
Supramolecular Motifs in the Crystal Structures of Triethylbenzene Derivatives Bearing Pyridinium Subunits in Combination with Pyrimidinyl or Pyridinyl Groups

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
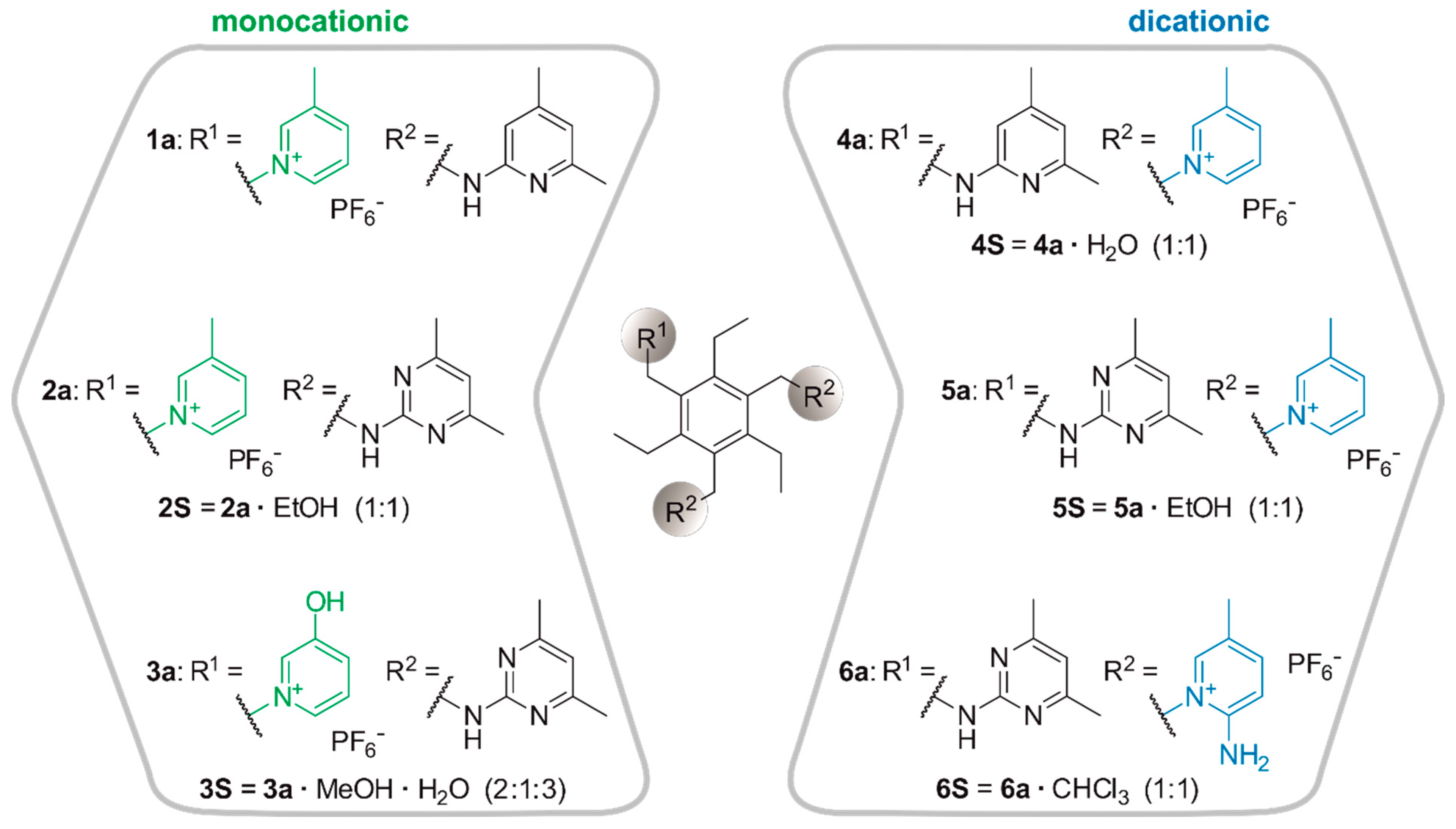
2.1. Synthesis
2.2. Crystallographic Studies
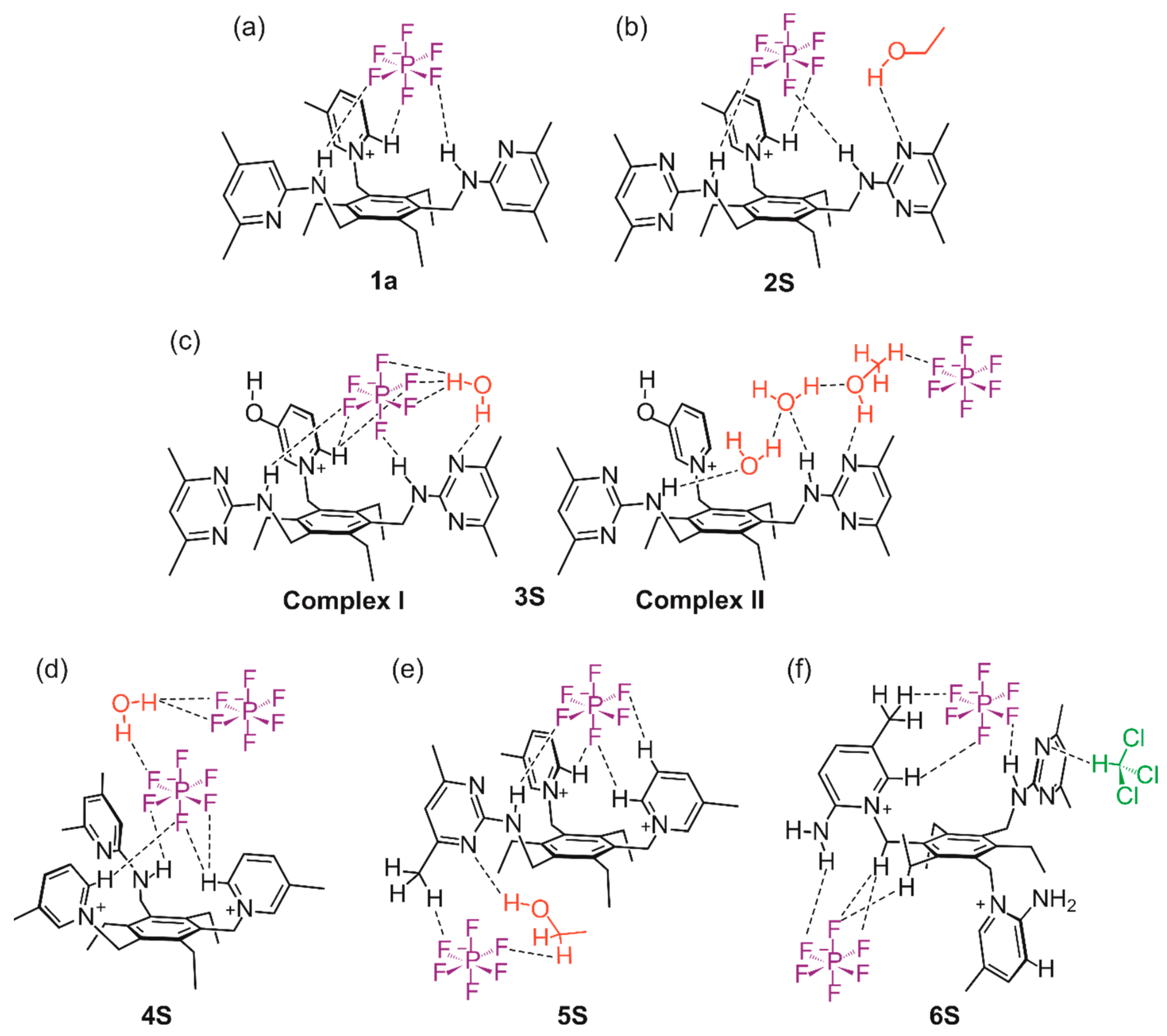
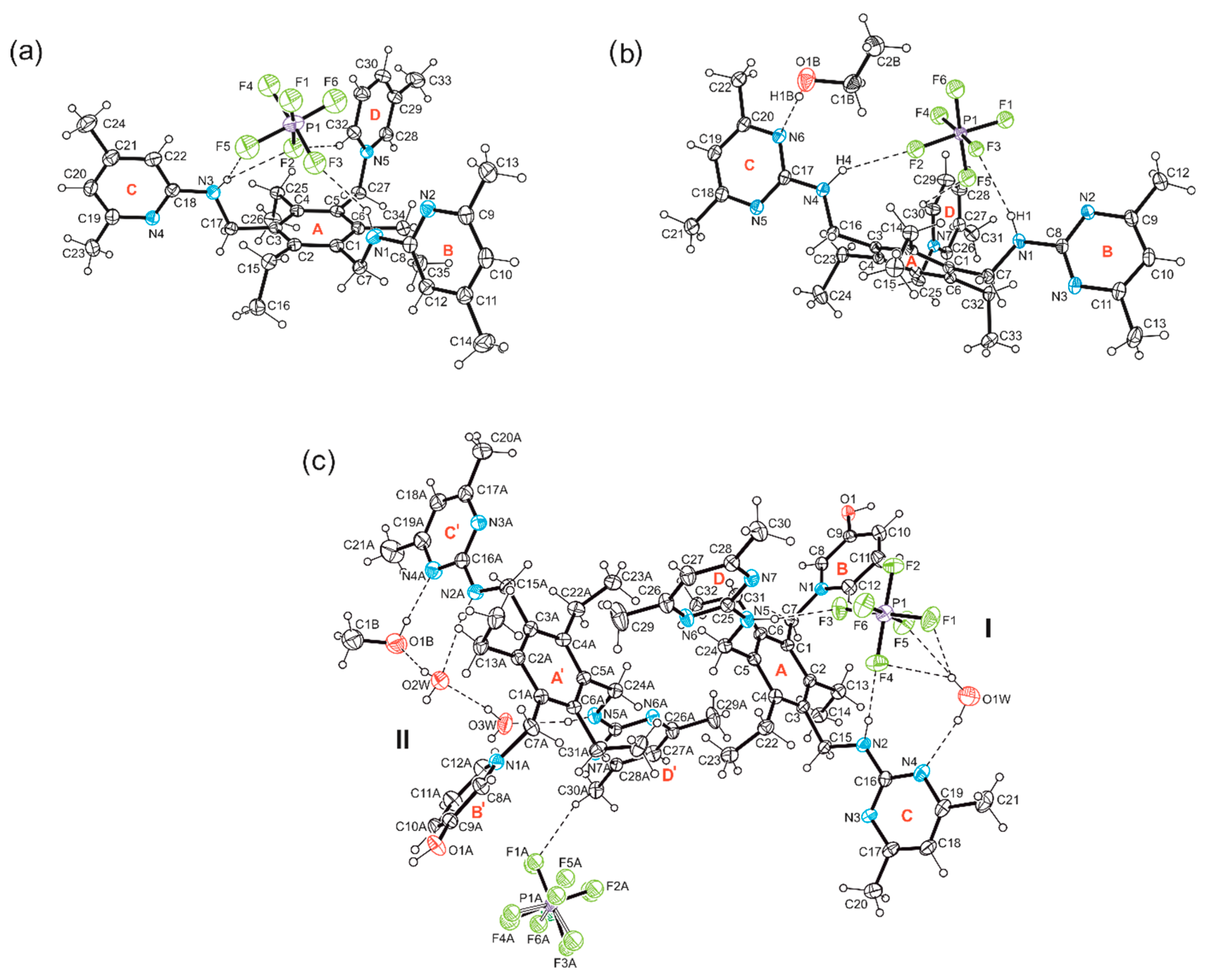
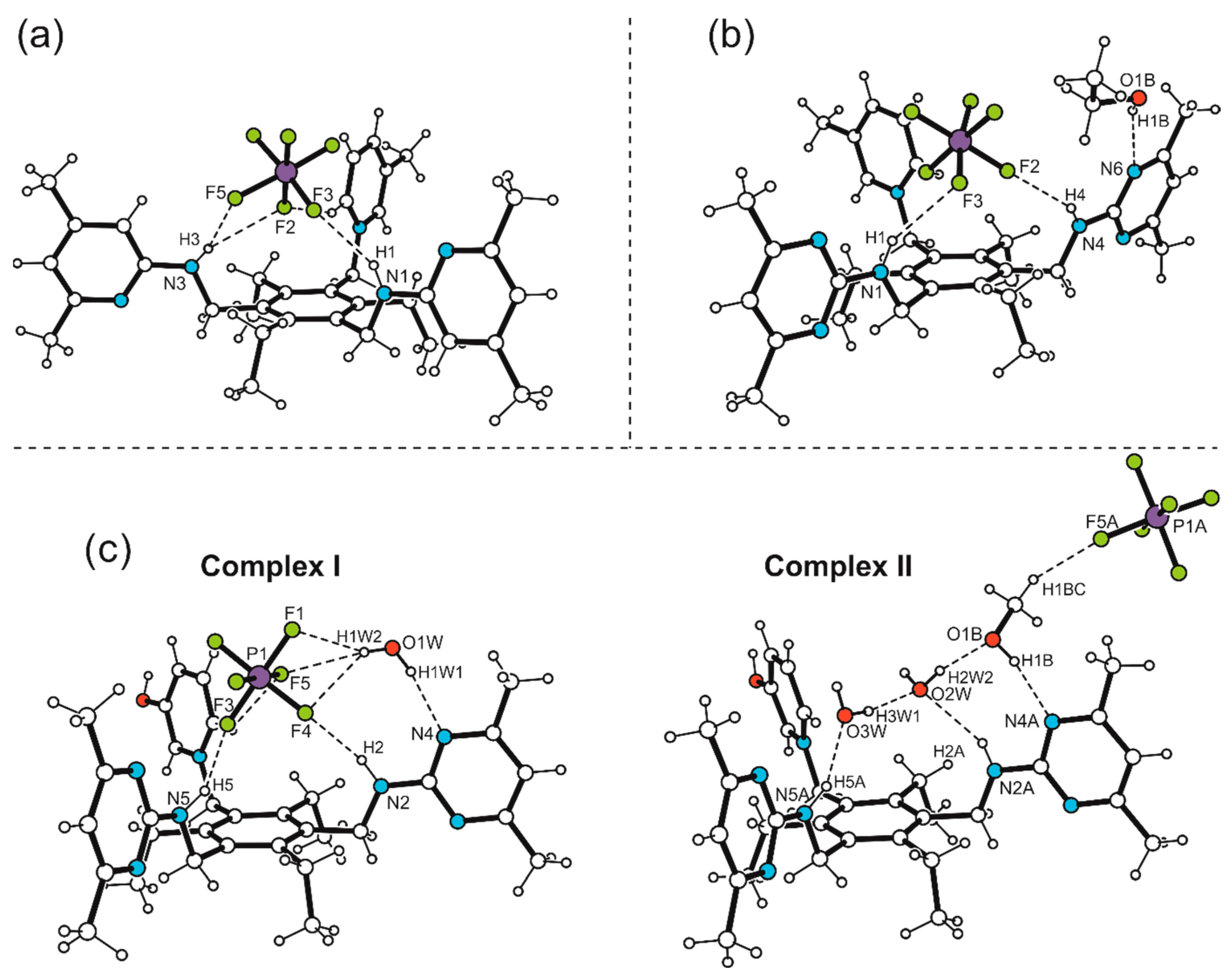
2.2.1. Hexafluorophosphate Salts of the Monocationic Tripodal Receptors: Solvent-Free Structure 1a and Solvates 2S and 3S
2.2.2. Hexafluorophosphate Salts of the Dicationic Compounds: Solvates 4S, 5S and 6S
3. Conclusions
4. Experimental Section
X-ray Crystallography
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Mazik, M. Molecular recognition of carbohydrates by acyclic receptors employing noncovalent interactions. Chem. Soc. Rev. 2009, 38, 935–956. [Google Scholar] [CrossRef] [PubMed]
- Mazik, M. Recent developments in the molecular recognition of carbohydrates by artificial receptors. RSC Adv. 2012, 2, 2630–2642. [Google Scholar] [CrossRef]
- Kaiser, S.; Geffert, C.; Mazik, M. Purine Unit as a Building Block of Artificial Receptors Designed for the Recognition of Carbohydrates. Eur. J. Org. Chem. 2019, 2019, 7555–7562. [Google Scholar] [CrossRef]
- Stapf, M.; Seichter, W.; Mazik, M. Cycloalkyl Groups as Subunits of Artificial Carbohydrate Receptors: Effect of Ring Size of the Cycloalkyl Unit on the Receptor Efficiency. Eur. J. Org. Chem. 2020, 2020, 4900–4915. [Google Scholar] [CrossRef]
- Geffert, C.; Kuschel, M.; Mazik, M. Molecular Recognition of N -Acetylneuraminic Acid by Acyclic Pyridinium- and Quinolinium-Based Receptors in Aqueous Media: Recognition through Combination of Cationic and Neutral Recognition Sites. J. Org. Chem. 2013, 78, 292–300. [Google Scholar] [CrossRef]
- Lippe, J.; Mazik, M. Carbohydrate receptors combining both a macrocyclic building block and flexible side arms as recognition units: Design, syntheses, and binding studies. J. Org. Chem. 2015, 80, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Amrhein, F.; Mazik, M. Compounds Combining a Macrocyclic Building Block and Flexible Side-Arms as Carbohydrate Receptors: Syntheses and Structure-Binding Activity Relationship Studies. Eur. J. Org. Chem. 2021, 2021, 6282–6303. [Google Scholar] [CrossRef]
- Leibiger, B.; Stapf, M.; Mazik, M. Cycloalkyl Groups as Building Blocks of Artificial Carbohydrate Receptors: Studies with Macrocycles Bearing Flexible Side-Arms. Molecules 2022, 27, 7630. [Google Scholar] [CrossRef]
- Gabius, H.-J. The Sugar Code: Fundamentals of Glycosciences; Wiley-VCH: Weinheim, Germany, 2011; ISBN 9783527644940. [Google Scholar]
- Lis, H.; Sharon, N. Lectins, 2nd ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003; ISBN 1402011725. [Google Scholar]
- Levonis, S.M.; Kiefel, M.J.; Houston, T.A. Boronolectin with divergent fluorescent response specific for free sialic acid. Chem. Commun. 2009, 2278–2280. [Google Scholar] [CrossRef]
- Abouderbala, L.O.; Belcher, W.J.; Boutelle, M.G.; Cragg, P.J.; Dhaliwal, J.; Fabre, M.; Steed, J.W.; Turner, D.R.; Wallace, K.J. Anion Sensing ‘Venus Flytrap’ Hosts: A Modular Approach. Chem. Commun. 2002, 2, 380–381. [Google Scholar] [CrossRef]
- Swinburne, A.N.; Paterson, M.J.; Beeby, A.; Steed, J.W. A Quinolinium-Derived Turn-off Fluorescent Anion Sensor. Org. Biomol. Chem. 2010, 8, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.R.; Paterson, M.J.; Steed, J.W. A Conformationally Flexible, Urea-Based Tripodal Anion Receptor: Solid-State, Solution, and Theoretical Studies. J. Org. Chem. 2006, 71, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Barnard, A.; Dickson, S.J.; Paterson, M.J.; Todd, A.M.; Steed, J.W. Enantioselective Lactate Binding by Chiral Tripodal Anion Hosts Derived from Amino Acids. Org. Biomol. Chem. 2009, 7, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Gong, W.T.; Chen, W.D.; Ye, J.W.; Lin, Y.; Ning, G.L. Colorimetric “naked-Eye” Sensor for Anions Based on Conformational Flexible Tripodal Receptor. J. Incl. Phenom. Macrocycl. Chem. 2011, 70, 115–119. [Google Scholar] [CrossRef]
- Amendola, V.; Bergamaschi, G.; Boiocchi, M.; Fusco, N.; La Rocca, M.V.; Linati, L.; Lo Presti, E.; Mella, M.; Metrangolo, P.; Miljkovic, A. Novel Hydrogen- and Halogen-Bonding Anion Receptors Based on 3-Iodopyridinium Units. RSC Adv. 2016, 6, 67540–67549. [Google Scholar] [CrossRef]
- Amendola, V.; Fabbrizzi, L.; Monzani, E. A Concave Fluorescent Sensor for Anions Based on 6-Methoxy-1-Methylquinolinium. Chem. Eur. J. 2004, 10, 76–82. [Google Scholar] [CrossRef]
- Saccone, M.; Cametti, M.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Systematic Study of Podand Molecules for Synergistic Halogen and Hydrogen Bond-Driven Anion Recognition in the Solid State. Chem.—Asian J. 2023, 18, e202201255. [Google Scholar] [CrossRef]
- Belcher, W.J.; Fabre, M.; Farhan, T.; Steed, J.W. Pyridinium CH⋯anion and π-Stacking Interactions in Modular Tripodal Anion Binding Hosts: ATP Binding and Solid-State Chiral Induction. Org. Biomol. Chem. 2006, 4, 781–786. [Google Scholar] [CrossRef]
- Bai, Y.; Zhang, B.-G.; Xu, J.; Duan, C.-Y.; Dang, D.-B.; Liu, D.-J.; Meng, Q.-J. Conformational Switching Fluorescent Chemosensor for Chloride Anion. New J. Chem. 2005, 29, 777. [Google Scholar] [CrossRef]
- Amendola, V.; Bergamaschi, G.; Boiocchi, M.; Legnani, L.; Presti, E.L.; Miljkovic, A.; Monzani, E.; Pancotti, F. Chloride-Binding in Organic-Water Mixtures: The Powerful Synergy of C-H Donor Groups within a Bowl-Shaped Cavity. Chem. Commun. 2016, 52, 10910–10913. [Google Scholar] [CrossRef]
- Faggi, E.; Porcar, R.; Bolte, M.; Luis, S.V.; García-Verdugo, E.; Alfonso, I. Chiral Imidazolium Receptors for Citrate and Malate: The Importance of the Preorganization. J. Org. Chem. 2014, 79, 9141–9149. [Google Scholar] [CrossRef] [PubMed]
- Howarth, J.; Al-Hashimy, N.A. A Homochiral Tripodal Receptor with Selectivity for Sodium (R)-2-Aminopropionate over Sodium (S)-2-Aminopropionate. Tetrahedron Lett. 2001, 42, 5777–5779. [Google Scholar] [CrossRef]
- Sato, K.; Okabe, Y.; Onitake, T.; Yamaguchi, M.; Arai, S.; Yamagishi, T. A Colorimetric Anion Sensing System Utilising Competitive CH· · ·X- Hydrogen Bonding. Supramol. Chem. 2011, 23, 249–251. [Google Scholar] [CrossRef]
- Yun, S.; Ihm, H.; Kim, H.G.; Lee, C.W.; Indrajit, B.; Oh, K.S.; Gong, Y.J.; Lee, J.W.; Yoon, J.; Lee, H.C.; et al. Molecular Recognition of Fluoride Anion: Benzene-Based Tripodal Imidazolium Receptor. J. Org. Chem. 2003, 68, 2467–2470. [Google Scholar] [CrossRef] [PubMed]
- Ihm, H.; Yun, S.; Kim, H.G.; Kim, J.K.; Kim, K.S. Tripodal Nitro-Imidazolium Receptor for Anion Binding Driven by (C-H)+⋯X- Hydrogen Bonds. Org. Lett. 2002, 4, 2897–2900. [Google Scholar] [CrossRef] [PubMed]
- Molina, P.; Zapata, F.; Caballero, A. Anion Recognition Strategies Based on Combined Noncovalent Interactions. Chem. Rev. 2017, 117, 9907–9972. [Google Scholar] [CrossRef]
- Steed, J.W. A Modular Approach to Anion Binding Podands: Adaptability in Design and Synthesis Leads to Adaptability in Properties. Chem. Commun. 2006, 2637–2649. [Google Scholar] [CrossRef]
- Langton, M.J.; Serpell, C.J.; Beer, P.D. Anion Recognition in Water: Recent Advances from a Supramolecular and Macromolecular Perspective. Angew. Chem. Int. Ed. 2016, 55, 1974–1987. [Google Scholar] [CrossRef]
- Macreadie, L.K.; Gilchrist, A.M.; McNaughton, D.A.; Ryder, W.G.; Fares, M.; Gale, P.A. Progress in Anion Receptor Chemistry. Chem 2022, 8, 46–118. [Google Scholar] [CrossRef]
- Busschaert, N.; Caltagirone, C.; Van Rossom, W.; Gale, P.A. Applications of Supramolecular Anion Recognition. Chem. Rev. 2015, 115, 8038–8155. [Google Scholar] [CrossRef]
- Kubik, S.; Reyheller, C.; Stüwe, S. Recognition of Anions by Synthetic Receptors in Aqueous Solution. J. Incl. Phenom. Macrocycl. Chem. 2005, 52, 137–187. [Google Scholar] [CrossRef]
- Kubik, S. Anion Recognition in Water. Chem. Soc. Rev. 2010, 39, 3648–3663. [Google Scholar] [CrossRef] [PubMed]
- Mazik, M.; Kuschel, M. Highly effective acyclic carbohydrate receptors consisting of aminopyridine, imidazole, and indole recognition units. Chem. Eur. J. 2008, 14, 2405–2419. [Google Scholar] [CrossRef]
- Lippe, J.; Mazik, M. Artificial receptors inspired by crystal structures of complexes formed between acyclic receptors and monosaccharides: Design, syntheses, and binding properties. J. Org. Chem. 2013, 78, 9013–9020. [Google Scholar] [CrossRef] [PubMed]
- Stack, T.D.P.; Hou, Z.; Raymond, K.N. Rational Reduction of the Conformational Space of a Siderophore Analog through Nonbonded Interactions: The Role of Entropy in Enterobactin. J. Am. Chem. Soc. 1993, 115, 6466–6467. [Google Scholar] [CrossRef]
- Wang, X.; Hof, F. (How) Does 1,3,5-Triethylbenzene Scaffolding Work? Analyzing the Abilities of 1,3,5-Triethylbenzene- and 1,3,5-Trimethylbenzene-Based Scaffolds to Preorganize the Binding Elements of Supramolecular Hosts and to Improve Binding of Targets. Beilstein J. Org. Chem. 2012, 8, 1–10. [Google Scholar] [CrossRef]
- Hennrich, G.; Anslyn, E. V 1,3,5-2,4,6-Functionalized, Facially Segregated Benzenes- Exploitation of Sterically Predisposed Systems in Supramolecular Chemistry. Chem. Eur. J. 2002, 8, 2218–2224. [Google Scholar] [CrossRef]
- Koch, N.; Seichter, W.; Mazik, M. Trimethyl-, Triethyl- and Trimethoxybenzene-Based Tripodal Compounds Bearing Pyrazole Groups: Conformations and Halogen-/Hydrogen-Bond Patterns in the Crystalline State. CrystEngComm 2017, 19, 3817–3833. [Google Scholar] [CrossRef]
- Schulze, M.M.; Schwarzer, A.; Mazik, M. Conformations of Benzene-Based Tripodal Isatin-Bearing Compounds in the Crystalline State. CrystEngComm 2017, 19, 4003–4016. [Google Scholar] [CrossRef]
- Grepioni, F.; Cojazzi, G.; Draper, S.M.; Scully, N.; Braga, D. Crystal Forms of Hexafluorophosphate Organometallic Salts and the Importance of Charge-Assisted C−H···F Hydrogen Bonds. Organometallics 1998, 17, 296–307. [Google Scholar] [CrossRef]
- Schneider, H.-J. Hydrogen Bonds with Fluorine. Studies in Solution, in Gas Phase and by Computations, Conflicting Conclusions from Crystallographic Analyses. Chem. Sci. 2012, 3, 1381–1394. [Google Scholar] [CrossRef]
- Dance, I. π-π Interactions: Theory and Scope. In Encyclopedia of Supramolecular Chemistry; Atwood, J.L., Steed, J.W., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 1076–1092. ISBN 9780429075728. [Google Scholar]
- James, S.L. π-π Stacking as a Crystal Engineering Tool. In Encyclopedia of Supramolecular Chemistry; Atwood, J.L., Steed, J.W., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 1093–1099. ISBN 9780429075728. [Google Scholar]
- Grell, J.; Bernstein, J.; Tinhofer, G. Graph-Set Analysis of Hydrogen-Bond Patterns: Some Mathematical Concepts. Acta Crystallogr. Sect. B Struct. Sci. 1999, 55, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-Set Analysis of Hydrogen-Bond Patterns in Organic Crystals. Acta Crystallogr. Sect. B Struct. Sci. 1990, 46, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Etter, M.C. Hydrogen Bonds as Design Elements in Organic Chemistry. J. Phys. Chem. 1991, 95, 4601–4610. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Köhler, L.; Hübler, C.; Seichter, W.; Mazik, M. Binding modes of methyl α-D-glucopyranoside to an artificial receptor in crystalline complexes. RSC Adv. 2021, 11, 22221–22229. [Google Scholar] [CrossRef]
- Köhler, L.; Seichter, W.; Mazik, M. Complexes Formed between Artificial Receptors and β-D-Glucopyranoside in the Crystalline State. Eur. J. Org. Chem. 2020, 2020, 7023–7034. [Google Scholar] [CrossRef]
- Fuhrmann, F.; Seichter, W.; Mazik, M. Selective Recognition of Ammonium over Potassium Ion with Acyclic Receptor Molecules Bearing 3,4,5-Trialkylpyrazolyl Groups. Org. Mater. 2022, 4, 61–72. [Google Scholar] [CrossRef]
- SAINT-NT, SADABS; Bruker AXS Inc.: Madison, WI, USA, 2014.
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- X-RED, X-SHAPE: Program for Data Reduction and Absorption Correction; Stoe & Cie GmbH: Darmstadt, Germany, 2002.
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal Structure Determination and Refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]

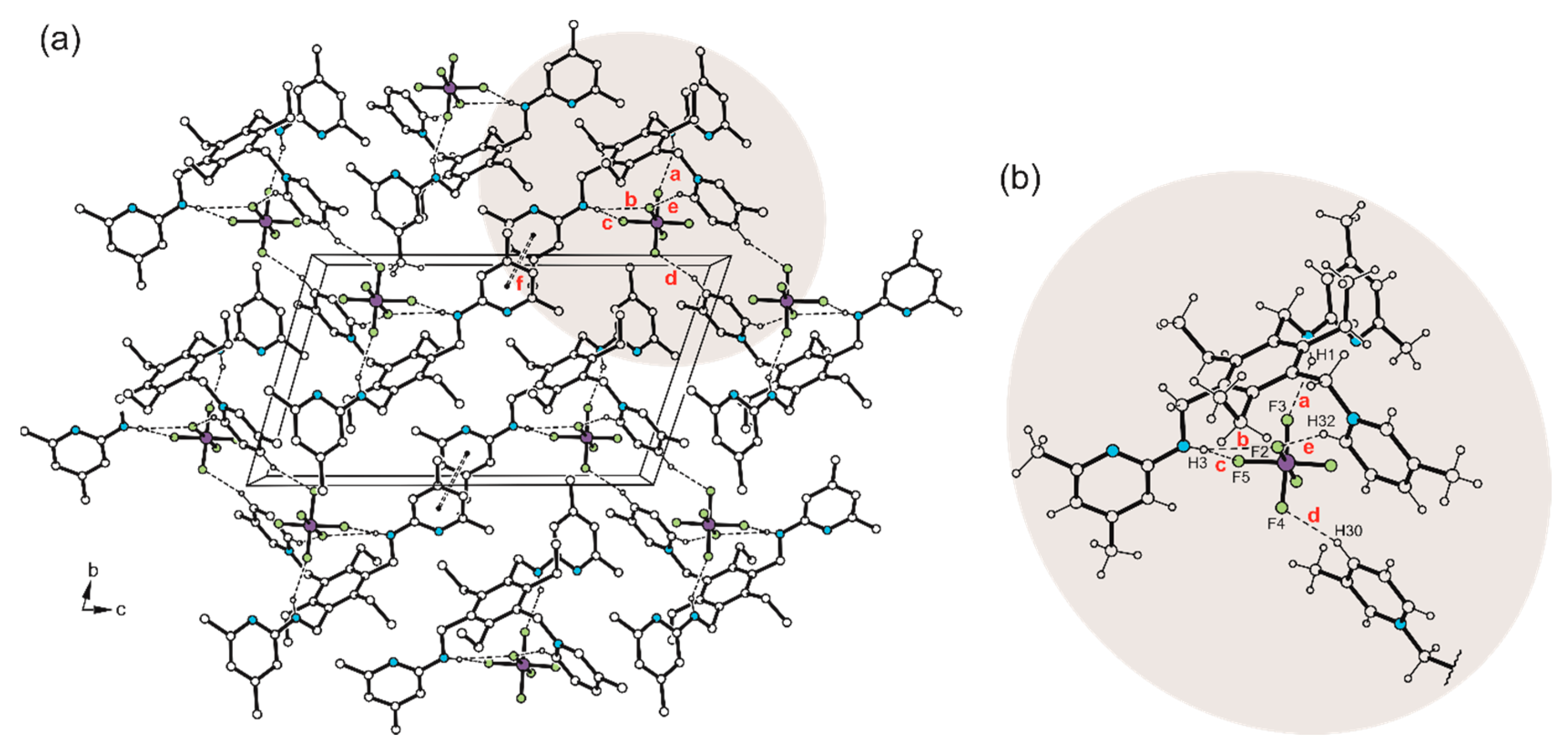
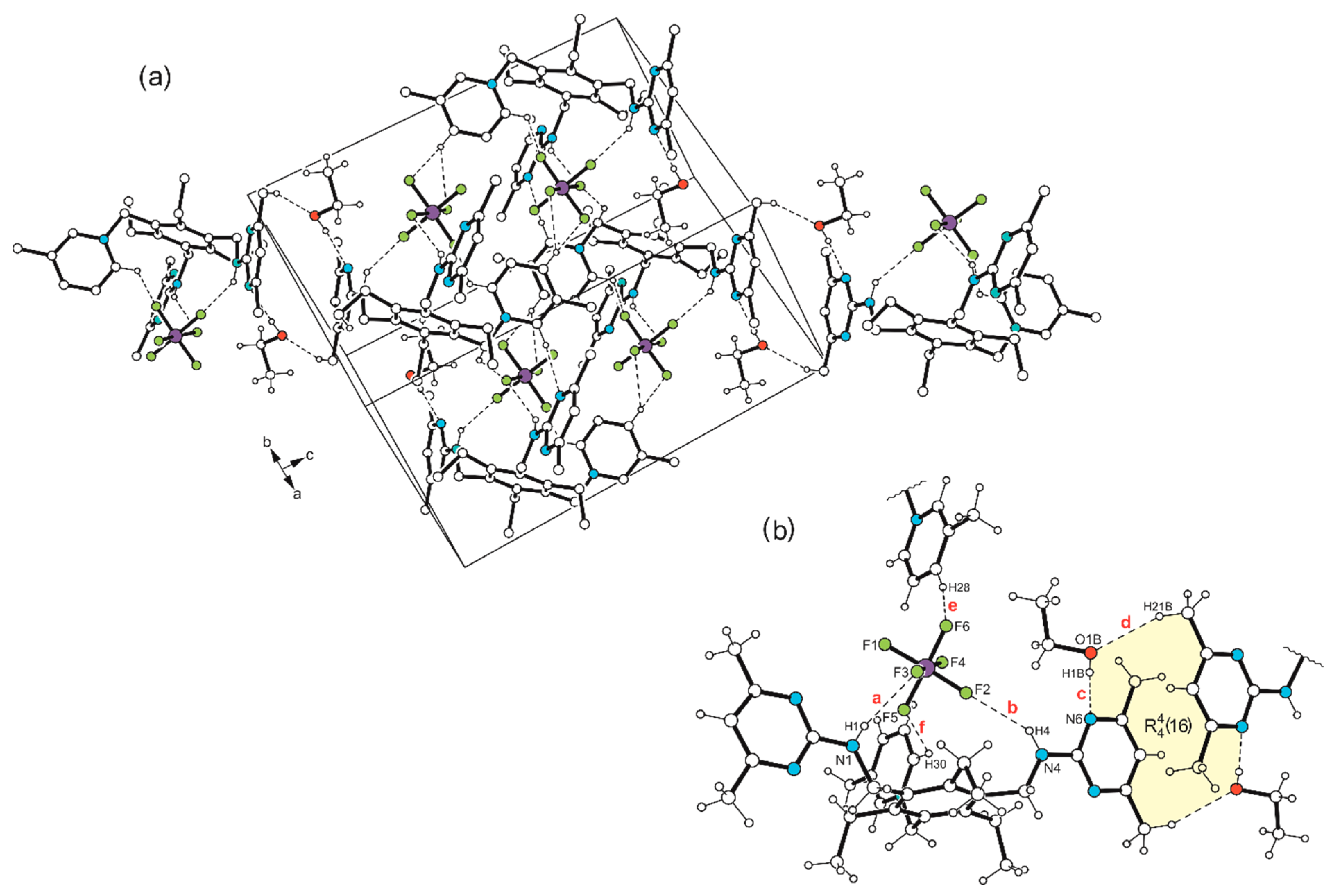



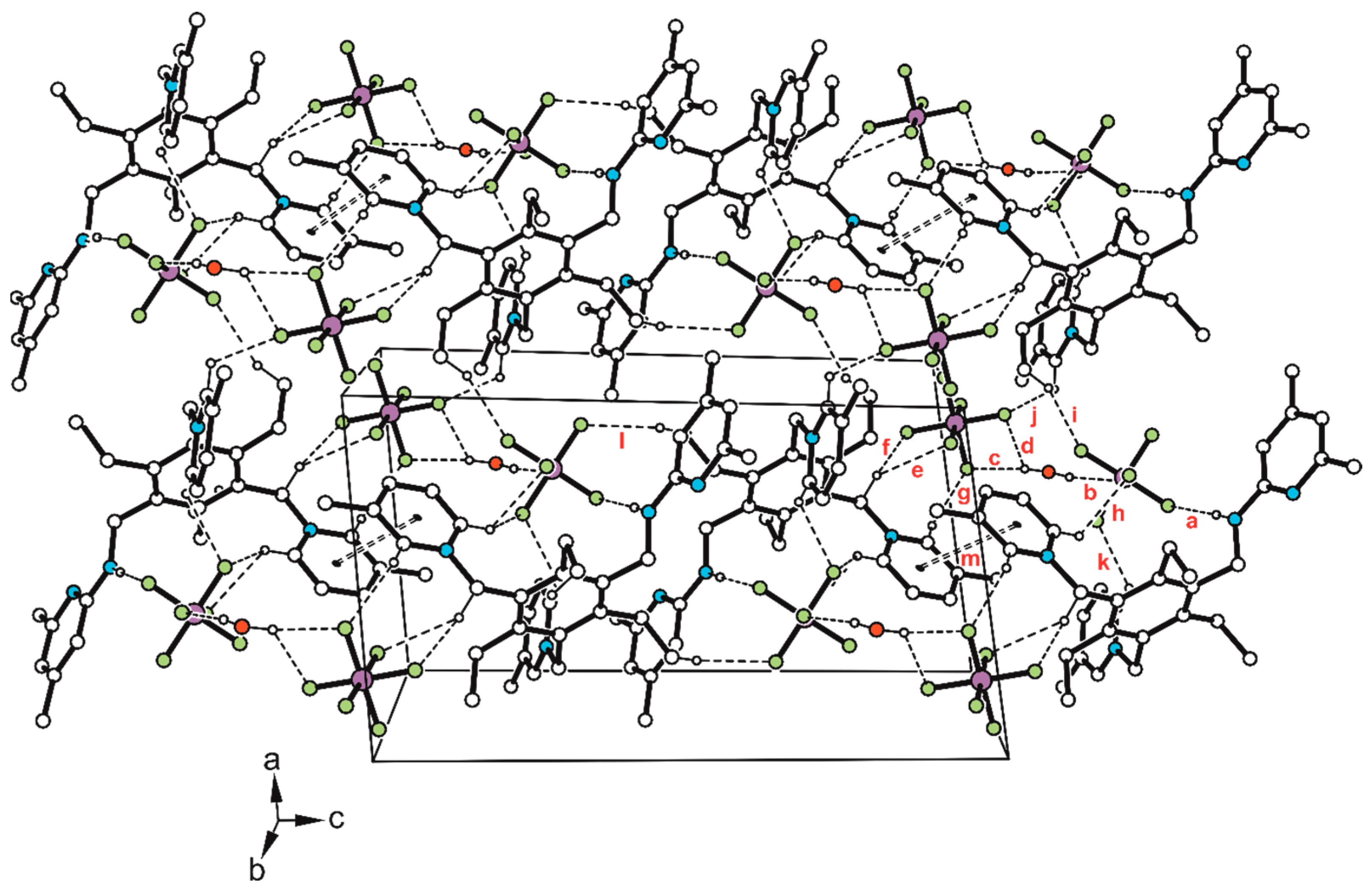

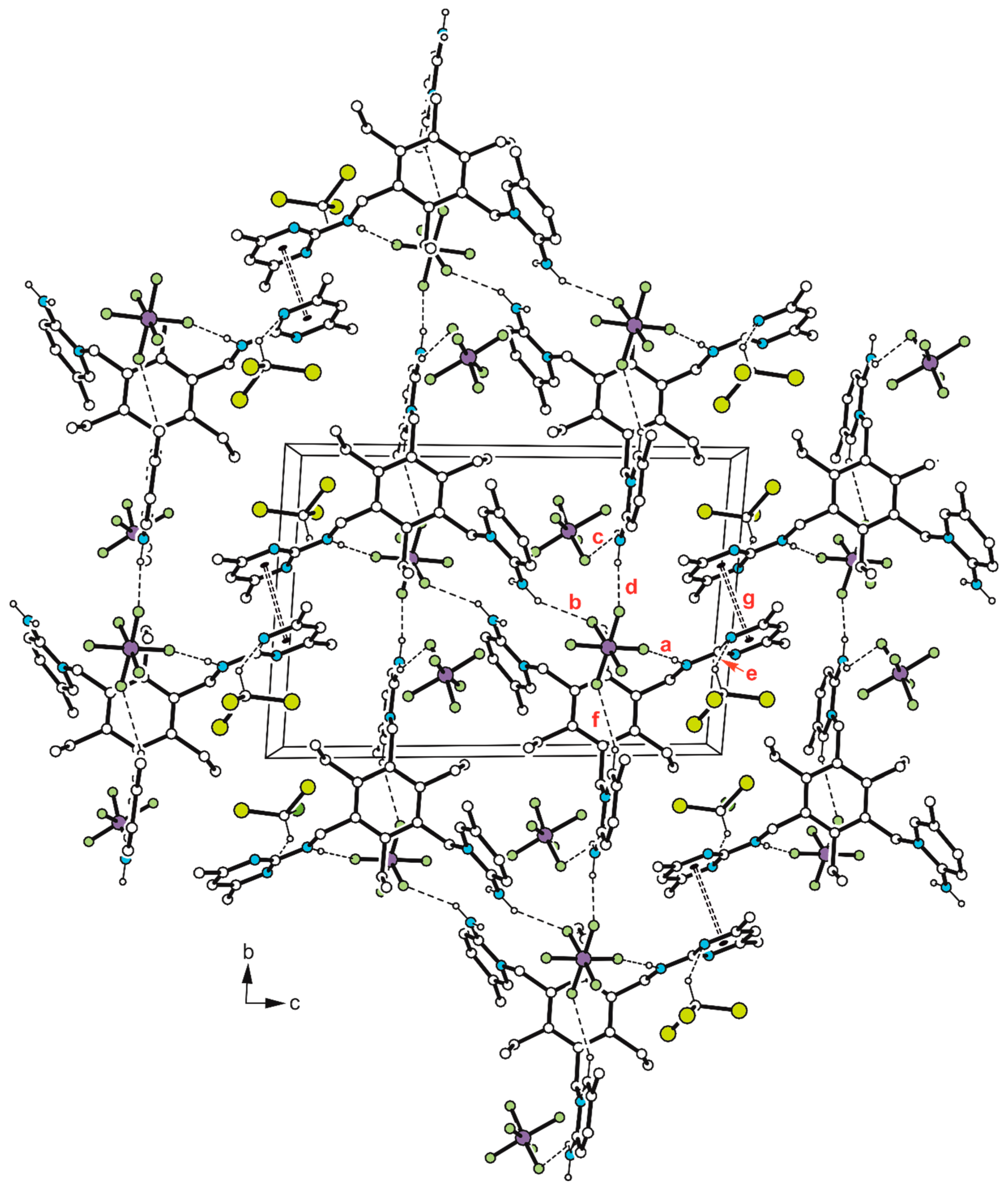
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiße, A.; Seichter, W.; Mazik, M. Supramolecular Motifs in the Crystal Structures of Triethylbenzene Derivatives Bearing Pyridinium Subunits in Combination with Pyrimidinyl or Pyridinyl Groups. Molecules 2023, 28, 6485. https://doi.org/10.3390/molecules28186485
Weiße A, Seichter W, Mazik M. Supramolecular Motifs in the Crystal Structures of Triethylbenzene Derivatives Bearing Pyridinium Subunits in Combination with Pyrimidinyl or Pyridinyl Groups. Molecules. 2023; 28(18):6485. https://doi.org/10.3390/molecules28186485
Chicago/Turabian StyleWeiße, Andrea, Wilhelm Seichter, and Monika Mazik. 2023. "Supramolecular Motifs in the Crystal Structures of Triethylbenzene Derivatives Bearing Pyridinium Subunits in Combination with Pyrimidinyl or Pyridinyl Groups" Molecules 28, no. 18: 6485. https://doi.org/10.3390/molecules28186485
APA StyleWeiße, A., Seichter, W., & Mazik, M. (2023). Supramolecular Motifs in the Crystal Structures of Triethylbenzene Derivatives Bearing Pyridinium Subunits in Combination with Pyrimidinyl or Pyridinyl Groups. Molecules, 28(18), 6485. https://doi.org/10.3390/molecules28186485