3035.
3.15. Synthesis of 3′-O-Acetyl-N3-benzoyl-2′-O-butyl-5-methyluridine (5c)
Compound 4c (120 mg, 0.16 mmol) was dissolved in CH2Cl2 (1.6 mL). TFA (0.4 mL) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting reaction mixture was concentrated in vacuo and diluted with CHCl3. The combined organic layers were washed with saturated NaHCO3 aq. and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 1/1 to 1/2) to afford compound 5c (57 mg, 0.12 mmol, 75%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.92 (dd, J = 1.2 Hz, 8.4 Hz, 2H), 7.68–7.63 (m, 2H), 7.49 (t, J = 7.8 Hz, 2H), 5.71 (d, J = 4.8 Hz, 1H), 5.26 (t, J = 5.0 Hz, 1H), 4.33 (t, J = 5.0 Hz, 1H), 4.23–4.21 (m, 1H), 3.96 (dd, J = 1.8 Hz, 12.5 Hz, 1H), 3.76 (dd, J = 2.0 Hz, 12.5 Hz, 1H), 3.62–3.56 (m, 1H), 3.53–3.47 (m, 1H), 2.14 (s, 3H), 1.96 (d, J = 1.1 Hz, 3H), 1.53–1.46 (m, 2H), 1.37–1.29 (m, 2H), 0.88 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 170.47, 168.67, 162.74, 149.39, 137.19, 135.17, 131.43, 130.51, 129.19, 111.14, 90.79, 82.99, 79.44, 71.20, 70.42, 61.50, 31.68, 20.81, 19.10, 13.84, 12.64. HRMS (MALDI): calcd for C23H28N2O8Na [M + Na]+ 483.1738, found 483.0741.
3.16. Synthesis of 2′-O-Butyl-5-methyluridine-5′-triphospahte (6c)
Compound 5c was dissolved in dry pyridine and the solution was evaporated to dryness in vacuo. Compound 5c (30 mg, 0.065 mmol) was dissolved in dry pyridine (0.25 mL) and 1,4-dioxane (0.25 mL). Then, 2-Chloro-4H-1,3,2-benzodioxaphosphorin-4-one (15 mg, 0.072 mmol) was added and the reaction mixture was stirred for 30 min at room temperature. Tris(tetrabutylammonium) hydrogen pyrophosphate (43 mg, 0.078 mmol) in DMF (0.5 mL) and tributylamine (52 µL, 0.22 mmol) were added and the reaction mixture was stirred for 1 h at room temperature. Next, 0.1 M I2 in pyridine/H2O (98:2, v/v) was added and the reaction mixture was stirred for 30 min at 0 °C. Then, 10% Na2S2O3 aq. was added and the reaction mixture was stirred for 10 min at room temperature. The reaction mixture was purified by ethanol precipitation. The precipitate was dissolved in 7% NH3 aq. and the reaction mixture was stirred for 2 h at room temperature. The solvent was removed in vacuo and the residue was purified by HPLC to give compound 6c (6 µmol, 10%). 1H NMR (400 MHz, D2O): δ7.79 (s, 1H), 6.03 (d, J = 5.4 Hz, 1H), 4.52 (t, J = 4.5 Hz, 1H), 4.27–4.24 (m, 3H), 4.18 (t, J = 5.4 Hz, 1H), 3.73–3.61 (m, 2H), 1.93 (s, 3H), 1.56–1.49 (m, 2H), 1.34–1.25 (m, 2H), 0.84 (t, J = 7.4 Hz, 3H). 31P NMR (120 MHz, D2O): δ −9.80 (d, J = 12.7 Hz), −12.65 (d, J = 14.5 Hz), −23.69 (t, J = 14.7 Hz). HRMS (MALDI): calcd for C14H24N2O15P3 [M – H]− 553.0395, found 553.0424.
3.17. Synthesis of N3-Benzoyl-2′-O-isopropyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-5-methyluridine (2d)
Compound 1 (1.5 g, 2.5 mmol) was dissolved in dry toluene (7.5 mL). Ag2O (1460 mg, 6.3 mmol) and 2-iodopropane (6.2 mL, 63 mmol) were added and the reaction mixture was stirred at 80 °C for 13 h. The resulting reaction mixture was filtered by celite pad and the filtrate was diluted with CHCl3. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 85/15) to afford compound 2d (372 mg, 0.58 mmol, 23%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.93 (dd, J = 1.2 Hz, 8.4 Hz, 2H), 7.76 (d, J = 1.2 Hz, 1H), 7.67–7.62 (m, 1H), 7.49 (t, J = 7.8 Hz, 2H), 5.64 (s, 1H), 4.27 (d, J = 13.7 Hz, 1H), 4.21–4.14 (m, 2 H), 4.01–3.95 (m, 2 H), 3.93 (d, J = 4.1 Hz, 1H), 1.95 (d, J = 1.2 Hz, 3H), 1.19–1.03 (m, 34H). 13C NMR (100 MHz, CDCl3): δ 169.19, 163.03, 149.01, 135.11, 134.93, 131.51, 130.59, 129.16, 109.91, 90.44, 82.01, 80.35, 72.83, 67.88, 59.41, 22.59, 22.35, 17.50, 17.42, 17.29, 17.21, 17.12, 17.05, 16.95, 13.58, 12.91, 12.83, 12.77, 12.73. HRMS (MALDI): calcd for C32H50N2O8NaSi2 [M + Na]+ 669.2998, found 669.2994.
3.18. Synthesis of N3-Benzoyl-2′-O-isopropyl-5-methyluridine (3d)
Compound 2d (277 mg, 0.43 mmol) was dissolved in dry THF (4.3 mL). 1 M TBAF in THF (1.3 mL, 1.3 mmol) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 99/1 to 97/3) to afford compound 3d (150 mg, 0.37 mmol, 86%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.91 (dd, J = 1.1 Hz, 8.4 Hz, 2H), 7.67–7.64 (m, 2H), 7.49 (t, J = 7.8 Hz, 2H), 5.68 (d, J = 2.9 Hz, 1H), 4.29–4.24 (m, 2H), 4.06–3.91 (m, 3H), 3.85–3.81 (m, 1H), 2.72 (d, J = 6.0 Hz, 1H), 2.59 (br, 1H), 1.95 (d, J = 1.1 Hz, 3H), 1.19 (dd, J = 3.9 Hz, 6.1 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 168.67, 162.75, 149.38, 137.23, 135.16, 131.42, 130.47, 129.17, 110.91, 91.17, 85.14, 78.75, 72.80, 68.66, 61.43, 22.89, 22.07, 12.58. HRMS (MALDI): calcd for C20H24N2O7Na [M + Na]+ 427.1476, found 427.1472.
3.19. Synthesis of 3′-O-Acetyl-N3-benzoyl-5′-O-dimethoxytrityl-2′-O-isopropyl-5-methyluridine (4d)
Compound 3d (150 mg. 0.37 mmol) was dissolved in dry pyridine (4.0 mL). DMTrCl (139 mg, 0.41 mmol) was added and the reaction mixture was stirred at room temperature for 6 h. DMAP (5 mg, 0.04 mmol) and Ac2O (173 µL, 1.9 mmol) were added and the reaction mixture was stirred at room temperature for 18 h. The resulting reaction mixture was concentrated in vacuo and diluted with ethyl acetate. The combined organic layers were washed with saturated NaHCO3 aq. and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 3/1 to 2/1) to afford compound 4d (204 mg, 0.27 mmol, 74%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.93 (dd, J = 1.2 Hz, 8.4 Hz, 2H), 7.74 (d, J = 1.2 Hz, 1H), 7.66–7.62 (m, 1H), 7.49 (t, J = 7.8 Hz, 2H), 7.41 (dd, J = 1.3 Hz, 8.3 Hz, 2H), 7.34–7.26 (m, 7H), 6.86 (dd, J = 2.1 Hz, 8.9 Hz, 4H), 6.00 (d, J = 5.0 Hz, 1H), 5.37 (t, J = 5.1 Hz, 1H), 4.38 (t, J = 5.1 Hz, 1H), 4.29–4.27 (m, 1H), 3.80 (d, J = 0.8 Hz, 6H), 3.60 (dd, J = 2.3 Hz, 11.0 Hz, 1H), 3.38 (dd, J = 2.3 Hz, 10.9 Hz, 1H), 2.10 (s, 3H), 1.38 (d, J = 1.0 Hz, 3H), 1.15 (d, J = 6.0 Hz, 3H), 1.09 (d, J = 6.0 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 170.12, 168.80, 162.77, 158.87, 158.85, 149.46, 144.07, 135.10, 135.08, 134.97, 131.61, 130.50, 130.15, 129.10, 128.18, 128.10, 127.34, 113.37, 113.36, 111.41, 87.74, 87.27, 81.27, 78.07, 72.44, 70.87, 62.42, 55.29, 22.33, 22.06, 20.73, 11.73. HRMS (MALDI): calcd for C43H44N2O10Na [M + Na]+ 771.2888, found 771.2894.
3.20. Synthesis of 3′-O-Acetyl-N3-benzoyl-2′-O-isopropyl-5-methyluridine (5d)
Compound 4d (167 mg, 0.22 mmol) was dissolved in CH2Cl2 (2.2 mL). TFA (0.6 mL) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting reaction mixture was concentrated in vacuo and diluted with CHCl3. The combined organic layers were washed with saturated NaHCO3 aq. and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 1/1 to 1/2) to afford compound 5d (67 mg, 0.15 mmol, 68%) as a white form. 1H NMR (400 MHz, CDCl3): δ 7.91 (dd, J = 1.1 Hz, 8.4 Hz, 2H), 7.67–7.63 (m, 1H), 7.56 (d, J = 1.0 Hz, 1H), 7.49 (t, J = 7.8 Hz, 2H), 5.57 (d, J = 5.4 Hz, 1H), 5.27 (t, J = 4.8 Hz, 1H), 4.49 (t, J = 5.3 Hz, 1H), 4.23–4.21 (m, 1H), 3.94 (dd, J = 1.9 Hz, 12.5 Hz, 1H), 3.79–3.70 (m, 2H), 2.14 (s, 3H), 1.96 (d, J = 1.0 Hz, 3H), 1.10 (dd, J = 6.0 Hz, 11.0 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 170.36, 168.55, 162.71, 149.45, 137.83, 135.19, 131.38, 130.50, 129.18, 111.11, 92.20, 83.21, 72.60, 70.70, 61.75, 22.24, 20.84, 12.60. HRMS (MALDI): calcd for C22H26N2O8Na [M + Na]+ 469.1581, found 469.1583.
3.21. Synthesis of 2′-O-Isopropyl-5-methyluridine-5′-triphospahte (6d)
Compound 5d was dissolved in dry pyridine and the solution was evaporated to dryness in vacuo. Compound 5d (47 mg, 0.10 mmol) was dissolved in dry pyridine (0.5 mL) and 1,4-dioxane (0.5 mL). Then, 2-Chloro-4H-1,3,2-benzodioxaphosphorin-4-one (22 mg, 0.11 mmol) was added and the reaction mixture was stirred for 30 min at room temperature. Tris(tetrabutylammonium) hydrogen pyrophosphate (66 mg, 0.12 mmol) in DMF (1.0 mL) and tributylamine (79 µL, 0.33 mmol) were added and the reaction mixture was stirred for 1 h at room temperature. Next, 0.1 M I2 in pyridine/H2O (98:2, v/v) was added and the reaction mixture was stirred for 30 min at 0 °C. Then, 10% Na2S2O3 aq. was added and the reaction mixture was stirred for 10 min at room temperature. The reaction mixture was purified by ethanol precipitation. The precipitate was dissolved in 7% NH3 aq. and the reaction mixture was stirred for 2 h at room temperature. The solvent was removed in vacuo and the residue was purified by HPLC to give compound 6d (16 µmol, 16%). 1H NMR (400 MHz, D2O): δ7.79 (s, 1H), 6.01 (d, J = 5.4 Hz, 1H), 4.57 (t, J = 4.5 Hz, 1H), 4.30–4.27 (m, 4H), 3.96–3.90 (m, 1H), 1.96 (s, 3H), 1.20 (d, J = 6.2 Hz, 3H), 1.15 (d, J = 6.2 Hz, 3H). 31P NMR (120 MHz, D2O): δ − 8.71 (d, J = 14.4 Hz), −12.22 (d, J = 13.8 Hz), −22.79 (t, J = 14.1 Hz). HRMS (MALDI): calcd for C13H22N2O15P3 [M − H]− 539.0239, found 539.0236.
3.22. Synthesis of 2,2′-Anhydro-5′-O-(tert-butyldimethylsilyl)-5-methyluridine (8)
Compound 7 (500 mg, 2.1 mmol) was dissolved in dry DMF (2.0 mL). Imidazole (357 mg, 5.3 mmol) and TBSCl (362 mg, 2.4 mmol) were added and the reaction mixture was stirred at room temperature for 5 h. The resulting reaction mixture was concentrated in vacuo and diluted with ethyl acetate. The combined organic layers were washed with saturated NaHCO3 aq. and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 99/1 to 95/5) to afford compound 8 (556 mg, 1.6 mmol, 76%) as a white form. 1H NMR (400 MHz, DMSO-d6): δ7.77 (d, J = 1.2 Hz, 1H), 6.28 (d, J = 5.7 Hz, 1H), 5.94 (d, J = 4.4 Hz, 1H), 5.19 (d, J = 7.7Hz, 1H), 4.32 (br, 1H), 4.09–4.05 (m, 1H), 3.47–3.37 (m, 2H), 1.78 (d, J = 1.1Hz, 3H), 0.78 (s, 9H), −0.06 (s, 3H), −0.08 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 171.52, 159.14, 132.25, 116.90, 89.97, 88.37, 88.05, 74.22, 62.29, 25.69, 18.00, 13.50, −5.52. HRMS (MALDI): calcd for C16H26N2O5NaSi [M + Na]+ 377.1503, found 377.1506.
3.23. Synthesis of 2′-O-Acetoxylethyl-3′-O-acetyl-5′-O-(tert-butyldimethylsilyl)-5-methyluridine (9)
Ethylene glycol (5.0 mL) was added to 0.96 M Boran in THF (2.6 mL, 2.5 mmol). Compound 8 (400 mg. 1.0 mmol) and NaHCO3 (1 mg) were added and the reaction mixture was stirred at 150 °C for 24 h. The resulting reaction mixture was diluted with CHCl3. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was dissolved in dry pyridine (10 mL). DMAP (12 mg, 0.1 mmol) and Ac2O (930 µL, 10 mmol) were added and the reaction mixture was stirred at room temperature for 16 h. The resulting reaction mixture was concentrated in vacuo and diluted with ethyl acetate. The combined organic layers were washed with saturated NaHCO3 aq. and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 1/1) to afford compound 9 (250 mg, 0.50 mmol, 50%) as a white form. 1H NMR (400 MHz, CDCl3): δ 8.85 (s, 1H), 7.48 (d, J = 1.0 Hz, 1H), 6.06 (d, J = 5.2 Hz, 1H), 5.13 (t, J = 4.8 Hz, 1H), 4.22–4.21 (m, 1H), 4.14 (t, J = 4.3 Hz, 2H), 4.08 (t, J = 5.4 Hz, 1H), 3.98 (dd, J = 1.7 Hz, 11.8 Hz, 1H), 3.82–3.70 (m, 3H), 2.14 (s, 3H), 2.03 (s, 3H),1.92 (d, J = 0.9 Hz, 3H), 0.94 (s, 9H), 0.13 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 170.88, 170.25, 163.59, 150.37, 134.84, 111.30, 86.64, 82.40, 80.96, 70.22, 69.15, 63.15, 62.34, 25.98, 20.85, 20.77, 18.47, 12.60, −5.32, −5.40. HRMS (MALDI): calcd for C22H36N2O9NaSi [M + Na]+ 523.2082, found 523.2082.
3.24. Synthesis of 2′-O-Acetoxylethyl-3′-O-acetyl-5-methyluridine (10)
Compound 9 (250 mg, 0.50 mmol) was dissolved in dry THF (5.0 mL). 1M TBAF in THF (1.5 mL, 1.5 mmol) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (Hexane/Ethyl acetate = 1/9) to afford compound 10 (170 mg, 0.44 mmol, 88%) as a white form. 1H NMR (400 MHz, CDCl3): δ 9.12 (s, 1H), 7.41 (d, J = 1.2 Hz, 1H), 5.62 (d, J = 5.5 Hz, 1H), 5.32 (dd, J = 4.1 Hz, 5.2 Hz, 1H), 4.49 (t, J = 5.4 Hz, 1H), 4.24–4.22 (m, 1H), 4.20–4.11 (m, 2H), 3.95 (dd, J = 1.9 Hz, 12.5 Hz, 1H), 3.81–3.73 (m, 3H), 3.25 (br, 1H), 2.15 (s, 3H), 2.03 (s, 3H), 1.92 (d, J = 1.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 170.90, 170.35, 163.74, 150.45, 137.94, 111.27, 91.21, 83.23, 79.28, 70.67, 69.19, 63.24, 61.78, 20.86, 20.82, 12.51. HRMS (MALDI): calcd for C16H22N2O9Na [M + Na]+ 409.1218, found 409.1219.
3.25. Synthesis of 2′-O-Hydroxylethyl-5-methyluridine-5′-triphospahte (11)
Compound 10 was dissolved in dry pyridine and the solution was evaporated to dryness in vacuo. Compound 10 (39 mg, 0.10 mmol) was dissolved in dry pyridine (0.5 mL) and 1,4-dioxane (0.5 mL). Then, 2-Chloro-4H-1,3,2-benzodioxaphosphorin-4-one (22 mg, 0.11 mmol) was added and the reaction mixture was stirred for 30 min at room temperature. Tris(tetrabutylammonium) hydrogen pyrophosphate (66 mg, 0.12 mmol) in DMF (1.0 mL) and tributylamine (133 µL, 0.56 mmol) were added and the reaction mixture was stirred for 1 h at room temperature. Next, 0.1 M I2 in pyridine/H2O (98:2, v/v) was added and the reaction mixture was stirred for 30 min at 0 °C. Then, 10% Na2S2O3 aq. was added and the reaction mixture was stirred for 10 min at room temperature. The reaction mixture was purified by ethanol precipitation. The precipitate was dissolved in 7% NH3 aq. And the reaction mixture was stirred for 2 h at room temperature. The solvent was removed in vacuo and the residue was purified by HPLC to give compound 11 (16 µmol, 16%). 1H NMR (400 MHz, D2O): δ7.80 (s, 1H), 6.08 (d, J = 4.7 Hz, 1H), 4.56 (d, J = 4.9 Hz, 1H), 4.31–4.24 (m, 4H), 3.85–3.75 (m, 4H), 1.96 (s, 3H). 31P NMR (120 MHz, D2O): δ −9.49 (d, J = 11.7 Hz), −11.84 (d, J = 14.2 Hz), −22.72 (s). HRMS (MALDI): calcd for C12H20N2O16P3 [M − H]− 541.0031, found 541.0044.
3.26. Synthesis of 2′-O-Methoxyethyl-5-methyluridine-5′-triphospahte (13)
Compound
12 [
36] was dissolved in dry pyridine and the solution was evaporated to dryness in vacuo. Compound
12 (100 mg, 0.28 mmol) was dissolved in dry pyridine (1 mL) and 1,4-dioxane (2.0 mL). Then 2-Chloro-4
H-1,3,2-benzodioxaphosphorin-4-one (69 mg, 0.34 mmol) was added and the reaction mixture was stirred for 1 h at room temperature. Tris(tetrabutylammonium) hydrogen pyrophosphate (307 mg, 0.56 mmol) in DMF (1.0 mL) and tributylamine (133 µL, 0.56 mmol) were added and the reaction mixture was stirred for 1.5 h at room temperature. Next, 0.1 M I
2 in pyridine/H
2O (98:2,
v/
v) was added and the reaction mixture was stirred for 1 h at room temperature. Then, 10% Na
2S
2O
3 aq. was added and the reaction mixture was stirred for 10 min at room temperature. The reaction mixture was purified by ethanol precipitation. The precipitate was dissolved in 7% NH
3 aq. and the reaction mixture was stirred for 1 h at room temperature. The solvent was removed in vacuo and the residue was purified by HPLC to give compound
13 (44 µmol, 16%).
1H NMR (400 MHz, D
2O): δ7.79 (s, 1H), 6.06 (d,
J = 5.0 Hz, 1H), 4.55 (t,
J = 4.8 Hz, 1H), 4.32–4.23 (m, 4H), 3.87–3.85 (m, 2H), 3.66–3.64 (m, 2H), 3.37 (s, 3H), 1.96 (s, 3H).
31P NMR (120 MHz, D
2O): δ −9.12(d,
J = 13.7 Hz), −11.96 (d,
J = 14.5 Hz), −22.69 (t,
J = 14.0 Hz). HRMS (MALDI): calcd for C
13H
22N
2O
15P
3 [M + Na]
+ 579.0153, found 579.0144.
3.27. Expression and Purification of KOD DGLNK
KOD DGLNK was expressed and purified according to a previous report [
23].
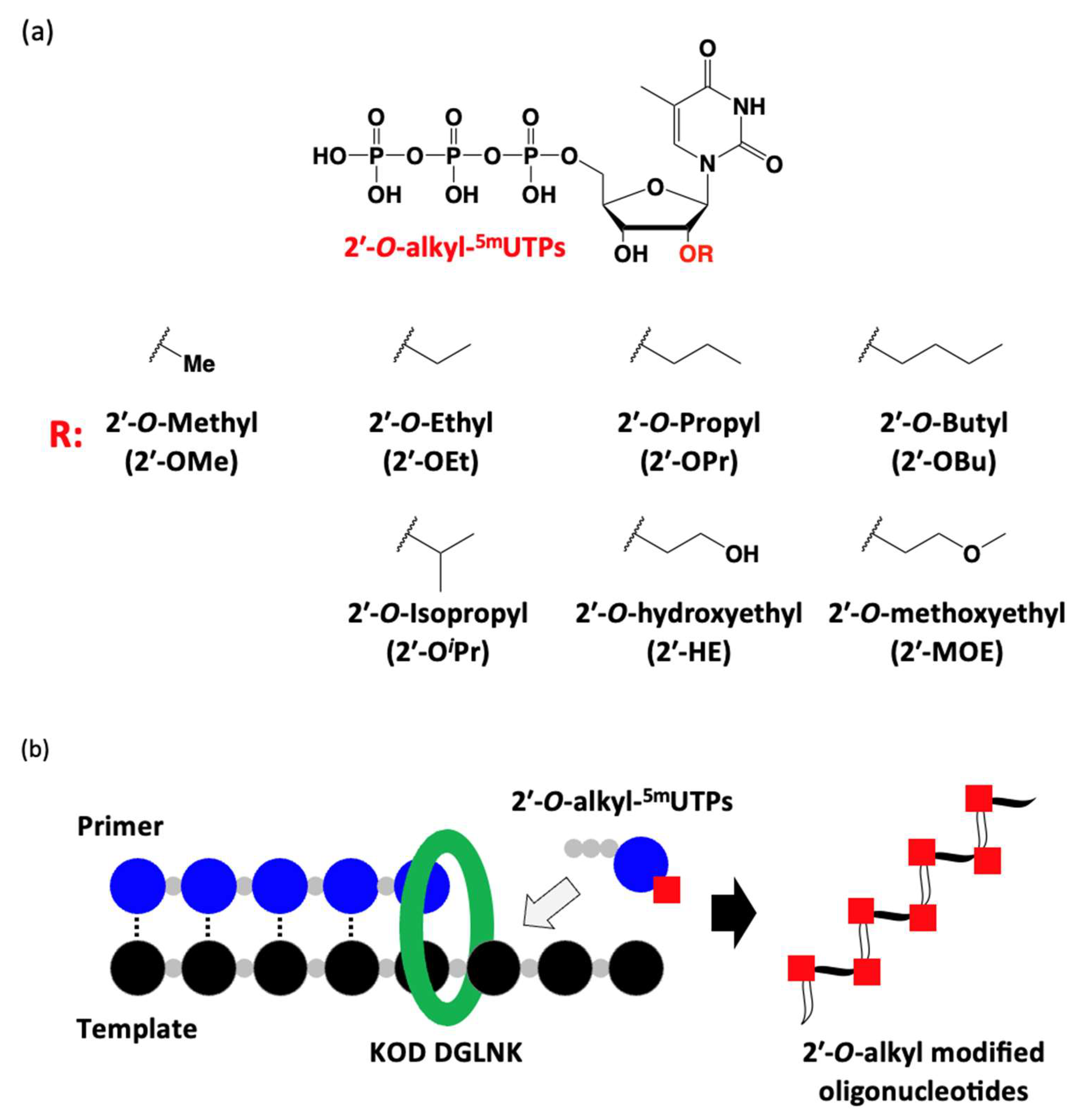
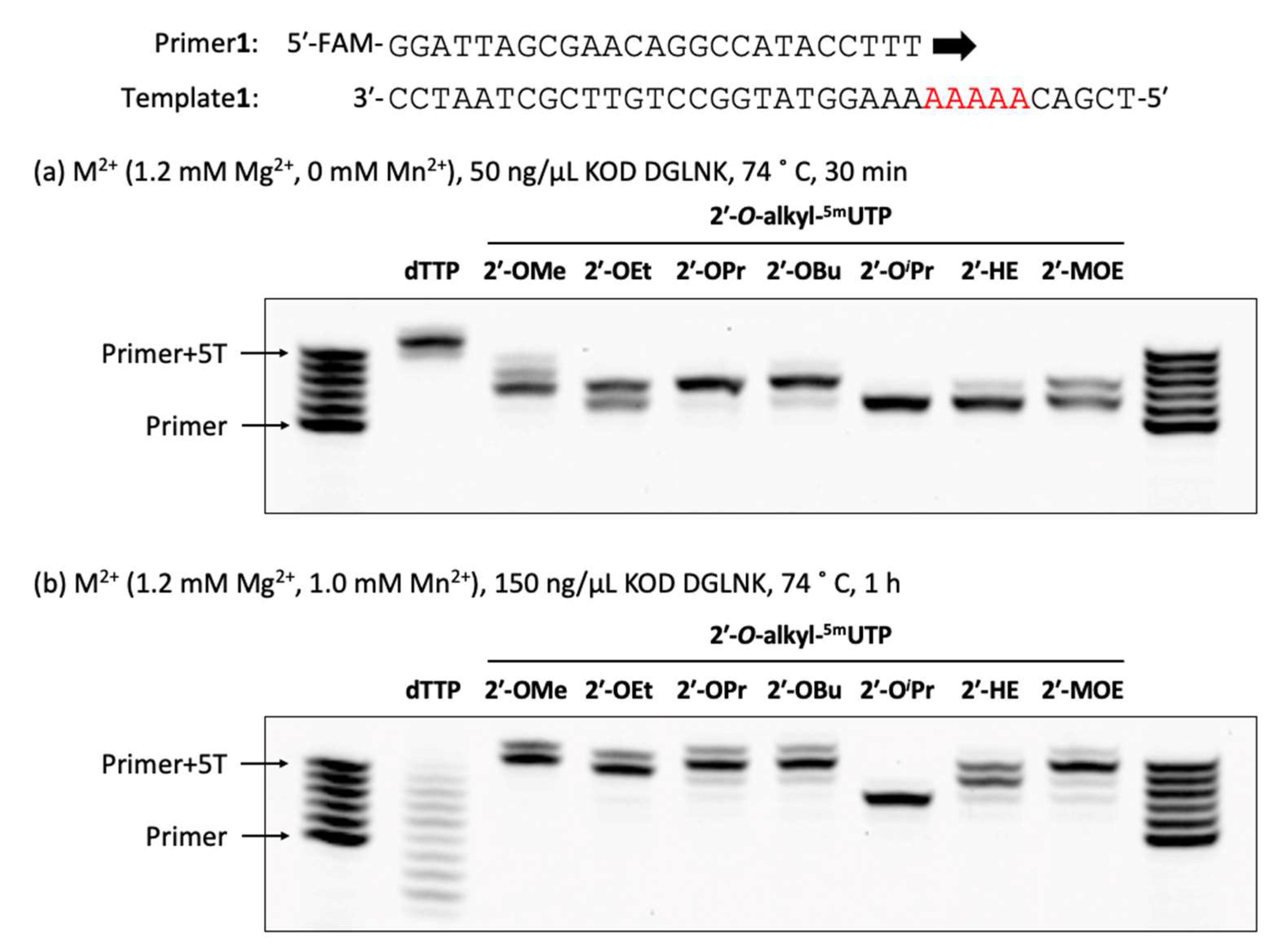
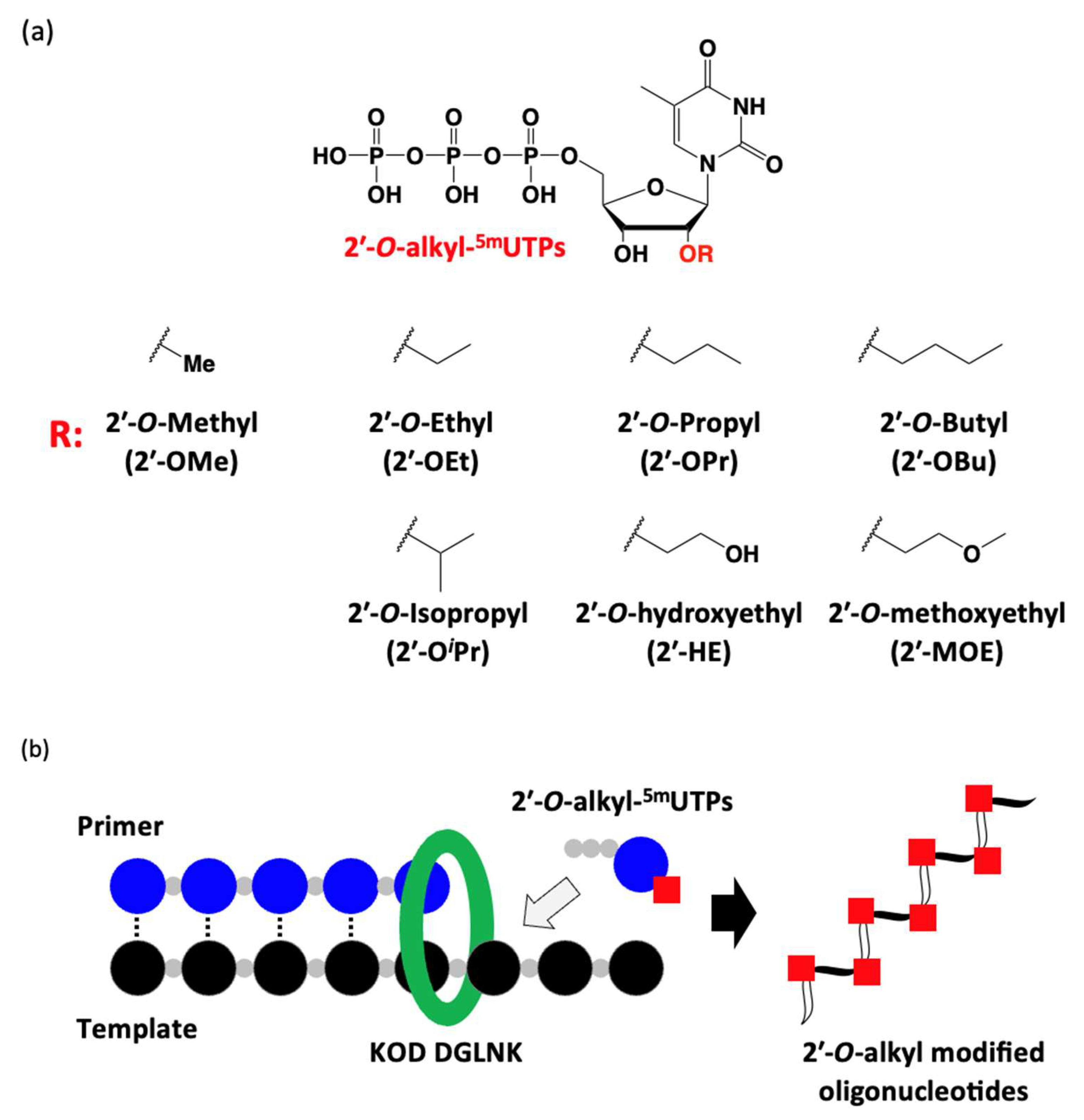
3.28. Polymerase Incorporation of 2′-O-Alkyl-5-methyluridine Triphosphates into Oligonucleotide
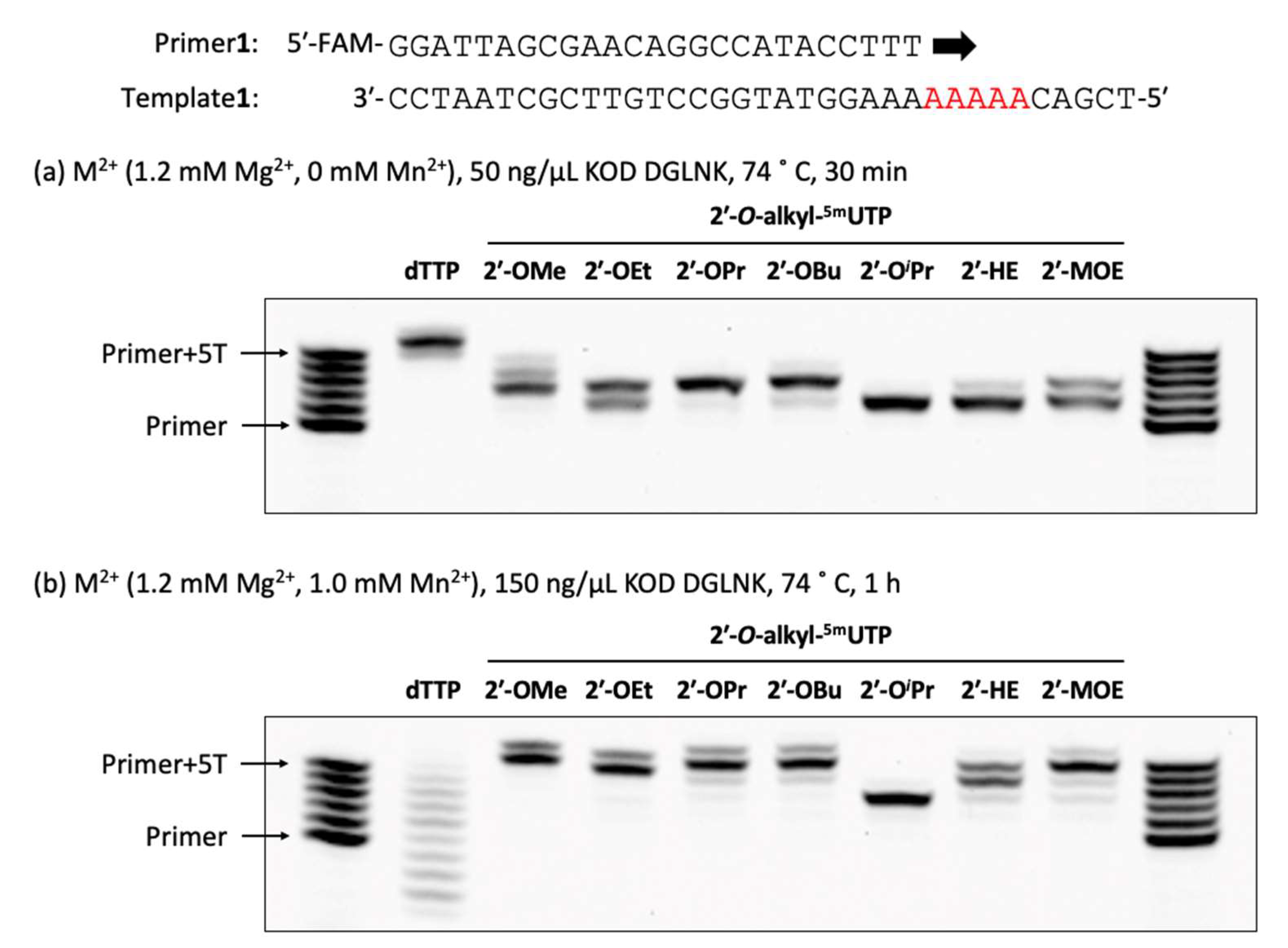
The reaction mixture [1 × KOD Dash® buffer (TOYOBO, Osaka, Japan), DNA or 2′-OMe primer (Primer1 or Primer2: 0.4 µM), DNA template (Template1: 0.5 µM), dTTP or 2′-O-alkyl-5mUTPs (0.2 mM), and MnSO4 (0 or 0.1 or 0.5 or 1.0 mM)] was prepared. Then, the reaction mixture was denatured at 94 °C for 1 min and annealed at 25 °C for 1 h (1.2 °C/min), KOD DGLNK (50 or 150 ng/µL) was added and incubated at 74 °C for 30 or 60 min. The reaction was stopped by stop buffer (aqueous 3 mM EDTA containing 0.1% bromophenol blue and aqueous 7 M Urea) and heated for 5 min at 95 °C. The reaction was analyzed by using a 20% denaturing urea-PAGE (70 min, 55 °C).
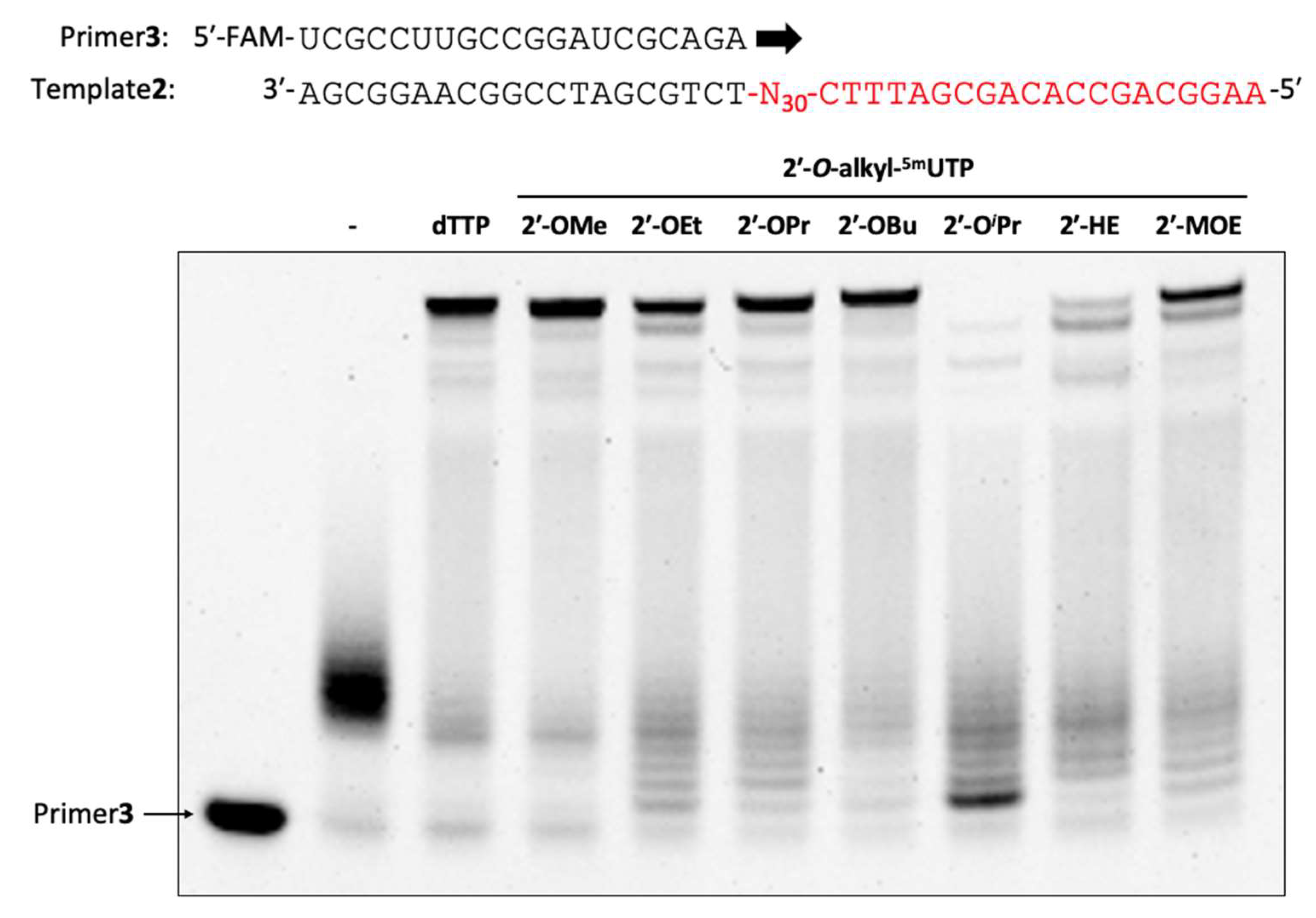
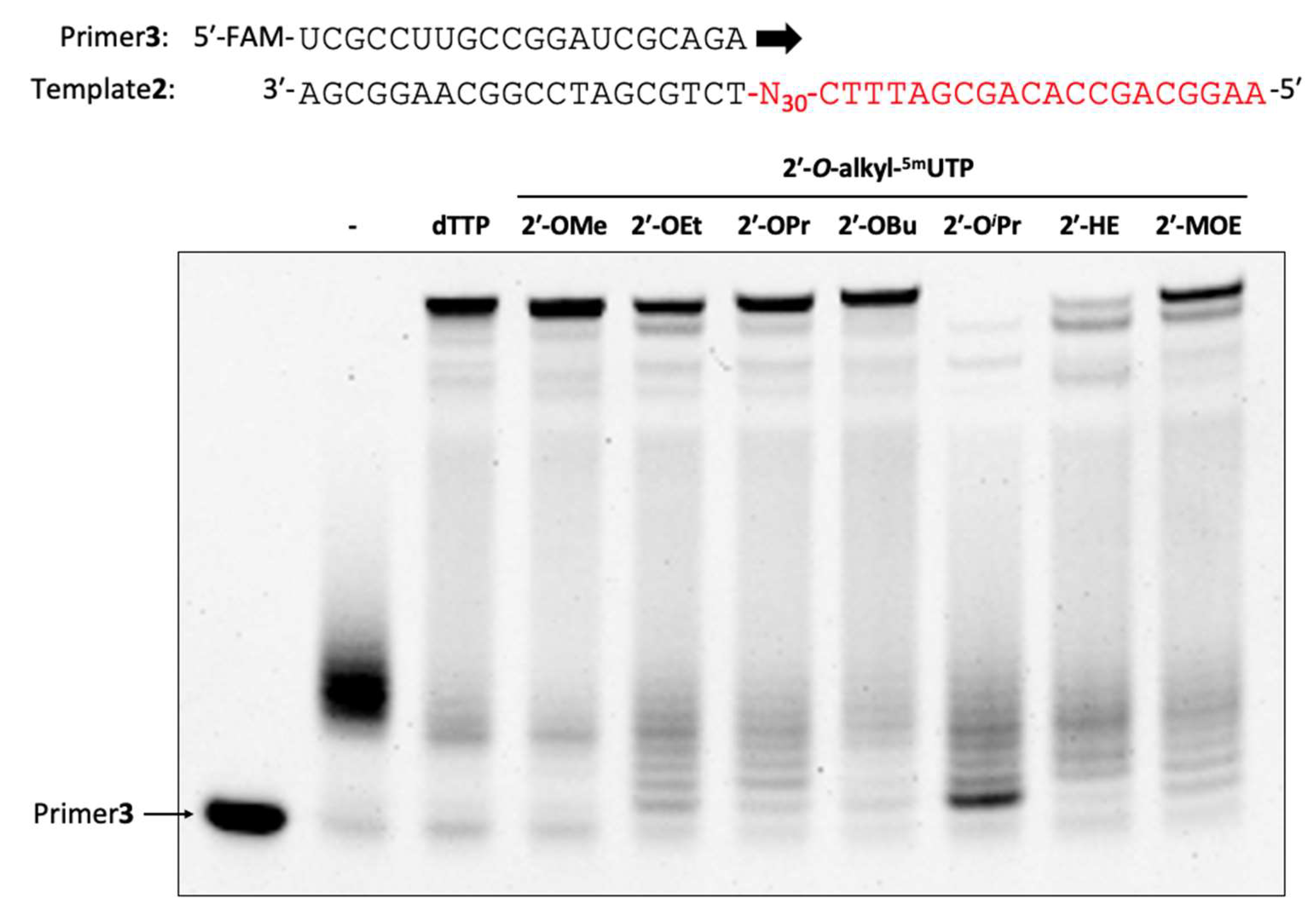
3.29. Enzymatic Synthesis of 2′-O-Alkyl-modified Oligonucleotide Libraries
The reaction mixture [1× KOD Dash® buffer (TOYOBO, Osaka, Japan), 2′-OMe primer (Primer3: 0.4 µM), DNA template (Template2: 0.5 µM), dTTP or 2′-O-alkyl-5mUTPs (0.2 mM), 2′-OMe-ATP (0.2 mM), 2′-OMe-GTP (0.2 mM), 2′-OMe-CTP (0.2 mM), MnSO4 (1.0 or 3.0 or 5.0 mM) and KOD DGLNK (300 ng/µL)] was prepared. Then, the reaction mixture was denatured at 94 °C for 1 min and annealed at 25 °C for 1 h (1.2 °C/min), KOD DGLNK was added and incubated from 60 °C for 1 h, 60 °C to 74 °C for 4 or 8 h, and 74 °C for 1 h. After polymerization, DNase1 (95 mU/µL) was added and incubated at 37 °C for 30 min. The reaction was stopped by stop buffer (aqueous 3 mM EDTA containing 0.1% bromophenol blue and aqueous 7 M Urea) and heated for 5 min at 95 °C. The reaction was analyzed by using a 10% denaturing urea-PAGE (32 min, 55 °C).
3.30. Preparation of 2′-O-ALKYL-modified Oligonucleotide Libraries for Analysis of Properties
The reaction mixture [1× KOD Dash® buffer (TOYOBO, Osaka, Japan), 2′-OMe primer (Primer3: 0.4 µM), DNA template (Template2: 0.5 µM), 2′-O-alkyl-5mUTPs (0.2 mM), 2′-OMe-ATP (0.2 mM), 2′-OMe-GTP (0.2 mM), 2′-OMe-CTP (0.2 mM), MnSO4 (1.0 or 3.0 mM) and KOD DGLNK (300 ng/µL)] was prepared. Then, the reaction mixture was denatured at 94 °C for 1 min and annealed at 25 °C for 1 h (1.2 °C/min), KOD DGLNK (300 ng/µL) was added and incubated from 60 °C for 1 h, 60 °C to 74 °C for 4 or 8 h, and 74 °C for 1 h. After polymerization, DNase1 (95 mU/µL) was added and incubated at 37 °C for 30 min. Then, Proteinase K (36 mU/µL) was added and incubated at 37 °C for 30 min. The reaction was stopped by stop buffer (0.1% bromophenol blue and aqueous 7 M Urea) and heated for 5 min at 95 °C. The oligonucleotides were purified by using a 10% denaturing urea-PAGE (120 min, 55 °C).
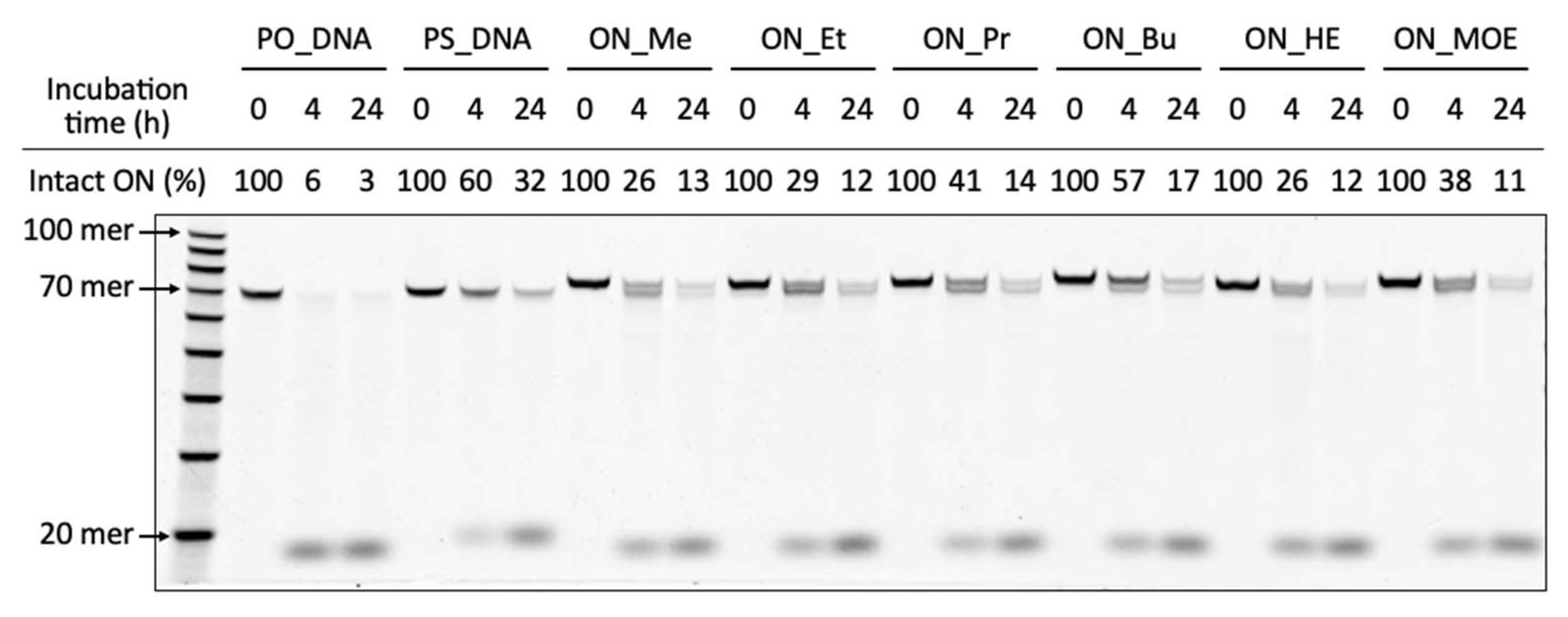
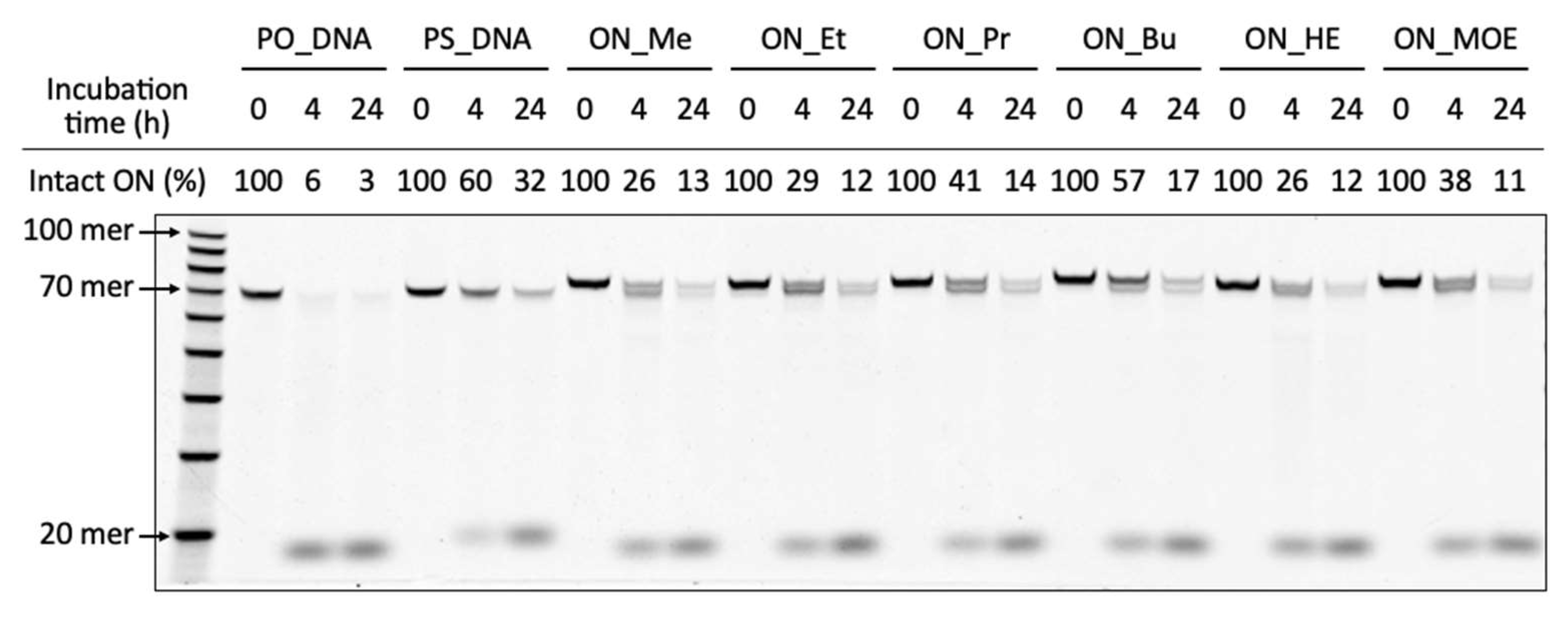
3.31. Enzymatic Stability of Oligonucleotide Libraries
The reaction mixture [oligonucleotide libraries (0.1 µM) and 50% FBS] was prepared. Then, the reaction mixture was incubated at 37 °C. After incubation, the reaction mixture was heated at 94 °C for 5 min. Then, Proteinase K (36 mU/µL) was added and incubated at 37 °C for 30 min. The reaction was stopped by stop buffer (aqueous 3 mM EDTA containing 0.1% bromophenol blue and aqueous 7 M Urea). After adding Cy5-labeled DNA (83 nM) as an internal standard to the reaction mixtures, they were heated for 5 min at 95 °C. The reaction was analyzed by using a 10% denaturing urea-PAGE (30 min, 55 °C).
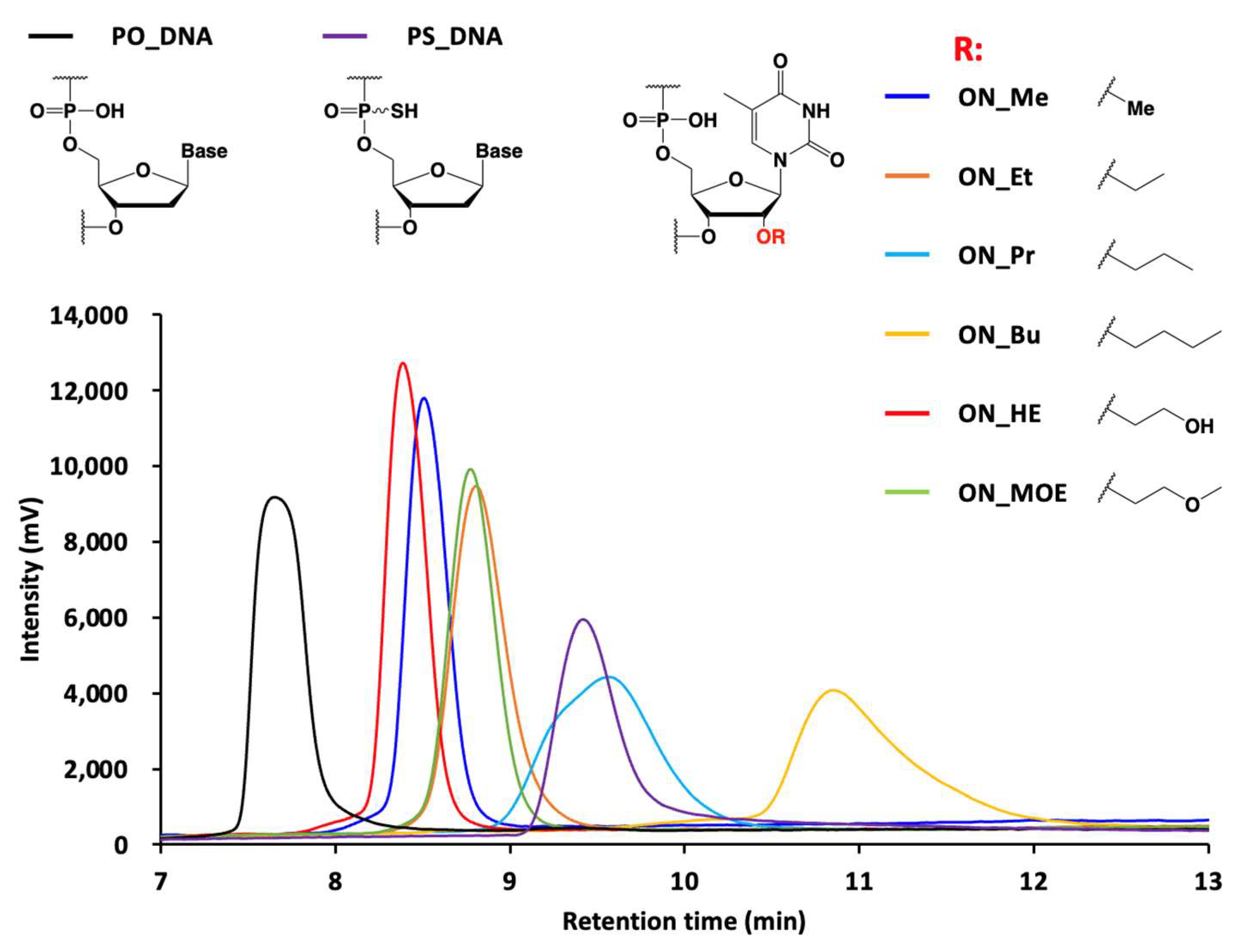
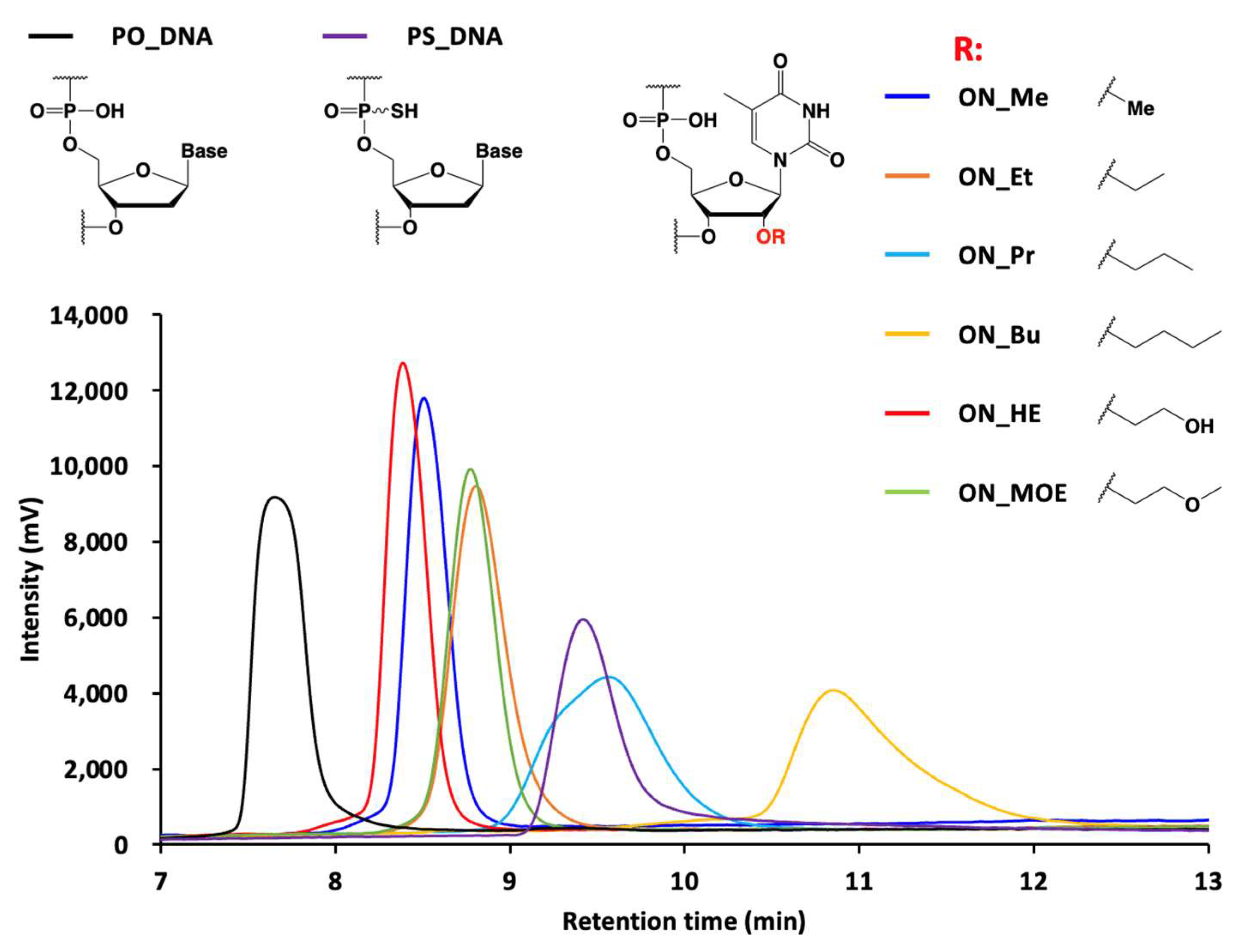
3.32. Ion Pair RP-HPLC Analysis of Oligonucleotide Libraries
Oligonucleotide libraries (5.0 pmol) were injected onto a XBridge Oligonucleotide BEH C18 column. The column temperature was 50 °C, with a flow rate of 1.0 mL/min, and detection at 260 nm. Mobile phase A consisted of 100 mM triethylamine acetic acid solution, pH 6.9, and mobile phase B was acetonitrile. The column was initially maintained at 10% mobile phase B, and then at a gradient of 10% to 40% over 15 min.
3.33. Ion Pair RP-HPLC Analysis of Nucleotide Triphosphates
Nucleotide triphosphates (700 pmol) were injected onto a XBridge Oligonucleotide BEH C18 column. The column temperature was 50 °C, with a flow rate of 1.0 mL/min, and detection at 260 nm. Mobile phase A consisted of 100 mM triethylamine acetic acid solution (pH 6.9), and mobile phase B was acetonitrile. The column was initially maintained at 5% mobile phase B, and then at a gradient of 5% to 20% over 15 min.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}