
Asymmetric Henry Reaction Using Cobalt Complexes with Bisoxazoline Ligands Bearing Two Fluorous Tags


Abstract
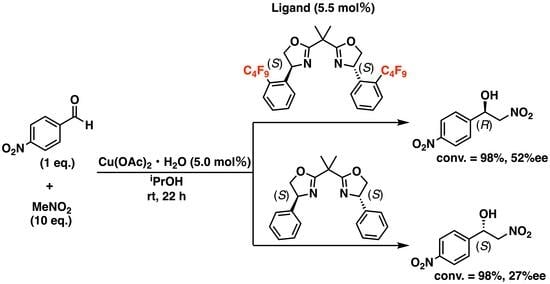
:
1. Introduction
2. Results and Discussions
3. Experimental Section
3.1. Materials and Reagents
3.2. Analytical Instruments
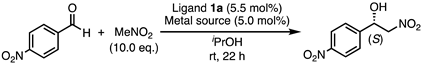
3.3. General Procedure for the Henry Reaction
3.4. Synthesis
- Methyl (S)-2-amino-3-(4-iodophenyl)propanoate hydrochloride (3) [29]. Under N2 atmosphere, a solution of 4-iodo-L-phenylalanine 2 (5.02 g, 17.23 mmol) in dry-MeOH (80 mL) was stirred at 0 °C, then SOCl2 (1.5 mL, 20.55 mmol) was added dropwise. The reaction mixture was warmed to room temperature and then stirred for 22 h. The reaction mixture was concentrated. After the addition of Et2O, the suspension was purified by filtration to give the desired product 3 (5.84 g, 17.10 mmol, 99%). White solid; mp 191–192 °C; 1H NMR (400 MHz, (CD3)2SO) δ 8.55 (br s, 3H), 7.70 (d, J = 8.4 Hz, 2H), 7.06 (d, J = 8.4 Hz, 2H), 4.30–4.26 (m, 1H), 3.69 (s, 3H), 3.14–3.04 (m, 2H).
- Methyl (S)-2-((tert-butoxycarbonyl)amino)-3-(4-iodophenyl)propanoate (4) [30]. A solution of methyl (S)-2-amino-3-(4-iodophenyl)propanoate hydrochloride 3 (252.3 mg, 0.74 mmol) and Et3N (213 μL, 1.53 mmol) in dry-DCM (4 mL) was stirred at 0 °C. A solution of Boc2O (167.7 mg, 0.77 mmol) in dry-DCM (3 mL) was slowly added to the mixture. Then the reaction temperature rose to room temperature and the mixture was stirred for a further 20 h. The mixture was diluted with brine. The reaction mixture was extracted with DCM. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:1) to give the desired product 4 (250.2 mg, 0.62 mmol, 84%). White solid; mp 74–75 °C; 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 8.4 Hz, 2H), 6.88 (d, J = 8.0 Hz, 2H), 4.98–4.96 (m, 1H), 4.58–4.56 (m, 1H), 3.72 (s, 3H), 3.10–2.95 (m, 2H), 1.42 (s, 9H).
- Methyl (S)-2-((tert-butoxycarbonyl)amino)-3-(4-(perfluorobutyl)phenyl)propanoate (5a). To a mixture of 4 (1.03 g, 2.54 mmol), Cu powder (1.60 g, 25.12 mmol), 2,2′-bipyridyl (38.8 mg, 0.25 mmol), and DMF (5 mL) was added a 1,1,1,2,2,3,3,4,4-nonafluoro-4-iodobutane (0.8 mL, 4.76 mmol). The mixture was stirred at 120 °C for 19 h. The mixture was filtered through Celite, and the solids were washed with MeOH. The filtrate was concentrated. Then 1 M HCl aq. was added to the suspension. The aqueous layer was extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:3) to give the desired product 5a (1.08 g, 2.18 mmol, 86%). Yellow solid; mp 40–41 °C; 1H NMR (270 MHz, CDCl3) δ 7.52 (d, J = 8.1 Hz, 2H), 7.30–7.27 (m, 2H), 5.06–5.03 (m, 1H), 4.65–4.62 (m, 1H), 3.72 (s, 3H), 3.25–3.04 (m, 2H), 1.40 (s, 9H); 19F NMR (376 MHz, CDCl3) δ −80.95 (3F), −110.79 (2F), −122.71 (2F), −125.51 (2F); 13C NMR (101 MHz, CDCl3) δ 172.03, 155.04, 140.77, 129.74, 127.64, 127.07,116.14–110.67, 80.26, 54.26, 52.44, 38.49, 28.31; HRMS-DART (m/z):[M + H]+ calcd for C19H21F9NO4: 498.1327, found: 498.1320.
- tert-Butyl (S)-(1-hydroxy-3-(4-(perfluorobutyl)phenyl)propan-2-yl)carbamate (6a). Lithium chloride (348.0 mg, 8.21 mmol) and sodium borohydride (312.5 mg, 8.26 mmol) were added to a solution of 5a (2.03 g, 4.07 mmol) in dry-THF (15 mL) and dry-EtOH (30 mL) and the white suspension was stirred for 23 h. Then, 1 M HCl aq. was added to the reaction mixture until pH = 4. The solution was concentrated. The aqueous layer was extracted with DCM. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:1) to give the desired product 6a (1.82 g, 3.88 mmol, 95%). White solid; mp 89 °C; 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 4.76 (d, J = 7.6 Hz, 1H), 3.90 (s, 1H), 3.71–3.54 (m, 2H), 2.93 (d, J = 6.8 Hz, 2H), 2.15 (s, 1H), 1.39 (s, 9H); 19F NMR (376 MHz, CDCl3) δ −80.95 (3F), −110.68 (2F), −122.69 (2F), −125.54 (2F); 13C NMR (101 MHz, CDCl3) δ 155.94, 142.58, 129.67, 127.41, 127.07, 118.99–115.88, 79.96, 64.13, 53.43, 39.49, 37.40, 28.33; HRMS-DART (m/z):[M + H]+ calcd for C18H21F9NO3: 470.1378, found: 470.1373.
- (S)-2-Amino-3-(4-(perfluorobutyl)phenyl)propan-1-ol hydrochloride (7a). To a solution of tert-butyl (S)-(1-hydroxy-3-(4-(perfluorobutyl)phenyl)propan-2-yl)carbamate 6a (0.85 g, 1.82 mmol) in ethyl acetate (6 mL) was added a conc. HCl (6 mL). The mixture was stirred at room temperature for 35 min. Excess water was separated as the toluene azeotrope. After the addition of Et2O, the suspension was purified by filtration to give the desired product 7a (0.71 g, 1.75 mmol, 95%). White solid; mp 166–167 °C; 1H NMR (400 MHz, (CD3)2SO) δ 8.06 (s, 3H), 7.57 (d, J = 8.0 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 5.28 (t, J = 4.4 Hz, 1H), 3.64–3.43 (m, 2H), 3.10–2.95 (m, 2H); 19F NMR (376 MHz, (CD3)2SO) δ −80.45 (3F), −109.43 (2F), −122.26 (2F), −125.11 (2F); 13C NMR (101 MHz, (CD3)2SO) δ 142.54, 130.75, 127.47, 126.10, 116.43–108.50, 60.17, 53.91, 35.02; HRMS-DART (m/z):[M − Cl]+ calcd for C13H13F9NO: 370.0853, found: 370.0844.
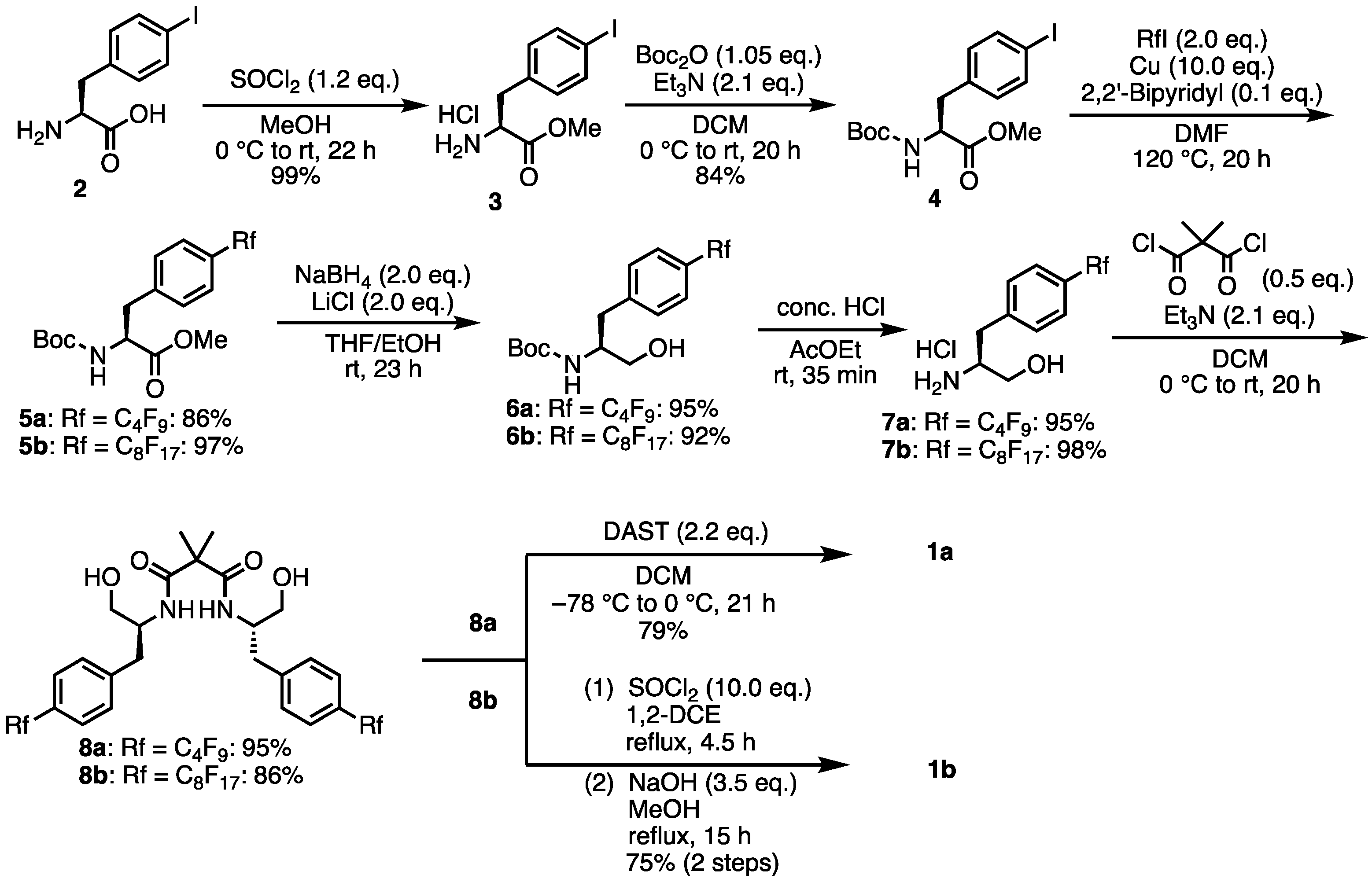
- N1,N3-Bis((S)-1-hydroxy-3-(4-(perfluorobutyl)phenyl)propan-2-yl)-2,2-dimethylmalonamide (8a). Under N2 atmosphere, to a solution of 7a (1.39 g, 3.44 mmol) and Et3N (1 mL, 7.18 mmol) in dry-DCM (30 mL) was added solution of dimethylmalonyl dichloride (227 μL, 1.72 mmol) in dry-DCM (11 mL) at 0 °C. Then, the reaction temperature rose to room temperature and the mixture was stirred for 20 h. The mixture was concentrated. The crude was purified by silica gel chromatography (MeOH:CHCl3 = 1:20) to give the desired product 8a (1.37 g, 1.64 mmol, 95%). White solid; mp 116–117 °C; 1H NMR (270 MHz, CDCl3) δ 7.49 (d, J = 8.1 Hz, 4H), 7.31 (d, J = 8.4 Hz, 4H), 6.49 (d, J = 8.4 Hz, 2H), 4.28 (br s, 2H), 3.74 (dd, J = 11.3, 3.2 Hz, 2H), 3.47 (dd, J = 11.3, 6.5 Hz, 2H), 2.95–2.73 (m, 4H), 1.16 (s, 6H); 19F NMR (376 MHz, CDCl3) δ −81.02 (6F), −110.81 (4F), −122.77 (4F), −125.54 (4F); 13C NMR (101 MHz, CDCl3) δ 173.98, 141.95, 129.42, 127.35, 127.01, 121.74–111.20, 64.16, 52.50, 49.78, 36.71, 23.26; HRMS-DART (m/z):[M + H]+ calcd for C31H29F18N2O4: 835.1840, found: 835.1824.
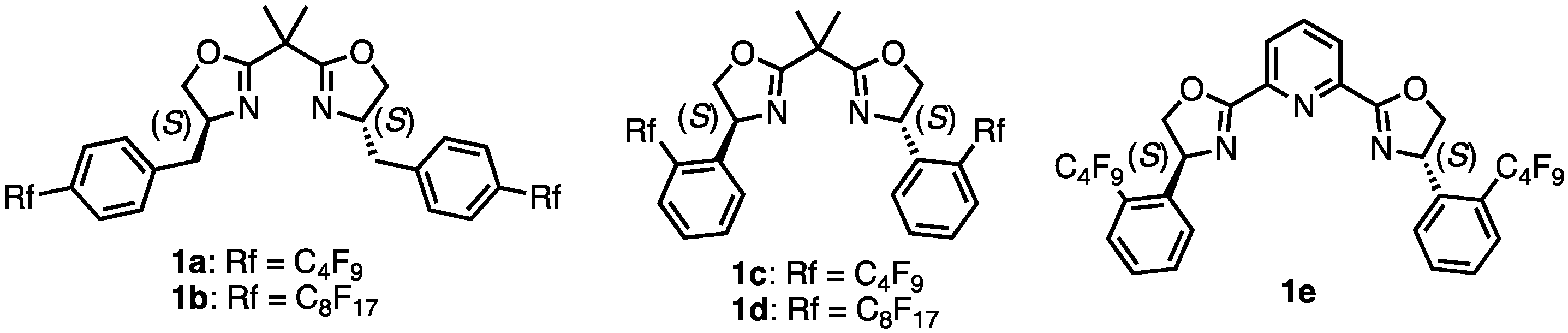
- (4S,4′S)-2,2′-(Propane-2,2-diyl)bis(4-(4-(perfluorobutyl)benzyl)-4,5-dihydrooxazole) (1a). Under N2 atmosphere, to a solution of 8a (444.2 mg, 0.53 mmol) in dry-DCM (30 mL) was added a solution of diethylaminosulfur trifluoride (155 μL, 1.17 mmol) in dry-DCM (15 mL) at −78 °C. After stirring at 0 °C for 11 h, the mixture was washed with saturated aqueous NaHCO3 and the aqueous layer was extracted with DCM. The combined organic layers were dried over Na2SO4 and concentrated. The crude was purified by silica gel chromatography (MeOH:CHCl3 = 1:20) to give the desired product 1a (337.3 mg, 0.42 mmol, 79%). Yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8.4 Hz, 4H), 7.34 (d, J = 8.0 Hz, 4H), 4.45–4.39 (m, 2H), 4.22 (t, J = 8.8 Hz, 2H), 3.97–3.94 (m, 2H), 3.05–3.00 (m, 2H), 2.85–2.80 (m, 2H), 1.41 (s, 6H); 19F NMR (376 MHz, CDCl3) δ −81.99 (6F), −110.76 (4F), −122.67 (4F), −125.54 (4F); 13C NMR (101 MHz, CDCl3) δ 169.70, 141.98, 129.96, 127.13, 126.88, 118.97–108.83, 71.79, 66.43, 41.06, 38.61, 24.08; HRMS-DART (m/z):[M + H]+ calcd for C31H25F18N2O: 799.1629, found: 799.1627.
- Methyl (S)-2-((tert-butoxycarbonyl)amino)-3-(4-(perfluorooctyll)phenyl)propanoate (5b). To a mixture of 4 (103.3 mg, 0.25 mmol), Cu powder (165.6 mg, 2.61 mmol), 2,2′-bipyridyl (4.0 mg, 0.03 mmol), and DMF (1 mL) was added a 1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8-heptadecafluoro-8-iodooctane (130 μL, 0.49 mmol). The mixture was stirred at 120 °C for 21 h. The mixture was filtered through Celite, and the solids were washed with MeOH. The filtrate was concentrated. Then, 1 M HCl aq. was added to the suspension. The aqueous layer was extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:3) to give the desired product 5b (0.17 g, 0.25 mmol, 97%). White solid; mp 70–71 °C; 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 5.02 (d, J = 7.2 Hz, 1H), 4.66–4.61 (m, 1H), 3.72 (s, 3H), 3.24–3.05 (m, 2H), 1.40 (s, 9H); 19F NMR (376 MHz, CDCl3) δ −80.83 (3F), −110.51 (2F), −121.17 (2F), −121.77 (6F), −122.63 (2F), −126.03 (2F); 13C NMR (101 MHz, CDCl3) δ 171.99, 154.99, 140.71, 129.68, 127.71, 127.04, 120.07–100.39, 80.21, 54.21,52.38, 38.46, 28.25; HRMS-DART (m/z):[M + H]+ calcd for C23H21F17NO4: 698.1199, found: 698.1188.
- tert-Butyl (S)-(1-hydroxy-3-(4-(perfluorooctyl)phenyl)propan-2-yl)carbamate (6b). Lithium chloride (211.3 mg, 4.98 mmol) and sodium borohydride (188.8 mg, 4.99 mmol) were added to a solution of 5b (1.70 g, 2.43 mmol) in dry-THF (15 mL) and dry-EtOH (30 mL) and the white suspension was stirred for 13 h. Then, 1 M HCl aq. was added to the reaction mixture until pH = 4. The solution was concentrated. The aqueous layer was extracted with DCM. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:2) to give the desired product 6b (1.50 g, 2.24 mmol, 92%). White solid; mp 186–187 °C; 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 4.74 (d, J = 7.2 Hz, 1H), 3.91 (s, 1H), 3.71–3.55 (m, 2H), 2.93 (d, J = 6.8 Hz, 2H), 2.10 (s, 1H), 1.39 (s, 9H); 19F NMR (376 MHz, CDCl3) δ −80.62 (3F), −110.32 (2F), −121.14 (2F), −121.71 (6F), −122.58 (2F), −125.97 (2F); 13C NMR (101 MHz, CDCl3) δ 155.95, 142.83, 129.67, 127.27, 127.14, 118.64–108.61, 79.94, 64.04, 53.59, 37.52, 28.57; HRMS-DART (m/z):[M + H]+ calcd for C22H21F17NO3: 670.1250, found: 670.1227.
- (S)-2-Amino-3-(4-(perfluorooctyl)phenyl)propan-1-ol hydrochloride (7b). To a solution of tert-butyl (S)-(1-hydroxy-3-(4-(perfluorooctyl)phenyl)propan-2-yl)carbamate 6b (1.44 g, 2.15 mmol) in ethyl acetate (8 mL) was added a conc. HCl (8 mL). The mixture was stirred at room temperature for 35 min. Excess water was separated as the toluene azeotrope. After the addition of Et2O, the suspension was purified by filtration to give the desired product 7b (1.28 g, 2.12 mmol, 98%). White solid; mp 98–99 °C; 1H NMR (400 MHz, (CD3)2SO) δ 8.10 (s, 3H), 7.65 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 5.41–5.40 (m, 1H), 3.54–3.37 (m, 2H), 3.05–2.92 (m, 2H); 19F NMR (376 MHz, (CD3)2SO) δ −80.13 (3F), −109.22 (2F), −120.95 (2F), −121.33 to −121.58 (m, 6F), −122.37 (2F), −125.63 (2F); 13C NMR (101 MHz, (CD3)2SO) δ 142.47, 130.74, 127.45, 126.47, 122.63–110.16, 60.22, 53.84, 35.08; HRMS-DART (m/z):[M − Cl]+ calcd for C17H13F17NO: 570.0726, found: 570.0722.
- N1,N3-bis((S)-1-hydroxy-3-(4-(perfluorooctyl)phenyl)propan-2-yl)-2,2-dimethylmalonamide (8b). Under N2 atmosphere, to a solution of 7b (1.23 g, 2.04 mmol) and Et3N (595 μL, 4.27 mmol) in dry-DCM (12 mL) was added a solution of dimethylmalonyl dichloride (135 μL, 1.02 mmol) in dry-DCM (12 mL) at 0 °C. Then, the reaction temperature rose to room temperature and the mixture was stirred for 20 h. The mixture was concentrated. The crude was purified by silica gel chromatography (MeOH:CHCl3 = 1:20) to give the desired product 8b (1.09 g, 0.88 mmol, 86%). White solid; mp 142 °C; 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8.0 Hz, 4H), 7.32 (d, J = 8.4 Hz, 4H), 6.44 (d, J = 8.4 Hz, 2H), 4.27–4.24 (m, 2H), 3.76–3.72 (m, 2H), 3.52–3.48 (m, 2H), 2.95–2.90 (m, 2H), 2.84–2.79 (m, 2H), 1.18 (s, 6H); 19F NMR (376 MHz, CDCl3) δ −80.69 (6F), −110.57 (4F), −121.17 (4F), −121.80 (12F), −122.63 (4F), −126.02 (4F); 13C NMR (101 MHz, CD3OD) δ 174.38, 143.64, 129.69, 126.46, 126.40, 118.58, 115.99, 115.71, 113.51, 110.92, 110.62, 110.29, 108.27, 63.14, 52.59, 49.79, 36.28, 22.72; HRMS-DART (m/z):[M + H]+ calcd for C39H29F34N2O4: 1235.1584, found: 1235.1556.
- (4S,4′S)-2,2′-(Propane-2,2-diyl)bis(4-(4-(perfluorohexyl)benzyl)-4,5-dihydrooxazole) (1b). The 8b (673.1 mg, 0.545 mmol) was dissolved in dry-1,2-DCE (110 mL) and heated to 80 °C. Thionyl chloride (4 mL, 5.48 mmol) was added and stirred at 80 °C for 5 h. The reaction mixture was concentrated and quenched with a saturated NaHCO3 solution. The mixture was extracted with DCM and the combined organic layers were dried over Na2SO4 filtered and the solvent was removed under reduced pressure. The residue was dissolved in a solution of NaOH (84.1 mg, 2.10 mmol) in MeOH (153 mL) and heated to 60 °C for 15 h. The solvent was removed under reduced pressure and the resulting residue was partitioned between DCM and H2O. The aqueous layer was extracted with DCM. The combined organic layers were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified by silica gel chromatography (MeOH:CHCl3 = 1:100) to give the desired product 1b (0.49 g, 0.41 mmol, 75%). White solid; mp 78–79 °C; 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8.4 Hz, 4H), 7.34 (d, J = 7.6 Hz, 4H), 4.46–4.39 (m, 2H), 4.24–4.19 (m, 2H), 3.97–3.93 (m, 2H), 3.02 (dd, J = 14.0, 5.2 Hz, 2H), 2.83 (dd, J = 13.6, 6.8 Hz, 2H), 1.41 (s, 6H); 19F NMR (376 MHz, CDCl3) δ −80.65 (6F), −110.36 (4F), −121.19 (4F), −121.80 (12F), −122.63 (4F), −126.02 (4F); 13C NMR (101 MHz, CDCl3) δ 169.62, 141.93, 129.93, 127.18, 126.91, 126.85, 126.79, 121.86–110.25, 71.75, 66.40, 40.95, 38.57, 24.02; HRMS-DART (m/z):[M + H]+ calcd for C39H25F34N2O2: 1199.1373, found: 1199.1360.
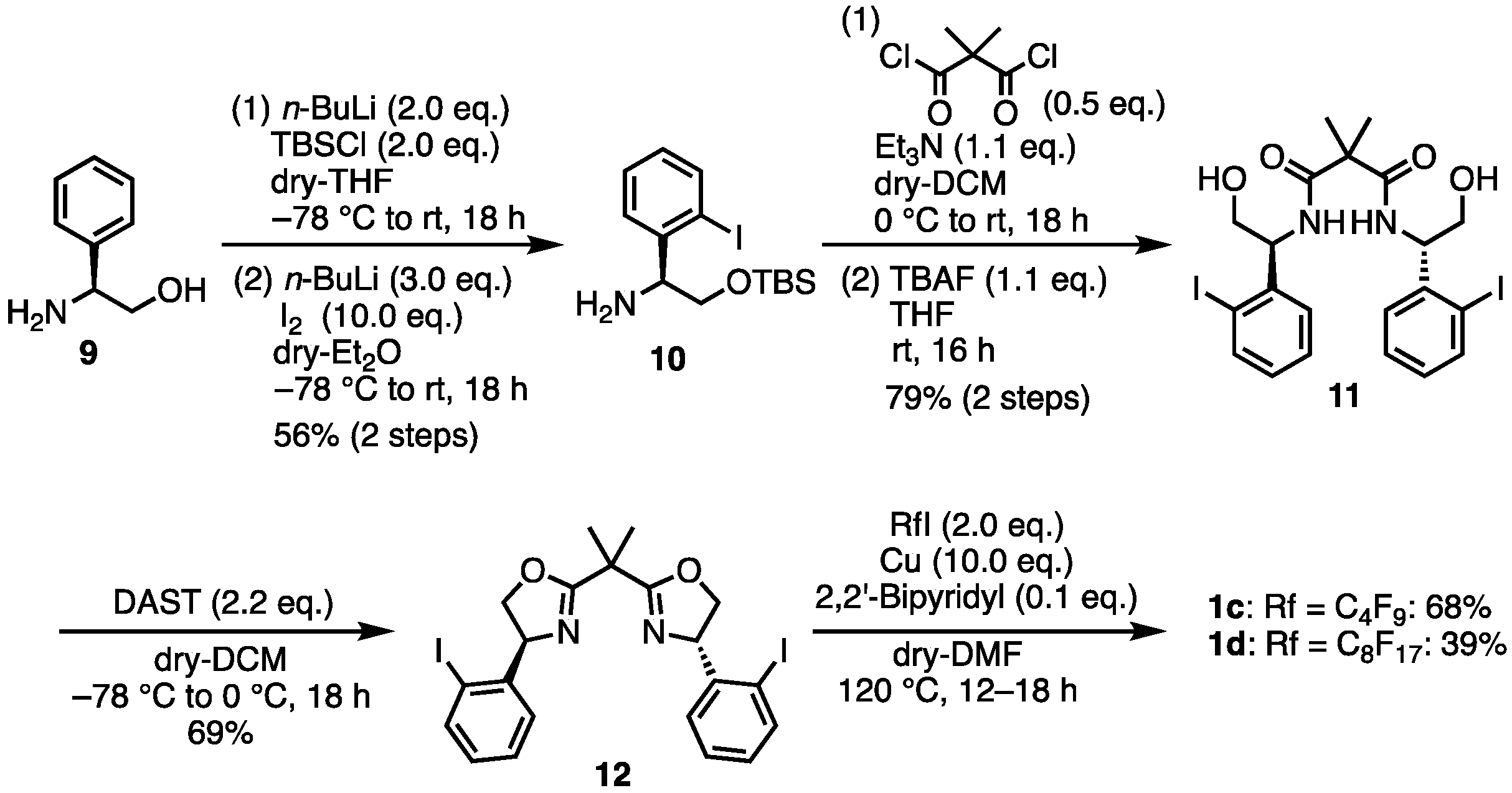
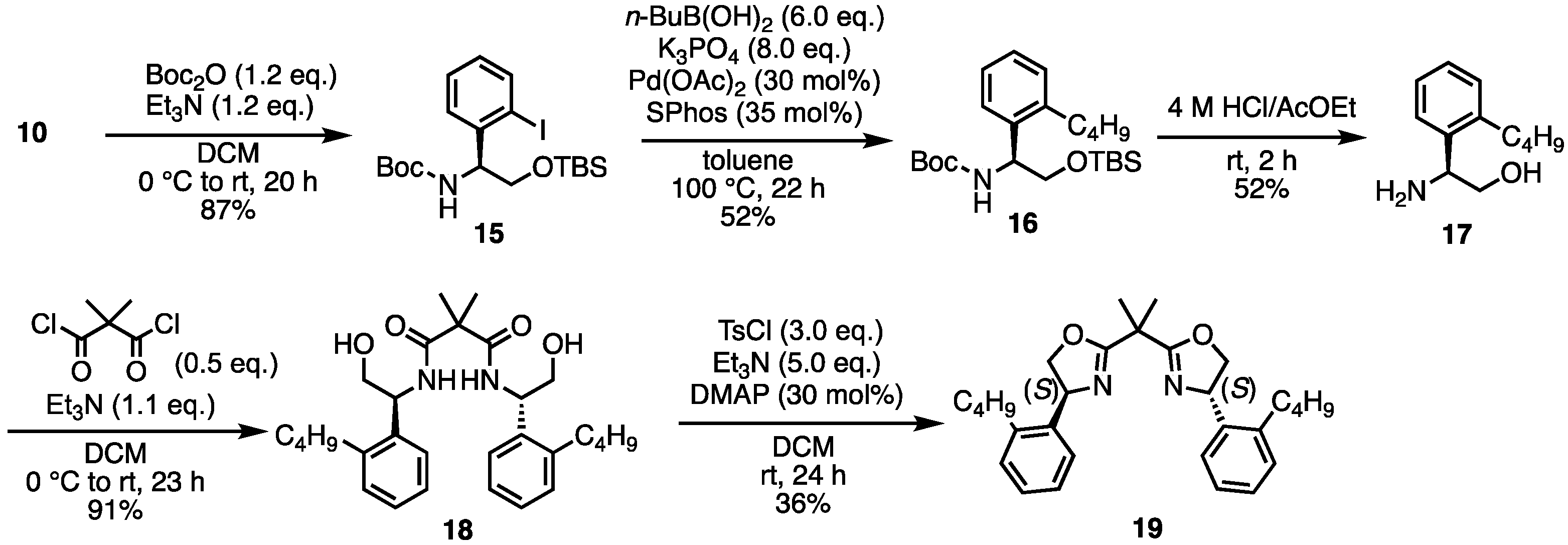
- (S)-2-((tert-Butyldimethylsilyl)oxy)-1-(2-iodophenyl)ethan-1-amine (10) [31]. To a suspension of (S)-2-amino-2-phenylethan-1-ol 9 (137.2 mg, 1.00 mmol) in THF (4 mL) at −78 °C was added n-BuLi (1.6 M solution in hexane, 1.25 mL) dropwise. The resulting purple solution was stirred at −78 °C for 30 min before a solution of TBSCl (tert-butyldimethylsilyl chloride) (317 mg, 2.10 mmol) in THF (2 mL) was added at the same temperature. The reaction mixture was allowed to warm to room temperature naturally and was stirred for 18 h. After removing the THF solvent under reduced pressure, the residue was redissolved in ether (5 mL). To this solution at −78 °C was added n-BuLi (1.6 M solution in hexane, 1.88 mL) dropwise. The reaction mixture was allowed to slowly warm to room temperature for 3 h and stirred at room temperature for 1 h. I2 (508 mg, 2.00 mmol) was added at −78 °C and the reaction mixture was allowed to warm to room temperature and stirred at room temperature for 18 h. Then, 10% Na2S2O3 solution (2 mL) was added and the resulting mixture was stirred vigorously for 10 min. To a suspension was added H2O (20 mL). The aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:5) to give the desired product 10 (212 mg, 0.56 mmol, 56%). Brown oil; 1H NMR (400 MHz, CDCl3) δ 7.80 (dd, J = 7.6, 1.2 Hz, 1H), 7.56 (dd, J = 7.6, 1.2 Hz, 1H), 7.33 (dt, J = 8.0, 1.6 Hz, 1H), 6.94 (dt, J = 7.2, 1.2 Hz, 1H), 4.32 (dd, J = 7.6, 3.6 Hz, 1H), 3.79 (dd, J = 9.6, 3.2 Hz, 1H), 3.41 (dd, J = 10.0, 7.6 Hz, 1H), 1.79 (br s, 2H), 0.90 (s, 9H), 0.06 (s, 3H), 0.02 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 144.40, 139.47, 129.11, 128.37, 128.30, 99.83, 67.53, 60.86, 26.04, 18.39, −5.14, −5.23; HRMS-DART (m/z):[M + H]+ calcd for C14H25INOSi: 378.0745, found: 378.0742.
- N1,N3-Bis((S)-2-Hydroxy-1-(2-iodophenyl)ethyl)-2,2-dimethylmalonamide (11). Under N2 atmosphere, to a solution of 10 (201 mg, 0.533 mmol) and Et3N (77 μL, 0.558 mmol) in dry-THF (2 mL) was added dimethylmalonyl dichloride (34 μL, 0.254 mmol) at 0 °C. Then, the reaction temperature rose to room temperature and the mixture was stirred for 18 h. To the mixture was added TBAF (1 M solution in THF, 553 μL). The mixture was stirred at room temperature for 16 h. To a suspension was added saturated aqueous NH4Cl (10 mL). The aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified by silica gel chromatography (MeOH:CHCl3 = 1:20) to give the desired product 11 (125 mg, 0.201 mmol, 79%). White solid; mp 75–76 °C; 1H NMR (400 MHz, CDCl3) δ 7.86 (dd, J = 8.4, 1.2 Hz, 2H), 7.27–7.23 (m, 8H), 7.15 (dd, J = 7.6, 1.2 Hz, 2H), 6.97 (dt, J = 7.6, 1.6 Hz, 2H), 5.33–5.28 (m, 2H), 3.94 (dd, J = 12.0, 4.4 Hz, 2H), 3.79 (dd, J = 11.6, 6.4 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 173.79, 140.77, 140.21, 129.62, 128.69, 127.78, 99.20, 64.59, 59.70, 50.00, 23.86; HRMS-DART (m/z):[M + H]+ calcd for C21H25I2N2O4: 622.9904, found: 622.9904.
- (4S,4′S)-2,2′-(Propane-2,2-diyl)bis(4-(2-iodophenyl)-4,5-dihydrooxazole) (12). Under N2 atmosphere, to a solution of 11 (1.92 g, 3.082 mmol) in dry-DCM (20 mL) was added a solution of diethylaminosulfur trifluoride (888 μL, 6.78 mmol) in dry-DCM (10 mL) at −78 °C. After stirring at 0 °C for 18 h, the mixture was washed with saturated aqueous NaHCO3 and the aqueous layer was extracted with DCM. The combined organic layers were dried over Na2SO4 and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:5) to give the desired product 12 (1.25 g, 2.217 mmol, 69%). Yellow solid; mp 135–136 °C; 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 8.4 Hz, 2H), 7.32 (d, J = 4.0 Hz, 4H), 7.00–6.96 (m, 2H), 5.49 (dd, J = 10.4, 7.6 Hz, 2H), 4.87 (dd, J = 10.0, 8.4 Hz, 2H), 3.98 (t, J = 8.0 Hz, 2H), 1.72 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 171.31, 145.54, 139.13, 129.28, 128.70, 127.68, 98.14, 74.95, 72.86, 39.41, 24.53; HRMS-DART (m/z):[M + H]+ calcd for C21H21I2N2O2: 586.9692, found: 586.9687.
- (4S,4′S)-2,2′-(Propane-2,2-diyl)bis(4-(2-(perfluorobutyl)phenyl)-4,5-dihydrooxazole) (1c). To a mixture of 12 (50 mg, 0.0853 mmol), Cu powder (54.2 mg, 0.853 mmol), 2,2′-bipyridyl (1.3 mg, 0.00853 mmoml), and DMF (1 mL) was added a 1,1,1,2,2,3,3,4,4-nonafluoro-4-iodobutane (29 μL, 0.171 mmol). The mixture was stirred at 120 °C for 12 h. The mixture was filtered through Celite, and the solids were washed with MeOH. The filtrate was concentrated. Then, 1 M HCl aq. was added to the suspension. The aqueous layer was extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:5) to give the desired product 1c (44.7 mg, 0.0580 mmol, 68%). White solid; mp 89–90 °C; 1H NMR (400 MHz, CDCl3) δ 7.58–7.50 (m, 6H), 7.43–7.39 (m, 2H), 5.64–5.58 (m, 2H), 4.70 (t, J = 8.8 Hz, 2H), 4.02 (t, J = 8.4 Hz, 2H), 1.74 (s, 6H); 19F NMR (376 MHz, CDCl3) δ −80.81 (6F), −102.04 to −105.02 (m, 4F), −121.34 (4F), −125.54 (4F); 13C NMR (101 MHz, CDCl3) δ 171.45, 142.55, 132.86, 128.37, 128.28, 127.62, 125.49, 117.35, 116.00, 112.30, 111.79, 76.16, 66.09, 39.27, 24.47; HRMS-DART (m/z):[M + H]+ calcd for C29H21F18N2O2: 771.1316, found: 771.1308.
- (4S,4′S)-2,2′-(Propane-2,2-diyl)bis(4-(2-(perfluorooctyl)phenyl)-4,5-dihydrooxazole) (1d). To a mixture of 12 (80 mg, 0.136 mmol), Cu powder (86.7 mg, 1.365 mmol), 2,2′-bipyridyl (2.1 mg, 0.0136 mmol), and DMF (1 mL) was added a 1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8-heptadecafluoro-8-iodooctane (72 μL, 0.273 mmol). The mixture was stirred at 120 °C for 18 h. The mixture was filtered through Celite, and the solids were washed with MeOH. The filtrate was concentrated. Then, 1 M HCl aq. was added to the suspension. The aqueous layer was extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:5) to give the desired product 1d (62.9 mg, 0.0537 mmol, 39%). White solid; mp 108–109 °C; 1H NMR (400 MHz, CDCl3) δ 7.57–7.50 (m, 6H), 7.43–7.39 (m, 2H), 5.65–5.59 (m, 2H), 4.70 (t, J = 8.4 Hz, 2H), 4.02 (t, J = 8.4 Hz, 2H), 1.74 (s, 6H); 19F NMR (376 MHz, CDCl3) δ −80.59 (6F), −101.81 to −104.80 (m, 4F), −120.35 (4F), −121.15 (4F), −121.61 to −121.76 (m, 8F), −122.58 (4F), −125.97 (4F); 13C NMR (101 MHz, CDCl3) δ 171.45, 142.53, 132.85, 128.39, 128.28, 127.61, 125.60, 121.25–107.94, 76.16, 66.08, 39.28, 24.46; HRMS-DART (m/z):[M + H]+ calcd for C37H21F34N2O2: 1171.1060, found: 1171.1079.
- N1,N3-Bis((S)-2-Hydroxy-1-phenylethyl)-2,2-dimethylmalonamide (13) [27]. Under N2 atmosphere, to a solution of 9 (600 mg, 4.374 mmol) and Et3N (635 μL, 4.582 mmol) in dry-DCM (6 mL) was added dimethylmalonyl dichloride (275 μL, 2.083 mmol) at 0 °C. Then, the reaction temperature rose to room temperature and the mixture was stirred for 18 h. The reaction mixture was concentrated. The crude was purified by silica gel chromatography (MeOH:CHCl3 = 1:5) to give the desired product 13 (765 mg, 2.064 mmol, 99%). Yellow solid; mp 128–130 °C; 1H NMR (400 MHz, CDCl3) δ 7.35–7.22 (m, 10H), 7.10 (d, J = 7.6 Hz, 2H), 5.14–5.10 (m, 2H), 3.96–3.90 (m, 2H), 3.83–3.77 (m, 2H), 2.79–2.76 (m, 2H), 1.61 (s, 6H).
- (4S,4′S)-2,2′-(Propane-2,2-diyl)bis(4-phenyl-4,5-dihydrooxazole) (14) [32]. To a solution of 13 (765 mg, 2.065 mmol), DMAP (25.2 mg, 0.207 mmol) and Et3N (1.46 mL, 10.532 mmol) in DCM (8 mL) was added tosyl chloride (826.7 mg, 4.337 mmol). After stirring at room temperature for 36 h, the mixture was washed with saturated aqueous NH4Cl and the aqueous layer was extracted with DCM. The combined organic layers were washed with saturated aqueous NaHCO3; the aqueous layer was extracted with DCM. The combined organic layers were dried over Na2SO4 and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:1) to give the desired product 14 (424.3 mg, 1.269 mmol, 61%). Colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.34–7.26 (m, 10H), 5.26–5.21 (m, 2H), 4.70–4.65 (m, 2H), 4.19–4.15 (m, 2H), 1.68 (s, 6H).
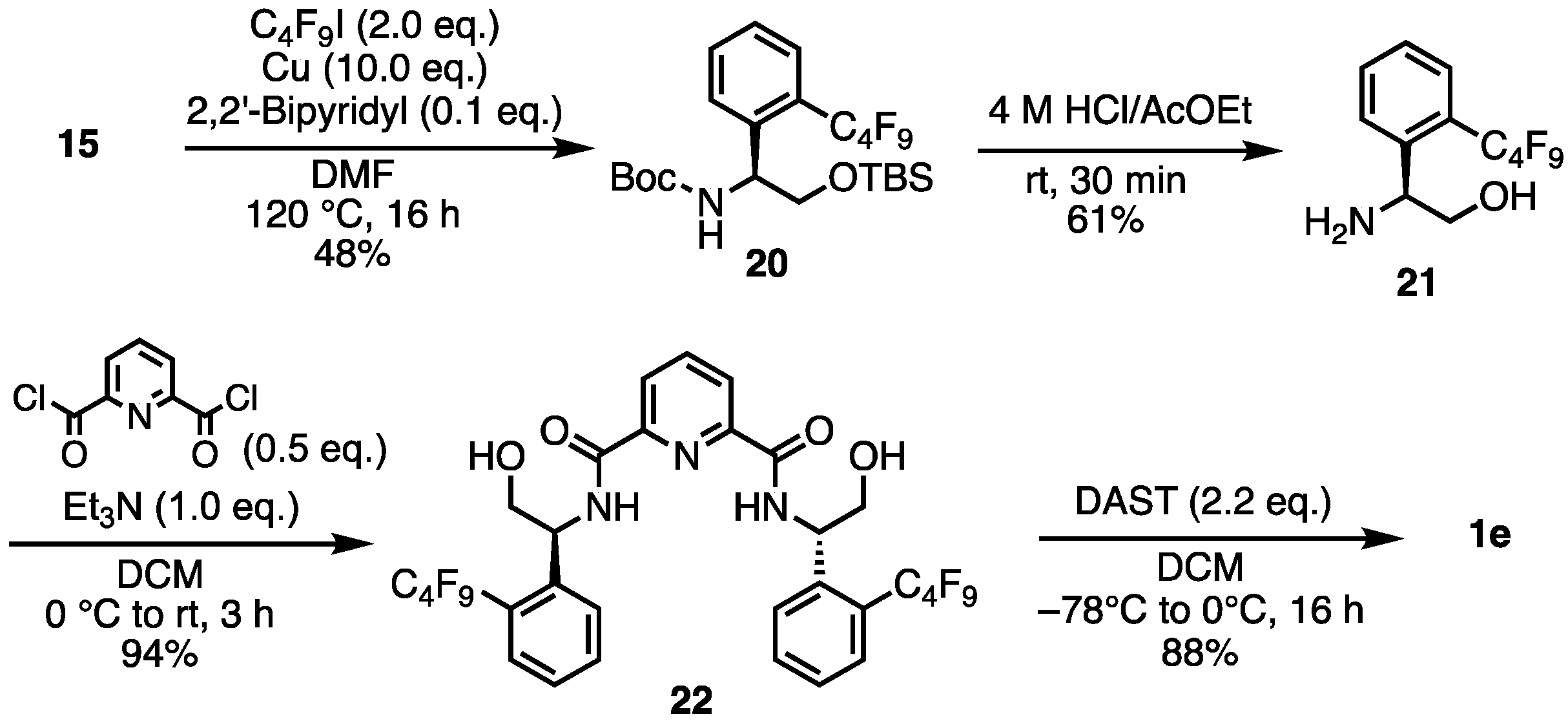
- tert-Butyl (S)-(2-((tert-butyldimethylsilyl)oxy)-1-(2-iodophenyl)ethyl)carbamate (15). A solution of 10 (639.7 mg, 1.695 mmol) and Et3N (285 μL, 2.048 mmol) in dry-DCM (8 mL) was stirred at 0 °C. A solution of Boc2O (449.2 mg, 2.058 mmol) in dry-DCM (4 mL) was slowly added to the mixture. Then the reaction temperature rose to room temperature and the mixture was stirred for a further 20 h. The mixture was diluted with brine. The reaction mixture was extracted with DCM. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:5) to give the desired product 15 (637 mg, 1.424 mmol, 84%). Yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 7.6 Hz, 1H), 7.32–7.29 (m, 2H), 6.96–6.92 (m, 1H), 5.51 (br s, 1H), 4.94 (br s, 1H), 3.88–3.71 (m, 2H), 1.43 (br s, 9H), 0.83 (s, 9H), −0.08 (s, 3H), −0.14 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 155.32, 142.31, 139.61, 129.02, 128.12, 127.97, 98.49, 79.73, 64.68, 59.64, 28.42, 25.90, 18.33, −5.55; HRMS-DART (m/z):[M + H]+ calcd for C19H33INO3Si: 478.1274, found: 478.1271.
- tert-Butyl (S)-(2-((tert-butyldimethylsilyl)oxy)-1-(2-butylphenyl)ethyl)carbamate (16). To a mixture of K3PO4 (216.4 mg, 1.019 mmol), toluene (15 mL), 15 (60.8 mg, 0.127 mmol), and butylboronic acid (68.2 mg, 0.669 mmol) was added Pd(OAc)2 (9.2 mg, 0.0410 mmol) and SPhos (19.7 mg, 0.0480 mmol). The mixture was stirred at 100 °C for 22 h. The mixture was filtered through Celite, and the solids were washed with MeOH. The filtrate was concentrated. The saturated aqueous NH4Cl was added to the suspension. The aqueous layer was extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:10) to give the desired product 16 (26.8 mg, 0.0657 mmol, 52%). Yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.31–7.27 (m, 1H), 7.18–7.13 (m, 3H), 5.14–4.99 (m, 2H), 3.85–3.81 (m, 1H), 3.69–3.67 (m, 1H), 2.70–2.66 (m, 2H), 1.68–1.37 (m, 13H), 0.95 (t, J = 7.6 Hz, 3H), 0.85 (s, 9H), −0.055 to −0.058 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 155.39, 140.31, 138.28, 129.43, 127.14, 126.41, 125.84, 79.23, 66.15, 60.40, 33.69, 32.35, 28.46, 25.77, 22.94, 18.26, 13.92, −5.45, −5.56; HRMS-DART (m/z):[M + H]+ calcd for C23H42NO3Si: 408.2934, found: 408.2933.
- (S)-2-Amino-2-(2-butylphenyl)ethan-1-ol (17). To 16 (59.3 mg, 0.145 mmol) was added 4 M HCl in AcOEt (2 mL). The mixture was stirred at room temperature for 2 h. The reaction mixture was concentrated. Then, 4 M NaOH aq. was added to the residue. The aqueous layer was extracted with ethyl acetate. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude was purified by silica gel chromatography (MeOH:CHCl3 = 1:5) to give the desired product 17 (14.6 mg, 0.0755 mmol, 52%). Yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.38–7.36 (m, 1H), 7.24–7.16 (m, 3H), 4.34–4.30 (m, 1H), 3.69 (dd, J = 10.8, 4.4 Hz, 1H), 3.56 (dd, J = 10.8, 8.8 Hz, 1H), 2.74–2.59 (m, 2H), 1.60–1.53 (m, 2H), 1.45–1.36 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 140.30, 140.07, 129.82, 127.37, 126.47, 125.59, 67.52, 52.43, 34.16, 32.54, 22.85, 14.07; HRMS-DART (m/z):[M + H]+ calcd for C12H20NO: 194.1545, found: 194.1540.
- N1,N3-Bis((S)-1-(2-butylphenyl)-2-hydroxyethyl)-2,2-dimethylmalonamide (18). Under N2 atmosphere, to a solution of 17 (17 mg, 0.088 mmol) and Et3N (13 μL, 0.097 mmol) in dry-DCM (2 mL) was added dimethylmalonyl dichloride (5.8 μL, 0.044 mmol) at 0 °C. Then, the reaction temperature rose to room temperature and the mixture was stirred for 3 h. The reaction mixture was concentrated. The crude was purified by silica gel chromatography (MeOH:CHCl3 = 1:20) to give the desired product 18 (19.4 mg, 0.040 mmol, 91%). Yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.22–7.17 (m, 10H), 6.96 (d, J = 7.6 Hz, 2H), 5.41–5.36 (m, 2H), 3.89–3.76 (m, 4H), 2.70–2.66 (m, 4H), 1.66–1.37 (m, 14H), 0.94 (t, J = 7.2 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 174.08, 140.75, 136.18, 130.11, 127.96, 126.53, 125.66, 66.54, 52.01, 49.89, 33.59, 32.50, 23.67, 22.79, 14.06; HRMS-DART (m/z):[M + H]+ calcd for C29H43N2O4: 483.3223, found: 483.3221.
- (4S,4′S)-2,2′-(Propane-2,2-diyl)bis(4-(2-butylphenyl)-4,5-dihydrooxazole) (19). To a solution of 18 (18.8 mg, 0.039 mmol), DMAP (1.4 mg, 0.0117 mmol) and Et3N (27 μL, 0.195 mmol) in DCM (0.5 mL) was added tosyl chloride (22.3 mg, 0.117 mmol). After stirring at room temperature for 24 h, the mixture was diluted with DCM. The mixture was washed with saturated aqueous NH4Cl and saturated aqueous NaHCO3. The organic layer was dried over Na2SO4 and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:5) to give the desired product 19 (6.2 mg, 0.0139 mmol, 36%). Colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.29–7.13 (m, 8H), 6.96 (d, J = 7.6 Hz, 2H), 5.53–5.48 (m, 2H), 4.75–4.70 (m, 2H), 4.04–4.00 (m, 2H), 2.66–2.53 (m, 4H), 1.72 (s, 6H), 1.59–1.34 (m, 8H), 0.94 (t, J = 7.2 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 170.47, 140.39, 139.41, 129.24, 127.30, 126.66, 126.32, 75.55, 65.65, 39.14, 33.68, 32.78, 24.64, 22.83, 14.05; HRMS-DART (m/z):[M + H]+ calcd for C29H39N2O2: 447.3012, found: 447.3008.
- tert-Butyl (S)-(2-((tert-butyldimethylsilyl)oxy)-1-(2-(perfluorobutyl)phenyl)ethyl)carbamate (20). To a mixture of 15 (3.5 g, 7.33 mmol), Cu powder (4.66 g, 73.31 mmol), 2,2′-bipyridyl (144.5 mg, 0.733 mmol), and DMF (10 mL) was added a 1,1,1,2,2,3,3,4,4-nonafluoro-4-iodobutane (2.46 mL, 14.661 mmol). The mixture was stirred at 120 °C for 16 h. The mixture was filtered through Celite, and the solids were washed with MeOH. The filtrate was concentrated. Then, 1 M HCl aq. was added to the suspension. The aqueous layer was extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:10) to give the desired product 20 (2.0 g, 3.51 mmol, 48%). Yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.62–7.51 (m, 3H), 7.39 (t, J = 7.6 Hz,1H), 5.34 (br s, 1H), 5.06 (br s, 1H), 3.94–3.90 (m, 1H), 3.64–3.62 (m, 1H), 1.37 (br s, 9H), 0.83 (s, 9H), −0.09 (s, 3H), −0.12 (s, 3H); 19F NMR (376 MHz, CDCl3) δ −80.75 (3F), −101.78 to −104.10 (m, 2F), −120.90 (2F), −125.42 (2F); 13C NMR (101 MHz, CDCl3) δ 159.41, 141.39, 136.25, 131.69, 131.69, 128.86, 128.72, 127.32, 119.91, 119.06, 117.73, 116.18, 77.29, 76.34, 66.67, 28.32, 25.85, 18.31, −5.65, −5.76; HRMS-DART (m/z):[M + H]+ calcd for C23H33F9NO3Si: 570.2086, found: 570.2088.
- (S)-2-Amino-2-(2-(perfluorobutyl)phenyl)ethan-1-ol (21). To 20 (2 g, 3.51 mmol) was added 4 M HCl in AcOEt (17.5 mL). The mixture was stirred at room temperature for 30 min. The reaction mixture was concentrated. Then, 4 M NaOH aq. was added to the residue. The aqueous layer was extracted with ethyl acetate. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude was purified by silica gel chromatography (MeOH:CHCl3 = 1:10) to give the desired product 21 (760.7 mg, 2.14 mmol, 61%). Yellow solid; mp 70–72 °C; 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 8.0 Hz, 1H), 7.61–7.56 (m, 2H), 7.42 (t, J = 7.4 Hz, 1H), 4.42–4.38 (m, 1H), 3.75–3.71 (m, 1H), 36.0–3.55 (m, 1H), 1.78 (br s, 3H); 19F NMR (376 MHz, CDCl3) δ −80.88 (3F), −102.09 to −104.25 (m, 2F), −120.61 to −122.47 (m, 2F), −125.54 (2F); 13C NMR (101 MHz, CDCl3) δ 143.70, 132.47, 128.54, 128.15, 127.57, 126.01, 119.97, 118.97, 117.39, 116.11, 67.66, 52.80; HRMS-DART (m/z):[M + H]+ calcd for C12H11F9NO: 356.0697, found: 356.0695.
- N2,N6-Bis((S)-2-hydroxy-1-(2-(perfluorobutyl)phenyl)ethyl)pyridine-2,6-dicarboxamide (22). Under N2 atmosphere, to a solution of 21 (400 mg, 1.126 mmol) and Et3N (157 μL, 1.126 mmol) in dry-DCM (5mL) was added pyridine-2,6-dicarbonyl dichloride (114.9 mg, 0.563 mmol) at 0 °C. Then, the reaction temperature rose to room temperature, stirring the mixture for 3 h. The reaction mixture was concentrated. The crude was purified by silica gel chromatography (MeOH:CHCl3 = 1:10) to give the desired product 22 (445.3 mg, 0.529 mmol, 94%). White solid; mp 95–96 °C; 1H NMR (400 MHz, CDCl3) δ 8.77 (d, J = 6.4 Hz, 2H), 8.33–8.29 (m, 2H), 8.04–8.00 (m, 1H), 7.69–7.44 (m, 8H), 5.54 (br s, 2H), 4.10–4.06 (m, 2H), 3.98–3.94 (m, 2H); 19F NMR (376 MHz, CDCl3) δ −80.67 (6F), −101.70 to −104.28 (m, 4F), −121.01 (4F), −125.34 (4F); 13C NMR (101 MHz, CDCl3) δ 163.13, 148.50, 139.60, 139.54, 132.50, 129.46, 128.13, 128.06, 126.26, 125.21, 119.78, 118.98, 117.90, 116.40, 66.75, 52.48; HRMS-DART (m/z):[M + H]+ calcd for C31H22F18N3O4: 842.1323, found: 842.1326.
- 2,6-Bis((S)-4-(2-(perfluorobutyl)phenyl)-4,5-dihydrooxazol-2-yl)pyridine (1e). Under N2 atmosphere, to a solution of 22 (416.7 mg, 0.495 mmol) in dry-DCM (5 mL) was added a solution of diethylaminosulfur trifluoride (195 μL, 1.486 mmol) in dry-DCM (2 mL) at −78 °C. After stirring at 0 °C for 16 h, the mixture was washed with saturated aqueous NaHCO3 and the aqueous layer was extracted with DCM. The combined organic layers were Na2SO4 and concentrate. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:3) to give the desired product 1e (350.9 mg, 0.436 mmol, 88%). Yellow solid; mp 63–64 °C; 1H NMR (400 MHz, CDCl3) δ 8.42 (d, J = 7.6 Hz, 2H), 7.99 (t, J = 8.0 Hz, 1H), 7.61–743 (m, 8H), 5.85–5.79 (m, 2H), 4.94–4.88 (m, 2H), 4.29–4.25 (m, 2H); 19F NMR (376 MHz, CDCl3) δ −80.80 (6F), −102.22 to −104.85 (m, 4F), −121.34 (4F), −125.54 (4F); 13C NMR (101 MHz, CDCl3) δ 164.60, 146.72, 141.96, 141.92, 137.75, 132.90, 128.52, 127.90, 126.66, 125.65, 119.88–116.11, 76.44, 66.88; HRMS-DART (m/z):[M + H]+ calcd for C31H18F18N3O2: 806.1112, found: 806.1113.
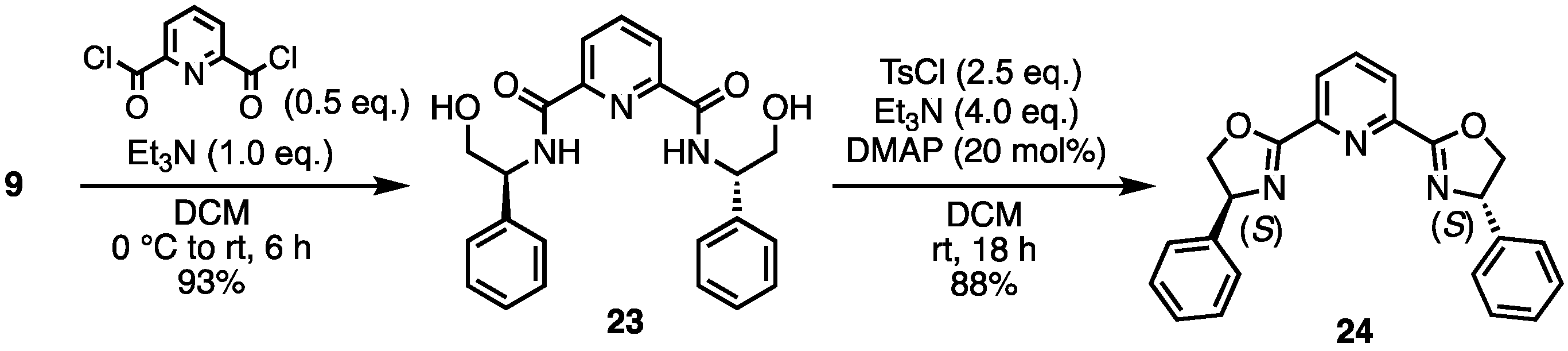
- N2,N6-Bis((S)-2-hydroxy-1-phenylethyl)pyridine-2,6-dicarboxamide (23) [33]. Under N2 atmosphere, to a solution of 9 (823 mg, 6.0 mmol) and Et3N (836 μL, 6.0 mmol) in dry-DCM (5 mL) was added pyridine-2,6-dicarbonyl dichloride (612 mg, 3.0 mmol) at 0 °C. Then, the reaction temperature rose to room temperature, stirring the mixture for 6 h. The reaction mixture was concentrated. The crude was purified by silica gel chromatography (MeOH:CHCl3 = 1:5) to give the desired product 23 (1.13 g, 2.79 mmol, 93%). White solid; mp 79–80 °C; 1H NMR (400 MHz, CDCl3) δ 8.71 (d, J = 7.2 Hz, 2H), 8.16 (d, J = 8.0 Hz, 2H), 7.83 (t, J = 7.6 Hz, 1H), 7.35–7.24 (m, 10H), 5.21–5.17 (m, 2H), 3.92–3.91 (m, 2H), 3.52 (br s, 2H).
- 2,6-Bis((S)-4-phenyl-4,5-dihydrooxazol-2-yl)pyridine (24) [33]. To a solution of 23 (1 g, 2.466 mmol), DMAP (60 mg, 0.493 mmol) and Et3N (1.37 mL, 9.866 mmol) in DCM (0.5 mL) was added tosyl chloride (1.18 g, 6.166 mmol). After stirring at room temperature for 18 h, water was added to the mixture. The aqueous layer was extracted with DCM. The combined organic layers were dried over Na2SO4 and concentrated. The crude was purified by silica gel chromatography (ethyl acetate:n-hexane = 1:1) to give the desired product 24 (801.8 mg, 2.170 mmol, 88%). White solid; mp 169 °C; 1H NMR (400 MHz, CDCl3) δ 8.35 (d, J = 8.4 Hz, 2H), 7.92 (t, J = 7.6 Hz, 1H), 7.39–7.29 (m, 10H), 5.49–5.44 (m, 2H), 4.96–4.91 (m, 2H), 4.45–4.41 (m, 2H).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ghosh, A.K.; Mathivanan, P.; Cappiello, J. C2-Symmetric Chiral Bis(oxazoline)–Metal Complexes in Catalytic Asymmetric Synthesis. Tetrahedron Asymmetry 1998, 9, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, K.A.; Johannsen, M.; Yao, S.; Audrain, H.; Thorhauge, J. Catalytic Asymmetric Addition Reactions of Carbonyls. A Common Catalytic Approach. Acc. Chem. Res. 1999, 32, 605–613. [Google Scholar] [CrossRef]
- Johnson, J.S.; Evans, D.A. Chiral Bis(oxazoline) Copper(II) Complexes: Versatile Catalysts for Enantioselective Cycloaddition, Aldol, Michael, and Carbonyl Ene Reactions. Acc. Chem. Res. 2000, 33, 325–335. [Google Scholar] [CrossRef] [PubMed]
- McManus, H.A.; Guiry, P.J. Recent Developments in the Application of Oxazoline-Containing Ligands in Asymmetric Catalysis. Chem. Rev. 2004, 104, 4151–4202. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Woerpel, K.A.; Hinman, M.M.; Faul, M.M. Bis(oxazolines) as Chiral Ligands in Metal-Catalyzed Asymmetric Reactions. Catalytic, Asymmetric Cyclopropanation of Olefins. J. Am. Chem. Soc. 1991, 113, 726–728. [Google Scholar] [CrossRef]
- Rechavi, D.; Lemaire, M. Enantioselective Catalysis Using Heterogeneous Bis(oxazoline) Ligands: Which Factors Influence the Enantioselectivity? Chem. Rev. 2002, 102, 3467–3494. [Google Scholar] [CrossRef]
- Fraile, J.M.; García, J.I.; Mayoral, J.A. Recent Advances in the Immobilization of Chiral Catalysts Containing Bis(oxazolines) and Related Ligands. Coord. Chem. Rev. 2008, 252, 624–646. [Google Scholar] [CrossRef]
- Fu, B.; Du, D.-M.; Wang, J. Synthesis of Novel C2-Symmetric Chiral Bis(oxazoline) Ligands and Their Application in the Enantioselective Addition of Diethylzinc to Aldehydes. Tetrahedron Asymmetry 2004, 15, 119–126. [Google Scholar] [CrossRef]
- Torres-Werlé, M.; Nano, A.; Maisse-François, A.; Bellemin-Laponnaz, S. Asymmetric Benzoylation and Henry Reaction Using Reusable Polytopic Bis(oxazoline) Ligands and Copper(II). New J. Chem. 2014, 38, 4748–4753. [Google Scholar] [CrossRef]
- Ollevier, T. Iron Bis(oxazoline) Complexes in Asymmetric Catalysis. Catal. Sci. Technol. 2016, 6, 41–48. [Google Scholar] [CrossRef]
- Wang, L.; Tang, Y. Side Arm Modified Chiral Bisoxazoline Ligands: Recent Development and Prospect in Asymmetric Catalysis. Tetrahedron 2022, 129, 133121. [Google Scholar] [CrossRef]
- Ginotra, S.K.; Singh, V.K. Enantioselective Henry Reaction Catalyzed by a C2-Symmetric Bis(oxazoline)–Cu(OAc)2·H2O Complex. Org. Biomol. Chem. 2007, 5, 3932–3937. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Seidel, D.; Rueping, M.; Lam, H.W.; Shaw, J.T.; Downey, C.W. A New Copper Acetate-Bis(oxazoline)-Catalyzed, Enantioselective Henry Reaction. J. Am. Chem. Soc. 2003, 125, 12692–12693. [Google Scholar] [CrossRef] [PubMed]
- Gaab, M.; Bellemin-Laponnaz, S.; Gade, L.H. “Catalysis in a Tea Bag”: Synthesis, Catalytic Performance and Recycling of Dendrimer-Immobilised Bis- and Trisoxazoline Copper Catalysts. Chem. Eur. J. 2009, 15, 5450–5462. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; MacMillan, D.W.C.; Campos, K.R. C2-Symmetric Tin(II) Complexes as Chiral Lewis Acids. Catalytic Enantioselective Anti Aldol Additions of Enolsilanes to Glyoxylate and Pyruvate Esters. J. Am. Chem. Soc. 1997, 119, 10859–10860. [Google Scholar] [CrossRef]
- Juhl, K.; Gathergood, N.; Jørgensen, K.A. Catalytic Asymmetric Direct Mannich Reactions of Carbonyl Compounds with α-Imino Esters. Angew. Chem. Int. Ed. 2001, 40, 2995–2997. [Google Scholar] [CrossRef]
- Juhl, K.; Jørgensen, K.A. Catalytic Asymmetric Direct α-Amination Reactions of 2-Keto Esters: A Simple Synthetic Approach to Optically Active syn-β-Amino-α-hydroxy Esters. J. Am. Chem. Soc. 2002, 124, 2420–2421. [Google Scholar] [CrossRef]
- Zhao, J.-L.; Liu, L.; Sui, Y.; Liu, Y.-L.; Wang, D.; Chen, Y.-J. Catalytic and Highly Enantioselective Friedel–Crafts Alkylation of Aromatic Ethers with Trifluoropyruvate under Solvent-Free Conditions. Org. Lett. 2006, 8, 6127–6130. [Google Scholar] [CrossRef]
- Horváth, I.T.; Curran, D.P.; Gladysz, J.A. Fluorous Chemistry: Scope and Definition. In The Handbook of Fluorous Chemistry, 1st ed.; Gladysz, J.A., Curran, D.P., Horvath, I.T., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp. 1–4. [Google Scholar]
- Bayardon, J.; Sinou, D. Fluorous Chiral Bisoxazolines. Synthesis and Applications to an Asymmetric Allylic Alkylation. Tetrahedron Lett. 2003, 44, 1449–1451. [Google Scholar] [CrossRef]
- Bayardon, J.; Holczknecht, O.; Pozzi, G.; Sinou, D. Asymmetric Cyclopropanation Catalyzed by Fluorous Bis(oxazolines)–Copper Complexes. Tetrahedron Asymmetry 2006, 17, 1568–1572. [Google Scholar] [CrossRef]
- Deng, T.; Cai, C. Fluorous Chiral Bis(oxazolines): Synthesis and Application in Asymmetric Henry Reaction. J. Fluor. Chem. 2013, 156, 183–186. [Google Scholar] [CrossRef]
- Zhang, W.; Curran, D.P. Synthetic Applications of Fluorous Solid-Phase Extraction (F-SPE). Tetrahedron 2006, 62, 11837–11865. [Google Scholar] [CrossRef] [PubMed]
- Gladysz, J.A.; Costa, R.C. Strategies for the Recovery of Fluorous Catalysts and Reagents: Design and Evaluation. In The Handbook of Fluorous Chemistry, 1st ed.; Gladysz, J.A., Curran, D.P., Horvath, I.T., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp. 24–40. [Google Scholar]
- Kobayashi, Y.; Obayashi, R.; Watanabe, Y.; Miyazaki, H.; Miyata, I.; Suzuki, Y.; Yoshida, Y.; Shioiri, T.; Matsugi, M. Unprecedented Asymmetric Epoxidation of Isolated Carbon–Carbon Double Bonds by a Chiral Fluorous Fe(III) Salen Complex: Exploiting Fluorophilic Effect for Catalyst Design. Eur. J. Org. Chem. 2019, 2019, 2401–2408. [Google Scholar] [CrossRef]
- Palomo, C.; Oiarbide, M.; Laso, A. Recent Advances in the Catalytic Asymmetric Nitroaldol (Henry) Reaction. Eur. J. Org. Chem. 2007, 2007, 2561–2574. [Google Scholar] [CrossRef]
- Corey, E.J.; Imai, N.; Zhang, H.-Y. Designed Catalyst for Enantioselective Diels–Alder Addition from a C2-Symmetric Chiral Bis(oxazoline)–Fe(III) Complex. J. Am. Chem. Soc. 1991, 113, 728–729. [Google Scholar] [CrossRef]
- Nishiyama, H.; Kondo, M.; Nakamura, T.; Itoh, K. Highly Enantioselective Hydrosilylation of Ketones with Chiral and C2-Symmetrical Bis(oxazolinyl)pyridine–Rhodium Catalysts. Organometallics 1991, 10, 500–508. [Google Scholar] [CrossRef]
- Rajput, S.; McLean, K.J.; Poddar, H.; Selvam, I.R.; Nagalingam, G.; Triccas, J.A.; Levy, C.W.; Munro, A.W.; Hutton, C.A. Structure−Activity Relationships of cyclo(L-Tyrosyl-L-tyrosine) Derivatives Binding to Mycobacterium tuberculosis CYP121: Iodinated Analogues Promote Shift to High-Spin Adduct. J. Med. Chem. 2019, 62, 9792–9805. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tao, X.-Z.; Dai, J.-J.; Li, C.-G.; Xu, J.; Xu, H.-M.; Xu, H.-J. Electrochemical Synthesis of 1,2-Diketones from Alkynes under Transition-Metal-Catalyst-Free Conditions. Chem. Commun. 2019, 55, 9208–9211. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Z.; Chen, C.; Yang, H.; Han, Z.; Dong, X.-Q.; Zhang, X. Iridium Catalysts with Modular Axial-Unfixed Biphenyl Phosphine–Oxazoline Ligands: Asymmetric Hydrogenation of α,β-Unsaturated Carboxylic Acids. Org. Chem. Front. 2017, 4, 627–630. [Google Scholar] [CrossRef]
- Fraile, J.M.; García, J.I.; Mayoral, J.A.; Roldán, M. Simple and Efficient Heterogeneous Copper Catalysts for Enantioselective C−H Carbene Insertion. Org. Lett. 2007, 9, 731–733. [Google Scholar] [CrossRef]
- Glöckner, S.; Tran, D.N.; Ingham, R.J.; Fenner, S.; Wilson, Z.E.; Battilocchio, C.; Ley, S.V. The Rapid Synthesis of Oxazolines and Their Heterogeneous Oxidation to Oxazoles under Flow Conditions. Org. Biomol. Chem. 2015, 13, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Alammari, A.S.; Al-Majid, A.M.; Barakat, A.; Alshahrani, S.; Ali, M.; Islam, M.S. Asymmetric Henry Reaction of Nitromethane with Substituted Aldehydes Catalyzed by Novel In Situ Generated Chiral Bis(β-Amino Alcohol-Cu(OAc)2·H2O Complex. Catalysts 2021, 11, 1208. [Google Scholar] [CrossRef]
- Alvarez-Casao, Y.; Marques-Lopez, E.; Herrera, R.P. Organocatalytic Enantioselective Henry Reactions. Symmetry 2011, 3, 220–245. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Metal Source | Conv. (%) 1 | ee (%) 2,3 |
|---|---|---|---|
| 1 | Cu(OAc)2·H2O | 97 | 69 |
| 2 | CuCl2 | no reaction | - |
| 3 | CuBr2 | no reaction | - |
| 4 | Cu(OTf)2 | no reaction | - |
| 5 | Co(OAc)2 | 95 | 9 |
| 6 | Zn(OAc)2 | 96 | 3 |
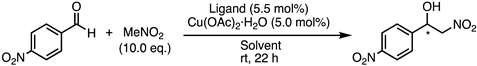
| Entry | Ligand | Solvent | Conv. (%) 1 | ee (%) 2 | Config. 3 |
|---|---|---|---|---|---|
| 1 | 1a | iPrOH | 97 | 69 | S |
| 2 | 1a | iPrOH/H2O (1/1) | 94 | 41 | S |
| 3 | 1a | THF | 79 | 41 | S |
| 4 | 1a | FC-72 | 93 | 45 | S |
| 5 | 1b | iPrOH | 91 | 58 | S |
| 6 | 1b | iPrOH/H2O (1/1) | 87 | 44 | S |
| 7 | 1b | FC-72 | 14 | 33 | S |
| 8 | 1c | iPrOH | 94 | 53 | R |
| 9 | 1c | iPrOH/H2O (1/1) | 95 | 36 | R |
| 10 | 1c | THF | 45 | 42 | R |
| 11 | 1c | FC-72 | 95 | 32 | R |
| 12 | 1d | iPrOH | 98 | 47 | R |
| 13 | 1d | THF | 37 | 35 | R |
| 14 | 14 | iPrOH | 98 | 27 | S |
| 15 | 19 | iPrOH | 98 | 14 | R |
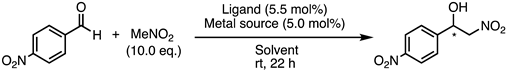
| Entry | Ligand | Metal Source | Solvent | Conv. (%) 1 | ee (%) 2 | Config. 3 |
|---|---|---|---|---|---|---|
| 1 | 1e | Co(OAc)2 | iPrOH | 97 | 25 | R |
| 2 | 1e | Cu(OAc)2·H2O | iPrOH | 98 | 10 | R |
| 3 | 1e | Zn(OAc)2 | iPrOH | 94 | 13 | R |
| 4 | 1e | Co(OAc)2 | iPrOH/H2O (1/1) | 96 | 2 | R |
| 5 | 1e | Co(OAc)2 | THF | 72 | 42 | R |
| 6 | 1e | Co(OAc)2 | FC-72 | no reaction | - | - |
| 7 | 24 | Co(OAc)2 | THF | 99 | 8 | R |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishihara, K.; Kato, Y.; Takeuchi, N.; Hayashi, Y.; Hagiwara, Y.; Shibuya, S.; Natsume, T.; Matsugi, M. Asymmetric Henry Reaction Using Cobalt Complexes with Bisoxazoline Ligands Bearing Two Fluorous Tags. Molecules 2023, 28, 7632. https://doi.org/10.3390/molecules28227632
Ishihara K, Kato Y, Takeuchi N, Hayashi Y, Hagiwara Y, Shibuya S, Natsume T, Matsugi M. Asymmetric Henry Reaction Using Cobalt Complexes with Bisoxazoline Ligands Bearing Two Fluorous Tags. Molecules. 2023; 28(22):7632. https://doi.org/10.3390/molecules28227632
Chicago/Turabian StyleIshihara, Kazuki, Yamato Kato, Narisa Takeuchi, Yuka Hayashi, Yuna Hagiwara, Shyota Shibuya, Tohya Natsume, and Masato Matsugi. 2023. "Asymmetric Henry Reaction Using Cobalt Complexes with Bisoxazoline Ligands Bearing Two Fluorous Tags" Molecules 28, no. 22: 7632. https://doi.org/10.3390/molecules28227632
APA StyleIshihara, K., Kato, Y., Takeuchi, N., Hayashi, Y., Hagiwara, Y., Shibuya, S., Natsume, T., & Matsugi, M. (2023). Asymmetric Henry Reaction Using Cobalt Complexes with Bisoxazoline Ligands Bearing Two Fluorous Tags. Molecules, 28(22), 7632. https://doi.org/10.3390/molecules28227632