


Deacetylated Sialic Acid Sensitizes Lung and Colon Cancers to Novel Cucurbitacin-Inspired Estrone Epidermal Growth Factor Receptor (EGFR) Inhibitor Analogs

 , ,
, ,
Abstract
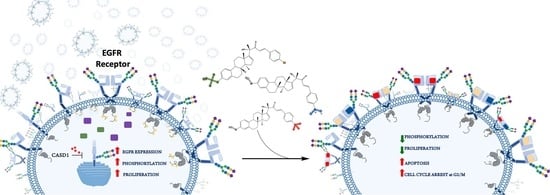
1. Introduction
2. Results and Discussion
2.1. Effects of Deacetylated Sia on Inhibitory Activity of CIEAs
2.2. Apoptotic Effects of CIEAs
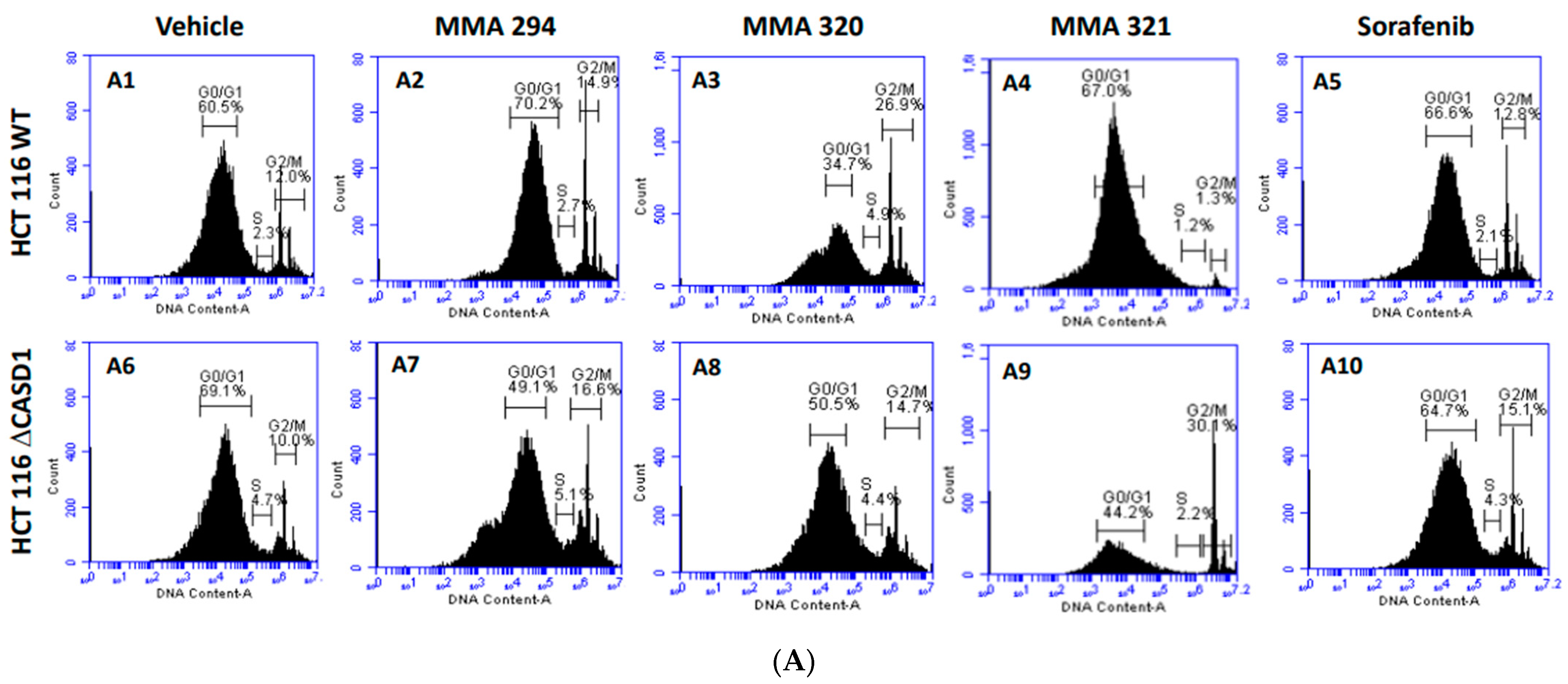
2.3. Effects of CIEAs on the Cell Cycle
2.4. Effects of Deacetylated Sia on Phosphorylated EGFR Expression
2.5. Effects of CIEAs on ERK and mTOR Phosphorylation Levels
3. Materials and Methods
3.1. Scientific Rigor
3.2. Chemicals and Reagents
3.3. Cell Lines and Cell Culture
3.4. Cell Viability Assay
3.5. Apoptosis Assay
3.6. Cell Cycle Analysis
3.7. Immunofluorescence Assay
3.8. In-Cell ELISA
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Grabenstein, S.; Barnard, K.N.; Anim, M.; Armoo, A.; Weichert, W.S.; Bertozzi, C.R.; Parrish, C.R.; Willand-Charnley, R. Deacetylated sialic acids modulates immune mediated cytotoxicity via the sialic acid-Siglec pathway. Glycobiology 2021, 31, 1279–1294. [Google Scholar] [CrossRef]
- Varki, A.; Schauer, R. Sialic Acids. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor: Long Island, NY, USA, 2009. [Google Scholar]
- Varki, N.M.; Varki, A. Diversity in cell surface sialic acid presentations: Implications for biology and disease. Lab. Investig. 2007, 87, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, V.S.; Pillai, S. Sialic acids and autoimmune disease. Immunol. Rev. 2016, 269, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, J.; Liu, Y.; Xu, D. Sialic acid metabolism as a potential therapeutic target of atherosclerosis. Lipids Health Dis. 2019, 18, 173. [Google Scholar] [CrossRef]
- Baker, A.N.; Richards, S.J.; Guy, C.S.; Congdon, T.R.; Hasan, M.; Zwetsloot, A.J.; Gallo, A.; Lewandowski, J.R.; Stansfeld, P.J.; Straube, A.; et al. The SARS-CoV-2 Spike Protein Binds Sialic Acids and Enables Rapid Detection in a Lateral Flow Point of Care Diagnostic Device. ACS Cent. Sci. 2020, 6, 2046–2052. [Google Scholar] [CrossRef] [PubMed]
- Barnard, K.N.; Wasik, B.R.; LaClair, J.R.; Buchholz, D.W.; Weichert, W.S.; Alford-Lawrence, B.K.; Aguilar, H.C.; Parrish, C.R. Expression of 9-O- and 7,9-O-Acetyl Modified Sialic Acid in Cells and Their Effects on Influenza Viruses. mBio 2019, 10, 10–1128. [Google Scholar] [CrossRef]
- Sjoberg, E.R.; Powell, L.D.; Klein, A.; Varki, A. Natural ligands of the B cell adhesion molecule CD22 beta can be masked by 9-O-acetylation of sialic acids. J. Cell Biol. 1994, 126, 549–562. [Google Scholar] [CrossRef]
- Tuffour, I.; Amuzu, S.; Bayoumi, H.; Surtaj, I.; Parrish, C.; Willand-Charnley, R. Early in vitro evidence indicates that deacetylated sialic acids modulate multi-drug resistance in colon and lung cancers via breast cancer resistance protein. Front. Oncol. 2023, 13, 1145333. [Google Scholar] [CrossRef] [PubMed]
- Mather, R.L.; Loveson, K.F.; Fillmore, H.L. Human Sialic acid O-acetyl esterase (SIAE)—Mediated changes in sensitivity to etoposide in a medulloblastoma cell line. Sci. Rep. 2019, 9, 8609. [Google Scholar] [CrossRef]
- Markman, M. The Promise and Perils of ‘Targeted Therapy’ of Advanced Ovarian Cancer. Oncology 2008, 74, 1–6. [Google Scholar] [CrossRef]
- Peters, G.J. From ‘Targeted Therapy’ to Targeted Therapy. Anticancer Res. 2019, 39, 3341–3345. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.-Y.; Liu, Y.-C.; Chen, N.-Y.; Tsai, C.-F.; Wang, Y.-T.; Chen, Y.-J.; Hsu, T.-L.; Yang, P.-C.; Wong, C.-H. Effect of sialylation on EGFR phosphorylation and resistance to tyrosine kinase inhibition. Proc. Natl. Acad. Sci. USA 2015, 112, 6955–6960. [Google Scholar] [CrossRef]
- Makin, G.; Hickman, J.A. Apoptosis and cancer chemotherapy. Cell Tissue Res. 2000, 301, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Wu, X.; Fang, W.; Zhao, Y.; Yang, Y.; Hu, Z.; Xue, C.; Zhang, J.; Zhang, J.; Ma, Y.; et al. Network meta-analysis of erlotinib, gefitinib, afatinib and icotinib in patients with advanced non-small-cell lung cancer harboring EGFR mutations. PLoS ONE 2014, 9, e85245. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Gijtenbeek, R.G.P.; van der Noort, V.; Aerts, J.; Staal-van den Brekel, J.A.; Smit, E.F.; Krouwels, F.H.; Wilschut, F.A.; Hiltermann, T.J.N.; Timens, W.; Schuuring, E.; et al. Randomised controlled trial of first-line tyrosine-kinase inhibitor (TKI) versus intercalated TKI with chemotherapy for EGFR-mutated nonsmall cell lung cancer. ERJ Open Res. 2022, 8, 00239–2022. [Google Scholar] [CrossRef]
- Herrera-Juárez, M.; Serrano-Gómez, C.; Bote-de-Cabo, H.; Paz-Ares, L. Targeted therapy for lung cancer: Beyond EGFR and ALK. Cancer 2023, 129, 1803–1820. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Halaweish, F.T. Cucurbitacins: Potential candidates targeting mitogen-activated protein kinase pathway for treatment of melanoma. J. Enzym. Inhib. Med. Chem. 2014, 29, 162–167. [Google Scholar] [CrossRef]
- Bartalis, J.; Halaweish, F.T. In Vitro and QSAR studies of cucurbitacins on HepG2 and HSC-T6 liver cell lines. Bioorg. Med. Chem. 2011, 19, 2757–2766. [Google Scholar] [CrossRef]
- Chen, X.; Bao, J.; Guo, J.; Ding, Q.; Lu, J.; Huang, M.; Wang, Y. Biological activities and potential molecular targets of cucurbitacins: A focus on cancer. Anticancer Drugs 2012, 23, 777–787. [Google Scholar] [CrossRef]
- Mahnashi, M.; Elgazwi, S.M.; Ahmed, M.S.; Halaweish, F.T. Cucurbitacins inspired organic synthesis: Potential dual inhibitors targeting EGFR—MAPK pathway. Eur. J. Med. Chem. 2019, 173, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M.; Laubli, H. Sialic acids in cancer biology and immunity. Glycobiology 2016, 26, 111–128. [Google Scholar] [CrossRef]
- Shen, Y.; Tiralongo, J.; Kohla, G.; Schauer, R. Regulation of sialic acid O-acetylation in human colon mucosa. Biol. Chem. 2004, 385, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Ghosh, S.; Mandal, C.; Kohla, G.; Brossmer, R.; Isecke, R.; Merling, A.; Schauer, R.; Schwartz-Albiez, R.; Bhattacharya, D.K.; et al. Purification and characterization of 9-O-acetylated sialoglycoproteins from leukemic cells and their potential as immunological tool for monitoring childhood acute lymphoblastic leukemia. Glycobiology 2004, 14, 859–870. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, Y.-C.; Yen, H.-Y.; Chen, C.-Y.; Chen, C.-H.; Cheng, P.-F.; Juan, Y.-H.; Chen, C.-H.; Khoo, K.-H.; Yu, C.-J.; Yang, P.-C.; et al. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11332–11337. [Google Scholar] [CrossRef]
- Nakanishi, T.; Ross, D.D. Breast cancer resistance protein (BCRP/ABCG2): Its role in multidrug resistance and regulation of its gene expression. Chin. J. Cancer 2012, 31, 73–99. [Google Scholar] [CrossRef]
- Koopman, G.; Reutelingsperger, C.P.; Kuijten, G.A.; Keehnen, R.M.; Pals, S.T.; van Oers, M.H. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 1994, 84, 1415–1420. [Google Scholar] [CrossRef]
- Verhoven, B.; Schlegel, R.A.; Williamson, P. Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J. Exp. Med. 1995, 182, 1597–1601. [Google Scholar] [CrossRef]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [CrossRef]
- Zhang, G.; Gurtu, V.; Kain, S.R.; Yan, G. Early detection of apoptosis using a fluorescent conjugate of annexin V. Biotechniques 1997, 23, 525–531. [Google Scholar] [CrossRef]
- Chambard, J.C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Duronio, R.J.; Xiong, Y. Signaling pathways that control cell proliferation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008904. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Zhang, Y.; Bunz, F. Checkpoint bypass and cell viability. Cell Cycle 2010, 9, 2102–2107. [Google Scholar] [CrossRef][Green Version]
- Wang, Z. Regulation of Cell Cycle Progression by Growth Factor-Induced Cell Signaling. Cells 2021, 10, 3327. [Google Scholar] [CrossRef]
- Plastaras, J.P.; Kim, S.H.; Liu, Y.Y.; Dicker, D.T.; Dorsey, J.F.; McDonough, J.; Cerniglia, G.; Rajendran, R.R.; Gupta, A.; Rustgi, A.K.; et al. Cell cycle dependent and schedule-dependent antitumor effects of sorafenib combined with radiation. Cancer Res. 2007, 67, 9443–9454. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Z.; Wang, K. Combining sorafenib with celecoxib synergistically inhibits tumor growth of non-small cell lung cancer cells in vitro and in vivo. Oncol. Rep. 2014, 31, 1954–1960. [Google Scholar] [CrossRef]
- Ormerod, M.G.; Orr, R.M.; Peacock, J.H. The role of apoptosis in cell killing by cisplatin: A flow cytometric study. Br. J. Cancer 1994, 69, 93–100. [Google Scholar] [CrossRef]
- Stark, G.R.; Taylor, W.R. Analyzing the G2/M checkpoint. Methods Mol. Biol. 2004, 280, 51–82. [Google Scholar] [CrossRef]
- Acheampong, F.; Ostlund, T.; Mahnashi, M.; Halaweish, F. Antiproliferation and apoptosis studies of estrone pharmacophores in triple-negative breast cancer. Chem. Biol. Drug Des. 2023; ahead of print. [Google Scholar] [CrossRef]
- Ahmed, M.S.; El-Senduny, F.; Taylor, J.; Halaweish, F.T. Biological screening of cucurbitacin inspired estrone analogs targeting mitogen-activated protein kinase (MAPK) pathway. Chem. Biol. Drug Des. 2017, 90, 478–484. [Google Scholar] [CrossRef]
- Hendler, F.J.; Ozanne, B.W. Human squamous cell lung cancers express increased epidermal growth factor receptors. J. Clin. Investig. 1984, 74, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.; Sainsbury, C.; Needham, G.; Farndon, J.; Malcolm, A.; Harris, A. Epidermal-growth-factor receptor status as predictor of early recurrence of and death from breast cancer. Lancet 1987, 329, 1398–1402. [Google Scholar] [CrossRef]
- Neal, D.E.; Marsh, C.; Bennett, M.K.; Abel, P.D.; Hall, R.R.; Sainsbury, J.R.; Harris, A.L. Epidermal-growth-factor receptors in human bladder cancer: Comparison of invasive and superficial tumours. Lancet 1985, 1, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Xiong, J.; Gu, X.; Chen, X.; Wu, S.; Wang, Z.; Wang, D.; Tu, J.; Xie, J. Novel recombinant immunotoxin of EGFR specific nanobody fused with cucurmosin, construction and antitumor efficiency in vitro. Oncotarget 2017, 8, 38568–38580. [Google Scholar] [CrossRef]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [CrossRef] [PubMed]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef]
- Ramos, J.W. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int. J. Biochem. Cell Biol. 2008, 40, 2707–2719. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Langereis, J.D.; de Jonge, M.I. Invasive Disease Caused by Nontypeable Haemophilus influenzae. Emerg. Infect Dis. 2015, 21, 1711–7188. [Google Scholar] [CrossRef] [PubMed]
- Osei-Safo, D.; Dziwornu, G.A.; Appiah-Opong, R.; Chama, M.A.; Tuffour, I.; Waibel, R.; Amewu, R.; Addae-Mensah, I. Constituents of the Roots of Dichapetalum pallidum and Their Anti-Proliferative Activity. Molecules 2017, 22, 532. [Google Scholar] [CrossRef] [PubMed]
- Tuffour, I.; Ayi, I.; Gwira, T.M.; Dumashie, E.; Ashong, Y.; Appiah-Opong, R. Schistosoma Egg Antigen Induces Oncogenic Alterations in Human Prostate Cells. Anal. Cell. Pathol. 2018, 2018, 4675380. [Google Scholar] [CrossRef] [PubMed]
- Ostlund, T.; Alotaibi, F.; Kyeremateng, J.; Halaweish, H.; Kasten, A.; Iram, S.; Halaweish, F. Triazole-estradiol analogs: A potential cancer therapeutic targeting ovarian and colorectal cancer. Steroids 2022, 177, 108950. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | |
|---|---|---|
| A549 WT | A549 ΔCASD1 | |
| MMA 294 | 19.07 ± 5.01 | 4.77 ± 2.60 (4.0) |
| MMA 320 | 9.02 ± 2.32 | 6.49 ± 1.67 (1.4) |
| MMA 321 | 20.97 ± 5.19 | 1.24 ± 0.09 (16.9) |
| Sorafenib | 26.74 ± 4.38 | 9.96 ± 3.41 (2.7) |
| HCT 116 WT | HCT 116 ΔCASD1 | |
| MMA 294 | 8.82 ± 0.87 | 4.29 ± 0.16 (2.1) |
| MMA 320 | 8.65 ± 1.14 | 3.14 ± 0.08 (2.8) |
| MMA 321 | 12.72 ± 1.07 | 6.17 ± 1.74 (2.1) |
| Sorafenib | 9.66 ± 1.58 | 7.79 ± 0.38 (1.2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anim, M.T.; Tuffour, I.; Willis, R.; Schell, M.; Ostlund, T.; Mahnashi, M.H.; Halaweish, F.; Willand-Charnley, R. Deacetylated Sialic Acid Sensitizes Lung and Colon Cancers to Novel Cucurbitacin-Inspired Estrone Epidermal Growth Factor Receptor (EGFR) Inhibitor Analogs. Molecules 2023, 28, 6257. https://doi.org/10.3390/molecules28176257
Anim MT, Tuffour I, Willis R, Schell M, Ostlund T, Mahnashi MH, Halaweish F, Willand-Charnley R. Deacetylated Sialic Acid Sensitizes Lung and Colon Cancers to Novel Cucurbitacin-Inspired Estrone Epidermal Growth Factor Receptor (EGFR) Inhibitor Analogs. Molecules. 2023; 28(17):6257. https://doi.org/10.3390/molecules28176257
Chicago/Turabian StyleAnim, Mathias T., Isaac Tuffour, Rylan Willis, Matthew Schell, Trevor Ostlund, Mater H. Mahnashi, Fathi Halaweish, and Rachel Willand-Charnley. 2023. "Deacetylated Sialic Acid Sensitizes Lung and Colon Cancers to Novel Cucurbitacin-Inspired Estrone Epidermal Growth Factor Receptor (EGFR) Inhibitor Analogs" Molecules 28, no. 17: 6257. https://doi.org/10.3390/molecules28176257
APA StyleAnim, M. T., Tuffour, I., Willis, R., Schell, M., Ostlund, T., Mahnashi, M. H., Halaweish, F., & Willand-Charnley, R. (2023). Deacetylated Sialic Acid Sensitizes Lung and Colon Cancers to Novel Cucurbitacin-Inspired Estrone Epidermal Growth Factor Receptor (EGFR) Inhibitor Analogs. Molecules, 28(17), 6257. https://doi.org/10.3390/molecules28176257