
Development of a Pure Certified Reference Material of D-Mannitol

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
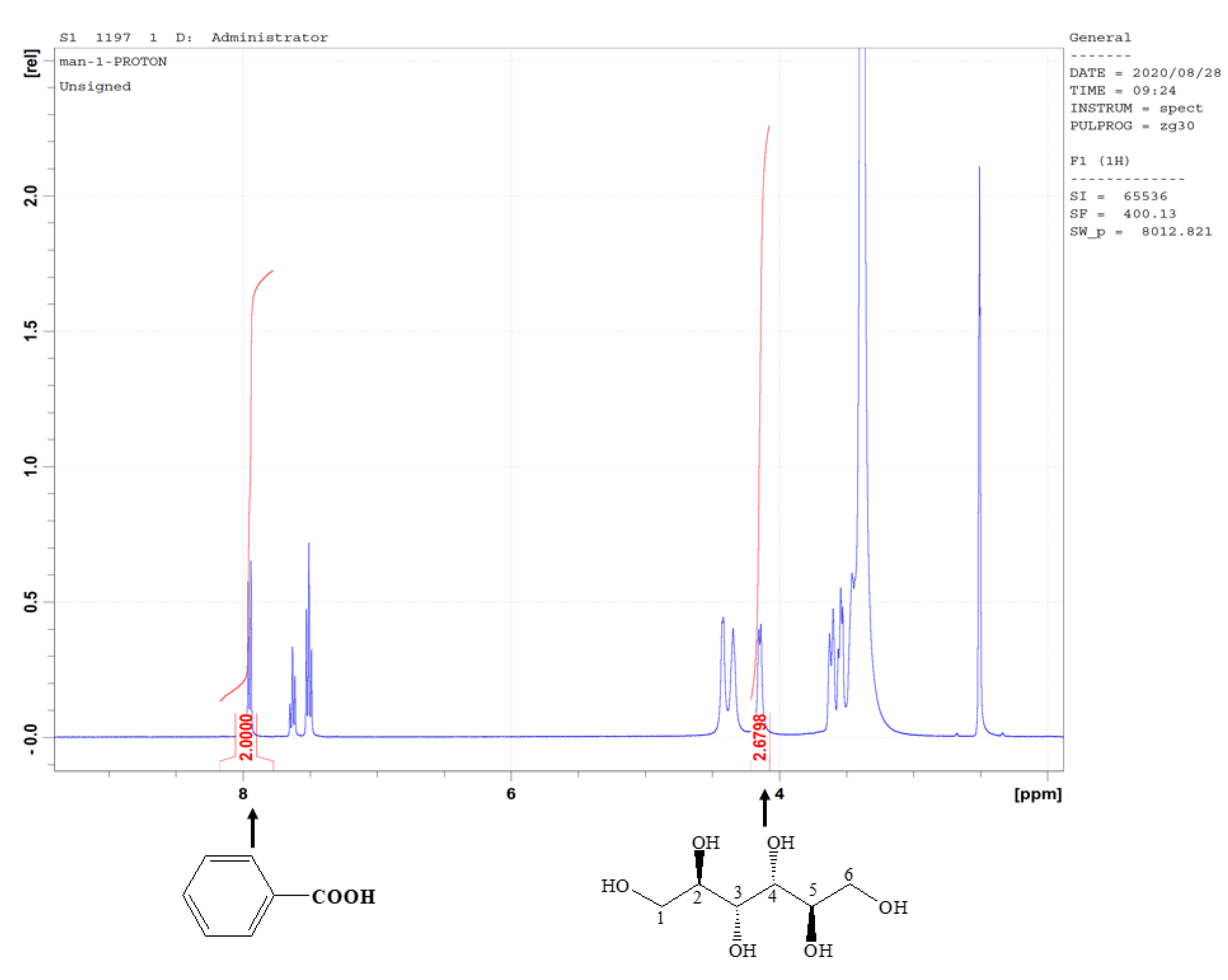
2.1. Structure Determination
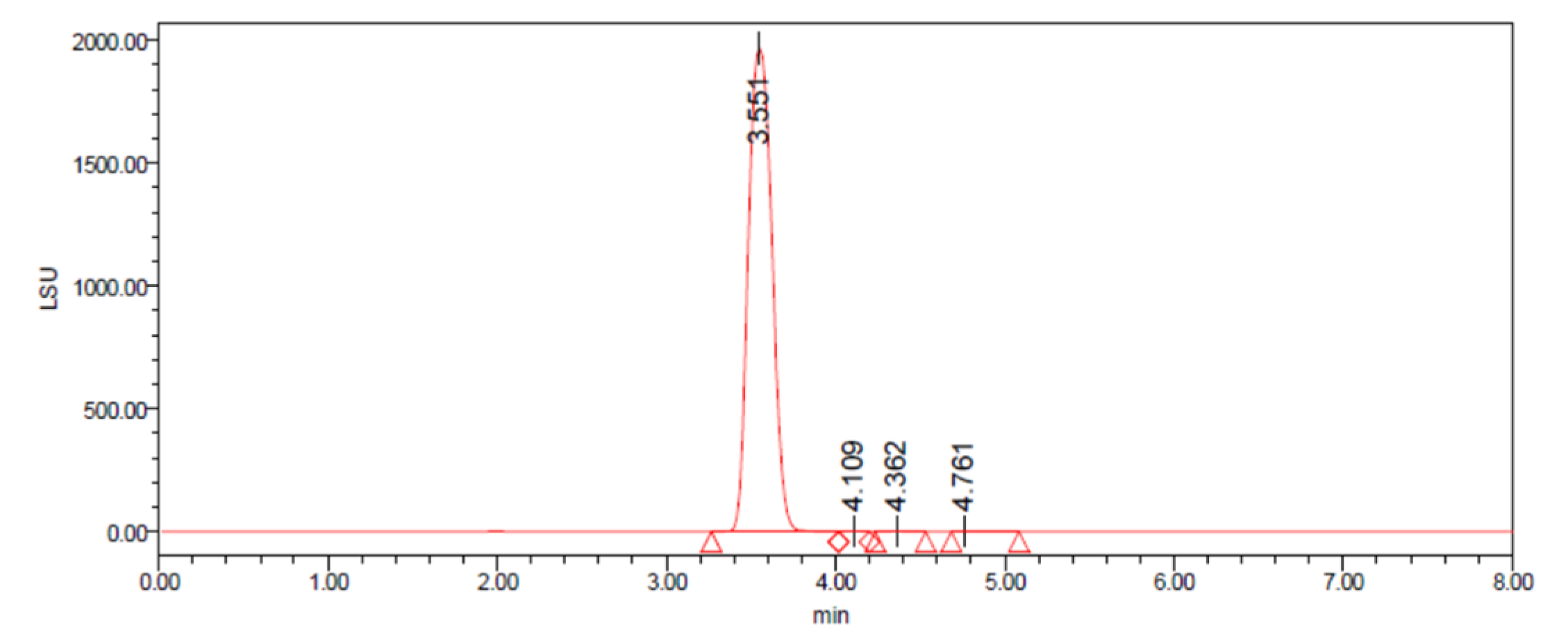
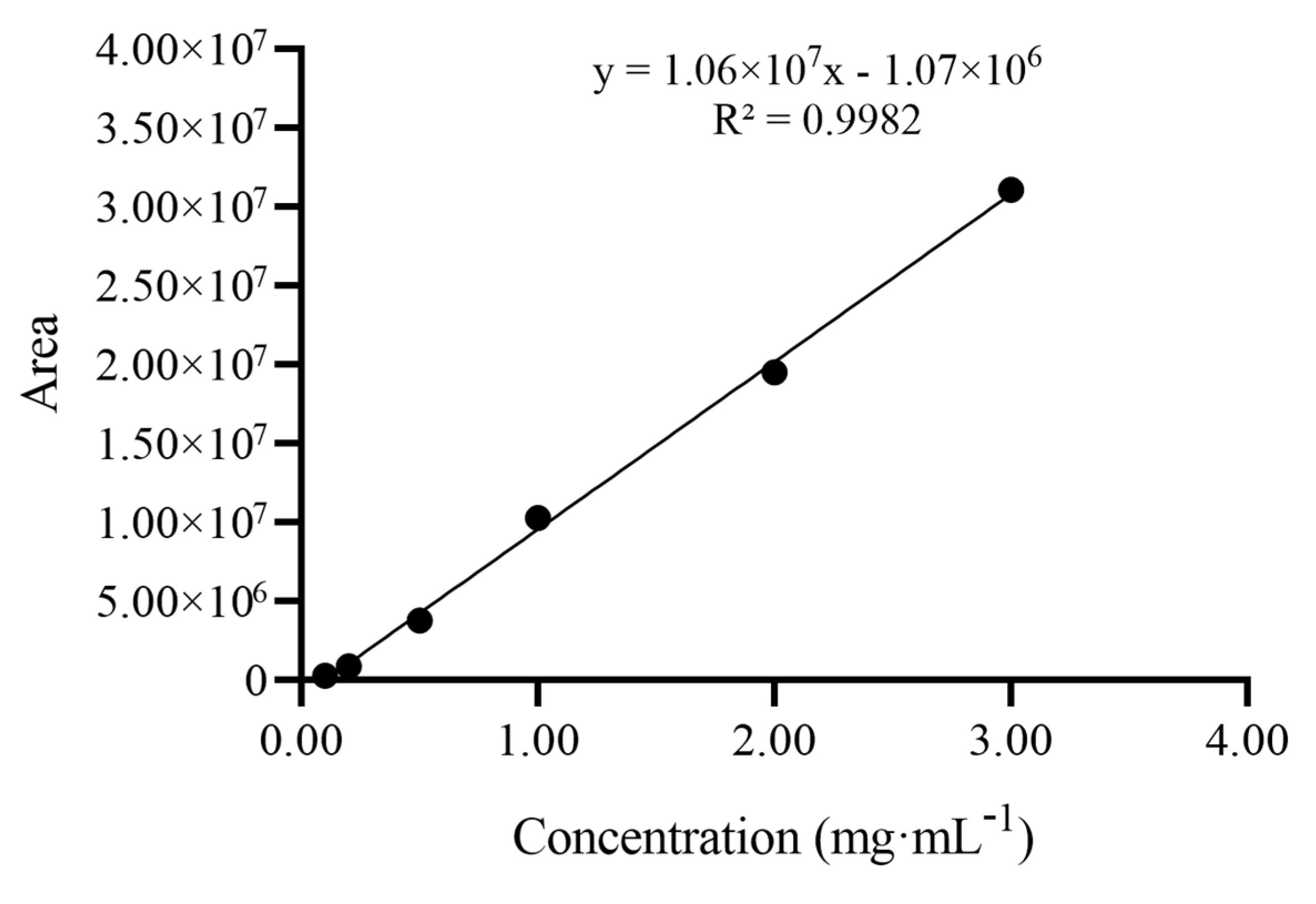
2.2. Development of the HPLC–ELSD Method
2.3. Homogeneity Test
2.4. Stability Studies
2.5. Mass Balance Approach
2.6. Quantitative NMR Approach
2.7. Purity Certification
2.8. Uncertainty Estimation
2.8.1. Uncertainty of the Mass Balance Approach
2.8.2. Uncertainty of the qNMR method
2.8.3. Uncertainty of Homogeneity
2.8.4. Uncertainty of Stability
2.8.5. Combined and Expanded Uncertainties
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Methods
3.2.1. Preparation of the Candidate CRM
3.2.2. Structural Analysis
High-Resolution Mass Spectra
Fourier-Transform Infrared (FT-IR) Spectra
Raman Spectra
Optical Rotation Value
Nuclear Magnetic Resonance Spectra
3.3. Characterization
3.3.1. Development and Method Validation of HPLC–ELSD
3.3.2. Mass Balance Method
3.3.3. Quantitative NMR Method
3.4. Homogeneity Test
3.5. Stability Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Yang, Y.X.; Liu, J.; Hu, A.; Nie, T.; Cheng, Z.N.; Liu, W.J. A Critical Review on Engineering of D-Mannitol Crystals: Properties, Applications, and Polymorphic Control. Crystals 2022, 12, 1080. [Google Scholar] [CrossRef]
- Martău, G.A.; Coman, V.; Vodnar, D.C. Recent advances in the biotechnological production of erythritol and mannitol. Crit. Rev. Biotechnol. 2020, 40, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Beluomini, M.A.; da Silva, J.L.; Sedenho, G.C. Nelson Ramos Stradiotto. D-mannitol sensor based on molecularly imprinted polymer on electrode modified with reduced graphene oxide decorated with gold nanoparticles. Talanta 2017, 165, 231–239. [Google Scholar] [CrossRef]
- Shawkat, H.; Westwood, M.M.; Mortimer, A. Mannitol: A review of its clinical uses. Continuing Education in An aesthesia. Crit. Care. Pain 2018, 12, 83–85. [Google Scholar] [CrossRef]
- Yang, D.Z.; Wang, F.F.; Zhang, L.; Gong, N.B.; Lv, Y. Development of a new ferulicacid certified reference material for use in clinical chemistry and pharmaceutic alanalysis. Acta. Pharm. Sin. B 2015, 5, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.H.; Kim, J.; Rosli, N.; Mok, I.; Oh, K.H.; Lee, H.; Hong, S.P.; Jin, Y.X.; Wu, L.Q.; Wang, J.; et al. Certification and stability assessment of recombinant human growth hormone as a certified reference material for protein quantification. J. Chromatog. B 2019, 1126–1127, 121732. [Google Scholar] [CrossRef]
- Tangpaisarnkul, N.; Tuchinda, P.; Wilairat, P.; Siripinyanonda, A.; Shiowattana, J.; Nobsathian, S. Development of pure certified reference material of stevioside. Food Chem. 2018, 255, 75–80. [Google Scholar] [CrossRef]
- Yang, M.R.; Wang, M.; Zhou, J.; Song, Y.Q.; Wang, T.T. Development of a new certified reference material of tinidazole for quality and safety of agro-products. Accred. Qual. Assur. 2016, 21, 341–349. [Google Scholar] [CrossRef]
- ISO 17034; General Requirements for the Competence of Reference Material Producers. International Standard Organization (ISO): Geneva, Switzerland, 2016. Available online: https://www.iso.org/standard/29357.html (accessed on 5 May 2019).
- ISO Guide 35; Reference Materials-Guidance for Characterization and Assessment of Homogeneity and Stability. International Standard Organization (ISO): Geneva, Switzerland, 2017. Available online: https://www.iso.org/standard/60281.html (accessed on 5 May 2019).
- JJF 1855-2020; Metrological Technical Specification for Purity Assessment of Certified Refence Materials-Organic Purity Certified Reference Materials. Station Administration for Market Regulation: Beijing, China, 2021.
- Spectral Database for Organic Compounds (SDBS). Available online: http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/cre_index.cgi?lang=eng (accessed on 10 July 2019).
- Cassanelli, M.; Norton, I.; Mills, T. Interaction of mannitol and sucrose with gellan gum in freeze-dried gel systems. Food Biophys. 2018, 13, 304–315. [Google Scholar] [CrossRef]
- Campbell Roberts, S.N.; Williams, A.C.; Grimsey, I.M.; Booth, S.W. Quantitative analysis of mannitol polymorphs. FT-Raman spectroscopy. J. Pharm. Biomed. Anal. 2002, 28, 1135–1147. [Google Scholar] [CrossRef]
- Shrivastva, A.; Sharma, S.; Kaurav, M.; Sharma, A. Characteristics and Analytical Methods of Mannitol: An Update. Int. J. App. Pharm. 2021, 13, 20–32. [Google Scholar] [CrossRef]
- Medeiros, P.M.; Simoneit, B.R.T. Analysis of sugars in environmental samples by gas chromatography-mass spectrometry. J. Chromatogr. A 2007, 1141, 271–278. [Google Scholar] [CrossRef]
- Molnár-Perl, I. Simultaneous quantitation of acids and sugars by chromatography: Gas or high-performance liquid chromatography. J. Chromatogr. A 1999, 845, 181–195. [Google Scholar] [CrossRef]
- Hanko, V.P.; Rohrer, J.S. Determination of carbohydrates, sugar alcohols, and glycols in cell cultures and fermentation broths using high-performance anion-exchange chromatography with pulsed amperometric detection. Anal. Biochem. 2000, 283, 192–199. [Google Scholar] [CrossRef]
- Andersen, R.; Sørensen, A. Separation and determination of alditols and sugars by high-pH anion-exchange chromatography with pulsed amperometric detection. J. Chromatogr. A 2000, 897, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Marsilio, R.; D’Antiga, L.; Zancan, L.; Dussini, N.; Zacchello, F. Simultaneous HPLC determination with light-scattering detection of lactulose and mannitol in studies of intestinal permeability in pediatrics. Clin. Chem. 1998, 44, 1685–1691. [Google Scholar] [CrossRef]
- Gervasoni, J.; Primiano, A.; Graziani, C.; Scaldaferri, F.; Gasbarrini, A.; Urbani, A.; Persichilli, S. Validation of UPLC-MS/MS Method for Determination of Urinary Lactulose/Mannitol. Molecules 2018, 23, 2705. [Google Scholar] [CrossRef] [PubMed]
- Nooran, B.; Chowdhury, E.A.; Alqahtani, F.; Ahn, Y.; Patel, D.; Al-Ahmad, A.; Mehvar, R.; Bickel, U. LC-MS/MS-based in vitro and in vivo investigation of blood-brain barrier integrity by simultaneous quantitation of mannitol and sucrose. Fluids Barriers CNS 2020, 17, 61. [Google Scholar] [CrossRef]
- Wong, Y.; Or, M.; Lau, H.; Wong, S.; Fung, W. Analysis of underivatized alditols in traditional chinese medicines by liquid chromatography–tandem mass spectrometry based on ligand exchange chromatography. Sep. Sci. Plus 2021, 4, 6–15. [Google Scholar] [CrossRef]
- Chen, W.Z.; Jin, W.H.; Fang, H.; Chen, H.; Chen, X.Y.; Zhang, Y.P.; Hong, Z. Development of a new taurine purity certified reference material. Microchem. J. 2022, 181, 107761. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W.; Li, X.; Zhou, S.; Tu, M.; Zhu, Y.; Li, H. Purity Assessment of Dinotefuran Using Mass Balance and Quantitative Nuclear Magnetic Resonance. Molecules 2023, 28, 3884. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Zhu, D.; Yu, J.; Fang, C.; Mou, R.; Hu, X.; Zhu, Z.; Chen, M. Development of Certified Reference Materials for the Determination of Apparent Amylose Content in Rice. Molecules 2022, 27, 4647. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | HPLC–ELSD (%) | Moisture (%) | Volatile Impurities (%) | Nonvolatile Impurities (%) | Determination Results | |
|---|---|---|---|---|---|---|
| MB (%) | qNMR (%) | |||||
| 1 | 99.91 | 0.0357 | <0.01 | <0.01 | 99.87 | 99.90 |
| 2 | 99.92 | 0.0341 | <0.01 | <0.01 | 99.89 | 99.88 |
| 3 | 99.92 | 0.0357 | <0.01 | <0.01 | 99.88 | 99.91 |
| 4 | 99.91 | 0.0342 | <0.01 | <0.01 | 99.88 | 99.89 |
| 5 | 99.91 | 0.0396 | <0.01 | <0.01 | 99.87 | 99.89 |
| 6 | 99.92 | 0.0368 | <0.01 | <0.01 | 99.88 | 99.90 |
| Mean | 99.91 | 0.0360 | - | - | 99.89 | 99.90 |
| SD | 0.01 | 0.0020 | - | - | 0.01 | 0.02 |
| 3.162 | ||||||
| 2.228 | ||||||
| Conclusion | < the means are coincident | |||||
| 3.66 | ||||||
| 5.05 | ||||||
| Conclusion | < , the standard deviations are coincident | |||||
| Uncertainties | Sources | Results (%) |
|---|---|---|
| Inhomogeneity among the bottles and within bottles | 0.03 | |
| Instability at long-term storage condition | 0.03 | |
| Instability at short-term transportation condition | 0.03 | |
| Purity characterization by | 0.49 | |
| Purity characterization by | 0.27 | |
| Combined and | 0.56 | |
| Combined and | 0.57 | |
| Relative expanded uncertainty, × k (k = 2) | 1.1 | |
| Expanded uncertainty, × P | 1.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Zhang, Y.; Chen, H.; Jin, W.; Chen, X.; Huang, X.; Xie, Y.; Fang, H.; Hong, Z. Development of a Pure Certified Reference Material of D-Mannitol. Molecules 2023, 28, 6794. https://doi.org/10.3390/molecules28196794
Chen W, Zhang Y, Chen H, Jin W, Chen X, Huang X, Xie Y, Fang H, Hong Z. Development of a Pure Certified Reference Material of D-Mannitol. Molecules. 2023; 28(19):6794. https://doi.org/10.3390/molecules28196794
Chicago/Turabian StyleChen, Weizhu, Yiping Zhang, Hui Chen, Wenhui Jin, Xiaoying Chen, Xiaoyan Huang, Yanrou Xie, Hua Fang, and Zhuan Hong. 2023. "Development of a Pure Certified Reference Material of D-Mannitol" Molecules 28, no. 19: 6794. https://doi.org/10.3390/molecules28196794
APA StyleChen, W., Zhang, Y., Chen, H., Jin, W., Chen, X., Huang, X., Xie, Y., Fang, H., & Hong, Z. (2023). Development of a Pure Certified Reference Material of D-Mannitol. Molecules, 28(19), 6794. https://doi.org/10.3390/molecules28196794