
Tuning Benzylic C−H Functionalization of (Thio)xanthenes with Electrochemistry
Abstract
:
1. Introduction
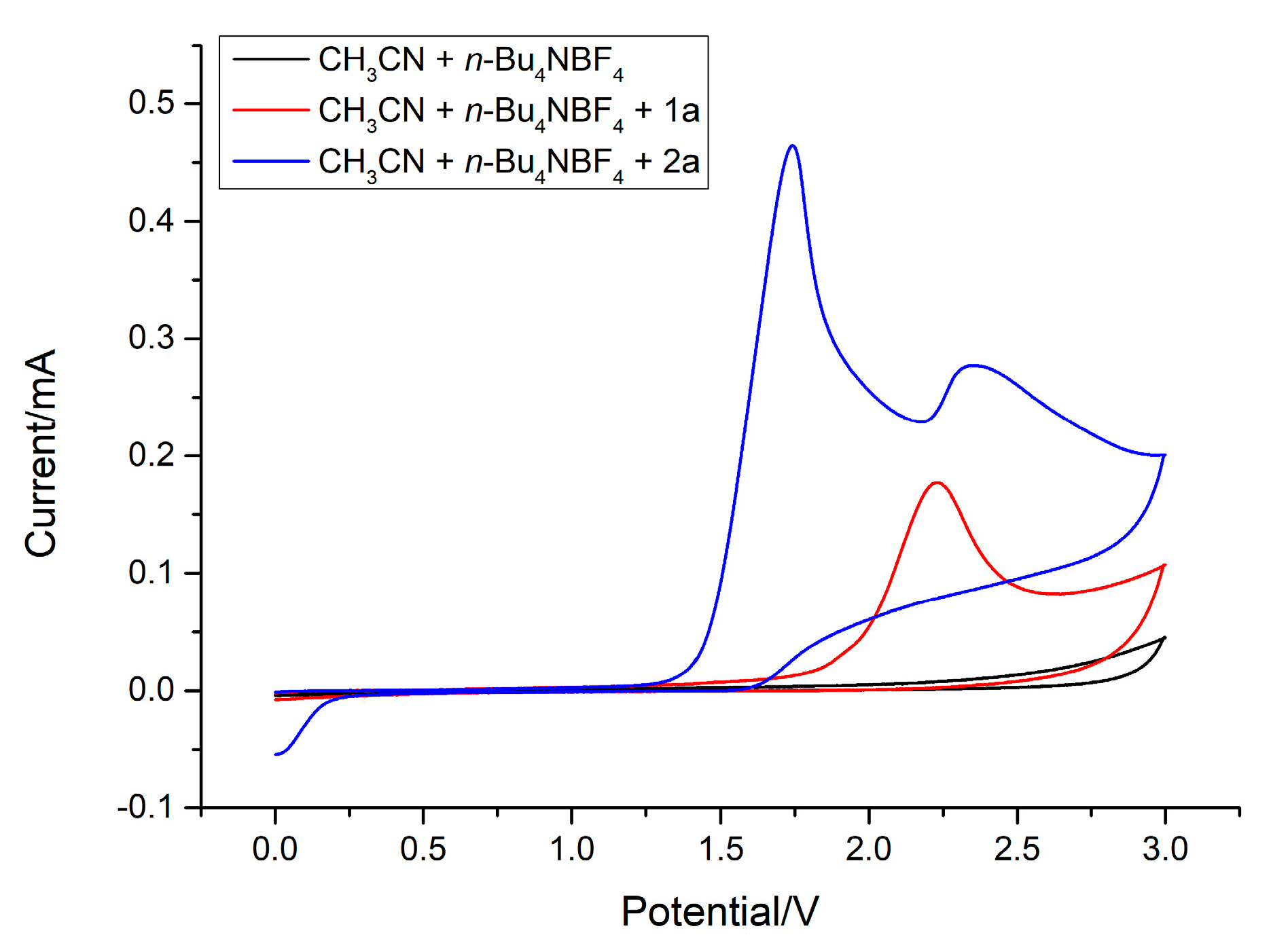
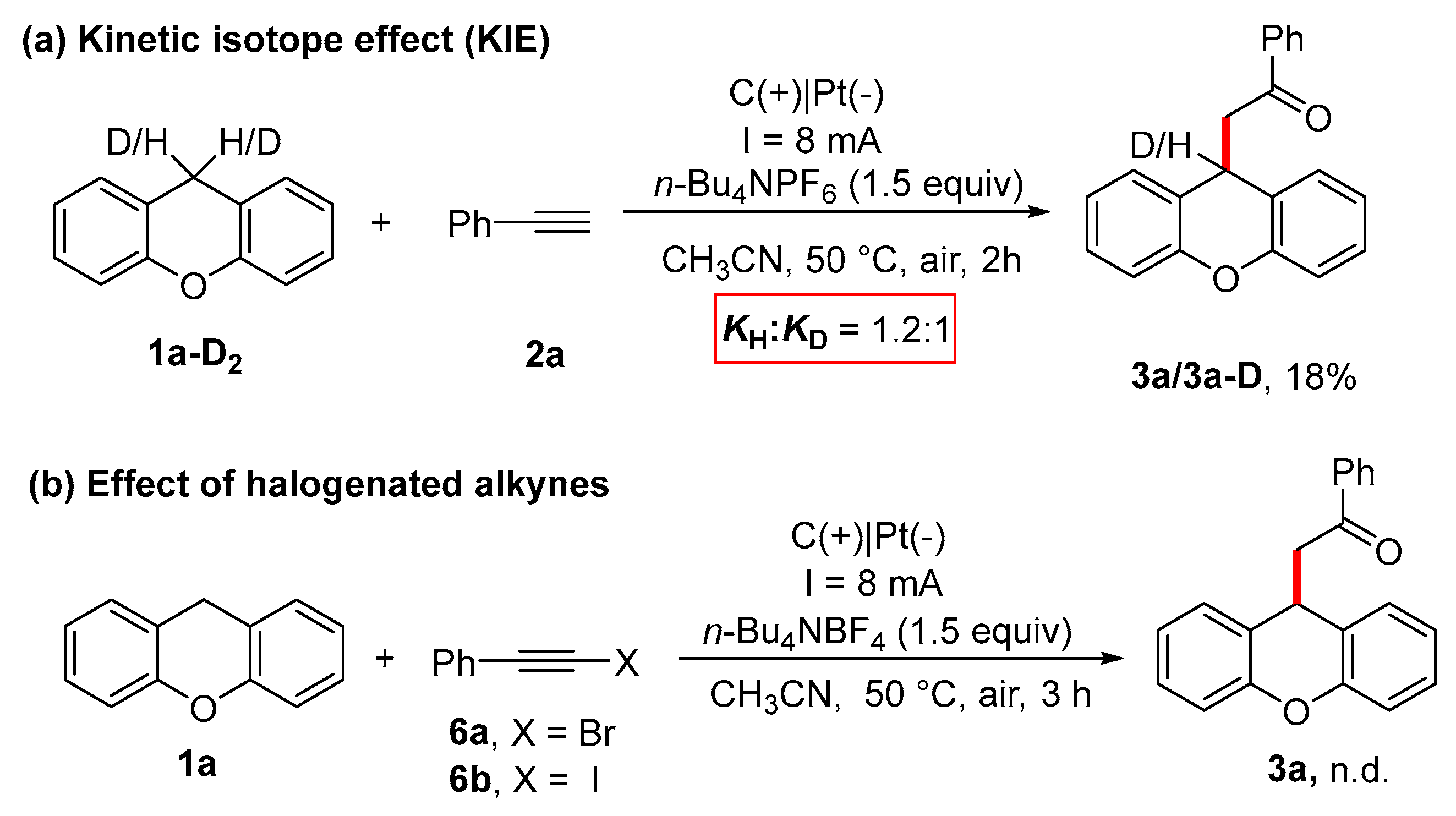
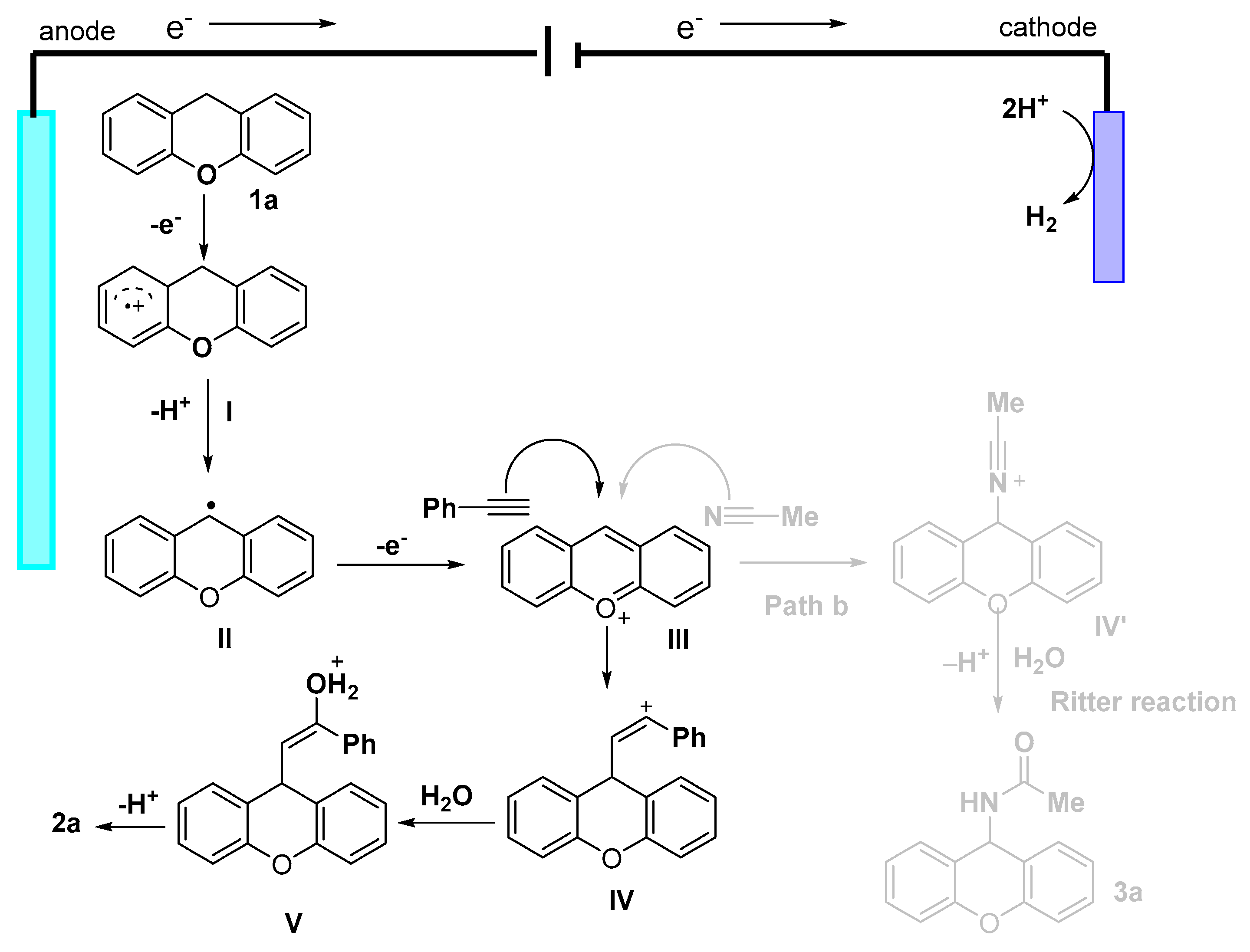
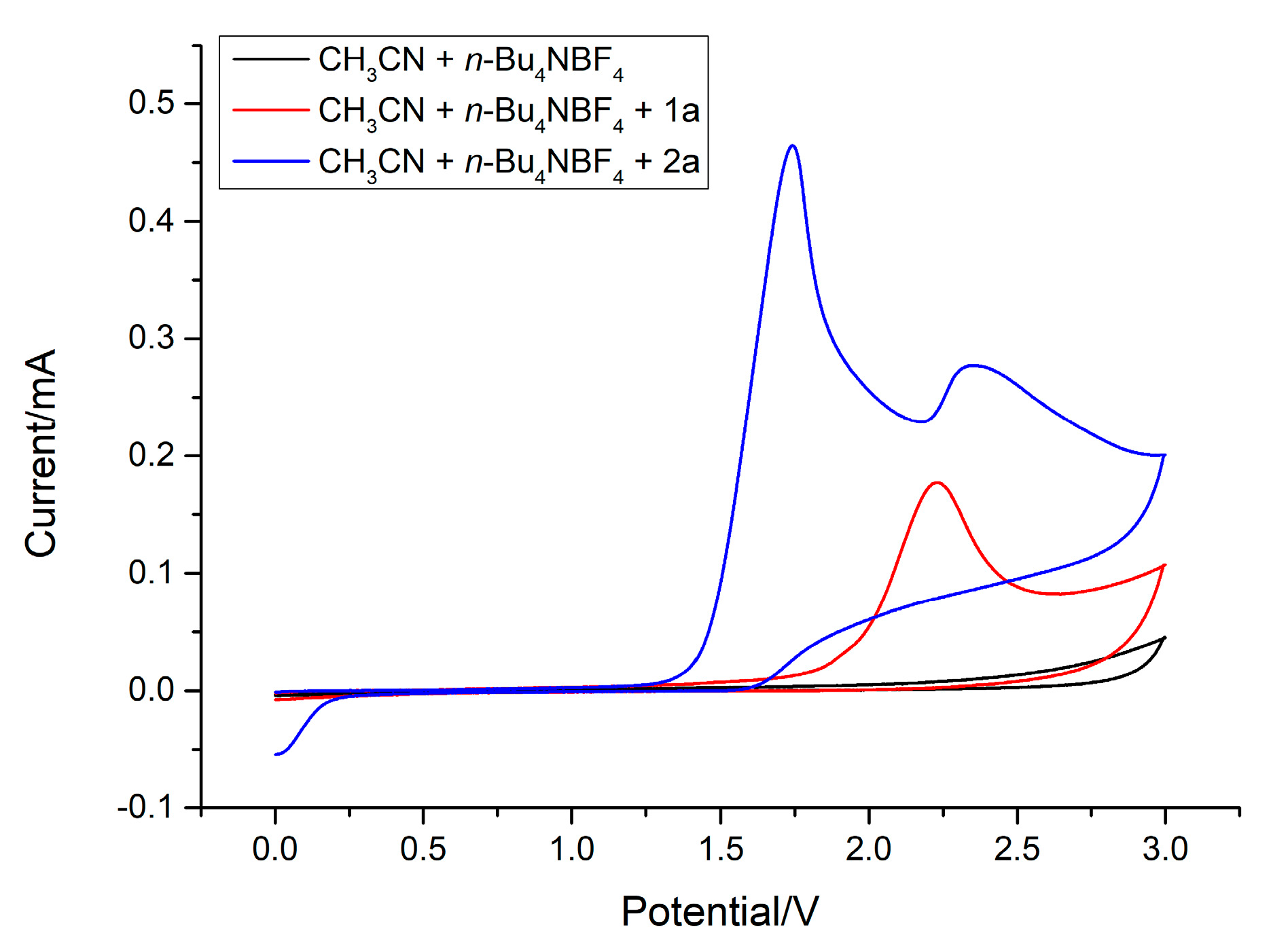
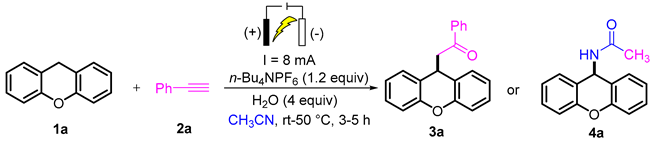
2. Results and Discussion
3. Materials and Methods
3.1. General Considerations
3.2. Typical Procedure for the Synthesis of 3a
- 1-Phenyl-2-(9H-xanthen-9-yl)ethan-1-one (3a) was prepared following general procedure [46], and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 100:1) to afford the product 3a (63.8 mg, 71% yield). 1H NMR (600 MHz, CDCl3) δ 7.81 (d, J = 7.2 Hz, 2H), 7.50 (t, J = 7.2 Hz, 1H), 7.37 (t, J = 7.8 Hz, 2H), 7.32 (dd, J = 7.2, 1.2 Hz, 2H), 7.23–7.19 (m, 2H), 7.12 (d, J = 8.4 Hz, 2H), 7.04–7.00 (m, 2H), 4.85 (t, J = 6.0 Hz, 1H), 3.35 (d, J = 6.6 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 197.9, 152.4, 137.0, 133.1, 128.8, 128.5, 128.1, 127.9, 125.5, 123.5, 116.6, 49.7, 34.7. HRMS (ESI) calcd. for C21H16NaO2+ ([M + Na]+): 323.1043, found: 323.1044.
- 1-(p-tolyl)-2-(9H-Xanthen-9-yl)ethan-1-one (3b) was prepared following general procedure, [47] and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 100:1) to afford the product 3b (59.2 mg, 63% yield). White solid; m.p.: 108~109 °C. 1H NMR (600 MHz, CDCl3) δ 7.71 (d, J = 7.8 Hz, 2H), 7.32 (dd, J = 7.8, 1.2 Hz, 2H), 7.22–7.19 (m, 2H), 7.17 (d, J = 8.4 Hz, 2H), 7.11 (dd, J = 7.8, 1.2 Hz, 2H), 7.03–7.00 (m, 2H), 4.85 (t, J = 6.6 Hz, 1H), 3.32 (d, J = 6.6 Hz, 2H), 2.36 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 197.69, 152.46, 144.09, 134.68, 129.33, 128.98, 128.36, 127.95, 125.75, 123.59, 116.65, 49.77, 34.81, 21.73. HRMS (ESI) calcd. for C22H18NaO2+ ([M + Na]+): 337.1199, found: 337.1203.
- 1-(4-Pentylphenyl)-2-(9H-xanthen-9-yl)ethan-1-one (3c) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 100:1) to afford the product 3c (82.2 mg, 75% yield). White solid; m.p.: 117~118 °C. 1H NMR (600 MHz, CDCl3) δ 7.73 (d, J = 8.4 Hz, 2H), 7.32 (dd, J = 7.8, 1.2 Hz, 2H), 7.22–7.16 (m, 2H), 7.17 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 8.4 Hz, 2H), 7.03–7.00 (m, 2H), 4.85 (t, J = 6.6 Hz, 1H), 3.33 (d, J = 6.6 Hz, 2H), 2.60 (t, J = 7.8 Hz, 2H), 1.58–1.32 (m, 2H), 1.31–1.25 (m, 4H), 0.88 (t, J = 7.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 197.5, 152.3, 148.9, 134.7, 128.9, 128.6, 128.3, 127.8, 125.7, 123.5, 116.5, 49.7, 34.6, 31.4, 30.8, 22.5, 22.3, 14.0. HRMS (ESI) calcd. for C26H26NaO2+ ([M + Na]+): 393.1825, found: 393.1826.
- 1-(4′-Ethyl-[1,1′-biphenyl]-4-yl)-2-(9H-xanthen-9-yl)ethan-1-one (3d) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 100:1) to afford the product 3d (78.6 mg, 65% yield). White solid; m.p.: 122~123 °C. 1H NMR (600 MHz, CDCl3) δ 7.86 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 7.8 Hz, 2H), 7.34 (dd, J = 7.8, 1.8 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H), 7.23–7.20 (m, 2H), 7.13 (dd, J = 7.8, 0.6 Hz, 2H), 7.03 (td, J = 7.2, 1.2 Hz, 2H), 4.88 (t, J = 6.6 Hz, 1H), 3.38 (d, J = 6.6 Hz, 2H), 2.70 (q, J = 7.8 Hz, 2H), 1.28 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 197.6, 152.5, 145.9, 144.8, 137.2, 135.6, 129.0, 128.8, 128.6, 128.0, 127.3, 127.0, 125.7, 123.6, 116.7, 49.9, 34.9, 28.7, 15.7. HRMS (ESI) calcd. for C29H24NaO2+ ([M + Na]+): 427.1669, found: 427.1668.
- 1-(o-tolyl)-2-(9H-Xanthen-9-yl)ethan-1-one (3e) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 100:1) to afford the product 3e (49.8 mg, 53% yield). White solid; m.p.: 105~106 °C. 1H NMR (600 MHz, CDCl3) δ 7.33 (d, J = 7.2 Hz, 2H), 7.30–7.27 (m, 2H), 7.21 (t, J = 7.8 Hz, 2H), 7.18 (d, J = 7.2 Hz, 1H), 7.12–7.09 (m, 3H), 7.04 (t, J = 7.2 Hz, 2H), 4.84 (t, J = 6.6 Hz, 1H), 3.26 (d, J = 6.6 Hz, 2H), 2.43 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 202.1, 152.5, 138.2, 132.0, 131.4, 128.9, 128.6, 128.0, 125.7, 125.7, 123.6, 116.7, 52.7, 35.0, 21.2. HRMS (ESI) calcd. for C22H18NaO2+ ([M + Na]+): 337.1199, found: 337.1201.
- 1-(m-tolyl)-2-(9H-Xanthen-9-yl)ethan-1-one (3f) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 100:1) to afford the product 3f (60.0 mg, 68% yield). White solid; m.p.: 111~112 °C. 1H NMR (600 MHz, CDCl3) δ 7.54–7.50 (m, 2H), 7.24 (t, J = 7.2 Hz, 2H), 7.20–7.16 (m, 2H), 7.13 (t, J = 7.8 Hz, 2H), 7.04 (d, J = 7.8 Hz, 2H), 6.95 (t, J = 7.2 Hz, 2H), 4.77 (t, J = 6.6 Hz, 1H), 3.26 (d, J = 6.6 Hz, 2H), 2.26 (s, 3H). 13C NMR (151 MHz, CDCl3) 198.3, 152.6, 138. 6, 137.2, 134.1, 129.1, 128.9, 128.6, 128.1, 125.8, 125.5, 123.7, 116.8, 50.0, 34.9, 21.5.
- 1-(Naphthalen-1-yl)-2-(9H-xanthen-9-yl)ethan-1-one (3g) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 100:1) to afford the product 3g (64.2 mg, 61% yield). White solid; m.p.: 116~117 °C. 1H NMR (600 MHz, CDCl3) δ 8.50 (d, J = 8.4 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.59–7.55 (m, 1H), 7.59–7.50 (m, 1H), 7.48 (dd, J = 7.2, 1.1 Hz, 1H), 7.38 (dd, J = 7.2, 1.2 Hz, 2H), 7.36–7.32 (m, 1H), 7.24–7.21 (m, 2H), 7.12 (dd, J = 8.4, 1.2 Hz, 2H), 7.06–7.03 (m, 2H), 4.94 (t, J = 6.6 Hz, 1H), 3.42 (d, J = 6.6 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 202.2, 152.5, 136.1, 134.0, 132.9, 130.2, 128.9, 128.6, 128.1, 127.9, 126.6, 125.8, 125.5, 124.4, 123.7, 53.2, 35.5. HRMS (ESI) calcd. for C25H18NaO2+ ([M+Na]+): 373.1199, found: 373.1200.
- 2-(2-Methyl-9H-xanthen-9-yl)-1-phenylethan-1-one (3h) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 100:1) to afford the product 3h (65.8 mg, 70% yield). White solid; m.p.: 102~103 °C. 1H NMR (600 MHz, CDCl3) δ 7.81 (d, J = 7.8 Hz, 2H), 7.50 (t, J = 7.2 Hz, 1H), 7.37 (t, J = 7.2 Hz, 2H), 7.31 (d, J = 7.8 Hz, 1H), 7.19 (t, J = 7.2 Hz, 1H), 7.10 (d, J = 7.8 Hz, 2H), 7.02–6.89 (m, 3H), 4.80 (t, J = 6.6 Hz, 1H), 3.35 (d, J = 6.6 Hz, 2H), 2.27 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 198.2, 152.6, 150.3, 137.2, 133.21, 133.0, 129.2, 129.0, 128.6, 128.6, 128.2, 127.9, 125.7, 125.3, 123.4, 116.6, 116.4, 49.9, 34.8, 20.8.
- 2-(4-Methyl-9H-xanthen-9-yl)-1-phenylethan-1-one (3i) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 100:1) to afford the product 3i (67.8 mg, 72% yield). White solid; m.p.: 107~108 °C. 1H NMR (600 MHz, CDCl3) δ 7.71 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 7.8 Hz, 2H), 7.23 –7.20 (m, 2H), 7.17 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 7.02 (t, J = 7.8 Hz, 2H), 4.86 (t, J = 6.6 Hz, 1H), 3.33 (d, J = 6.6 Hz, 2H), 2.36 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 197.5, 152.3, 143.9, 134.5, 130.0, 129.8, 129.5, 129.5, 129.2, 128.8, 128.3, 128.2, 127.8, 127.3, 125.6, 123.4, 116.5, 49.6, 34.7, 21.6. HRMS (ESI) calcd. for C22H18NaO2+ ([M + Na]+): 337.1199, found: 337.1196.
- 2-(4-(tert-Butyl)-9H-xanthen-9-yl)-1-phenylethan-1-one (3j) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 100:1) to afford the product 3j (74.0 mg, 70% yield). White solid; m.p.: 113~114 °C. 1H NMR (600 MHz, CDCl3) δ 7.78–7.75 (m, 2H), 7.41–7.38 (m, 2H), 7.32 (dd, J = 7.8, 1.2 Hz, 2H), 7.22–7.19 (m, 2H), 7.12 (dd, J = 8.4, 1.2 Hz, 2H), 7.02 (td, J = 7.2, 1.2 Hz, 2H), 4.87 (t, J = 6.6 Hz, 1H), 3.34 (d, J = 6.6 Hz, 2H), 1.30 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 196.4, 155.9, 151.3, 133.4, 128.8, 128.7, 127.8, 127.3, 127.0, 126.8, 126.3, 124.7, 124.6, 124.4, 122.4, 121.8, 115.5, 48.7, 34.0, 33.5, 29.9. HRMS (ESI) calcd. for C25H24NaO2+ ([M + Na]+): 379.1669, found: 379.1670.
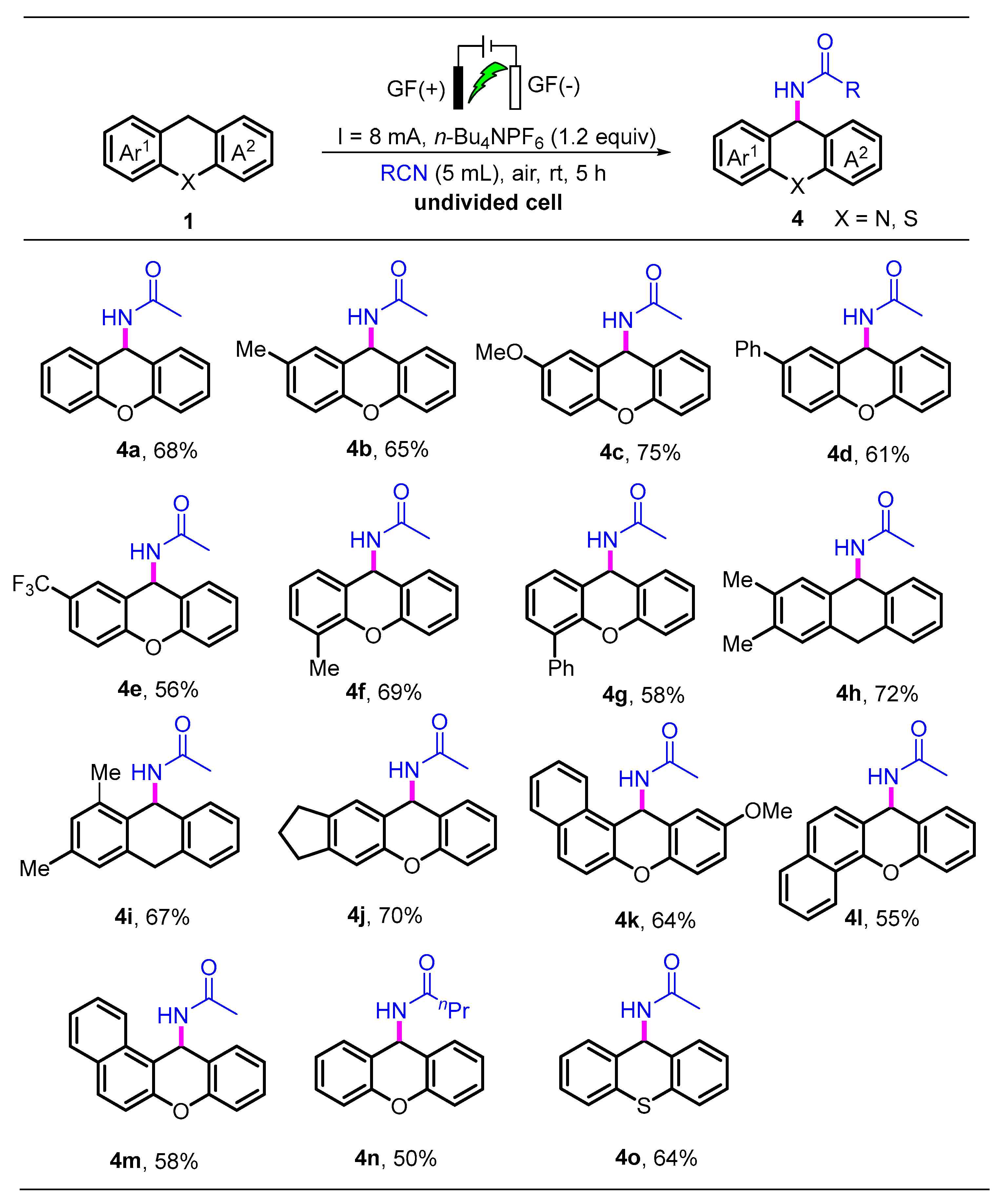
3.3. Typical Procedure for the Synthesis of 4a
- N-(9H-xanthen-9-yl)acetamide (4a) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4a (48.7 mg, 68% yield). White solid; m.p.: 238~239 °C. 1H NMR (600 MHz, CDCl3) δ 7.48–7.46 (m, 2H), 7.32–7.28 (m, 2H), 7.13–7.10 (m, 4H), 6.49 (d, J = 9.0 Hz, 1H), 6.05 (br, d, J = 9.0 Hz, 1H), 2.00 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.2, 151.3, 129.8, 129.4, 123.7, 121.3, 116.8, 44.0, 23.5. HRMS (ESI) calcd. for C15H13NNaO2+ ([M + Na]+): 262.0838, found: 262.0839.
- N-(2-Methyl-9H-xanthen-9-yl)acetamide (4b) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4b (49.5 mg, 65% yield). White solid; m.p.: 242~243 °C. 1H NMR (600 MHz, CDCl3) δ 7.46 (d, J = 7.8 Hz, 1H), 7.31–7.27 (m, 1H), 7.11–7.08 (m, 3H), 7.00 (d, J = 8.4 Hz, 1H), 6.46 (d, J = 9.0 Hz, 1H), 6.00 (br, d, J = 9.0 Hz, 1H), 2.32 (s, 3H), 2.01 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.2, 151.4, 149.2, 133.2, 130.2, 129.8, 129.8, 129.3, 123.5, 121.3, 120.8, 116.7, 116.5, 44.1, 23.6, 20.8. HRMS (ESI) calcd. for C16H15NNaO3+ ([M + Na]+): 276.0095, found: 276.0092.
- N-(2-Methoxy-9H-xanthen-9-yl)acetamide (4c) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4c (60.3 mg, 75% yield). White solid; m.p.: 251~252 °C. 1H NMR (600 MHz, CDCl3) δ 7.46 (d, J = 7.2 Hz, 1H), 7.31–7.27 (m, 1H), 7.11–7.07 (m, 2H), 7.04 (d, J = 9.0 Hz, 1H), 6.98 (d, J = 3.0 Hz, 1H), 6.87 (dd, J = 9.0, 3.0 Hz, 1H), 6.47 (d, J = 9.0 Hz, 1H), 6.01 (br, d, J = 9.0 Hz, 1H), 3.79 (s, 3H), 2.01 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.2, 155.7, 151.5, 145.4, 129.8, 129.4, 123.5, 121.7, 120.7, 117.7, 116.7, 116.4, 112.8, 55.9, 44.5, 23.6. HRMS (ESI) calcd. for C16H15NNaO3+ ([M + Na]+): 292.0944, found 292.0947.
- N-(2-Phenyl-9H-xanthen-9-yl)acetamide (4d) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4d (57.7 mg, 61% yield). White solid; m.p.: 247~248 °C. 1H NMR (600 MHz, CDCl3) δ 7.70 (d, J = 1.8 Hz, 1H), 7.58–7.56 (m, 2H), 7.54 (dd, J = 9.0, 2.4 Hz, 1H), 7.51–7.49 (m, 1H), 7.43 (t, J = 7.2 Hz, 2H), 7.36–7.30 (m, 2H), 7.18 (d, J = 8.4 Hz, 1H), 7.14–7.11 (m, 2H), 6.57 (d, J = 9.6 Hz, 1H), 6.06 (br, J = 9.6 Hz, 1H), 2.01 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.2, 151.2, 150.8, 140.2, 136.9, 129.9, 129.5, 129.0, 128.2, 128.2, 127.4, 127.0, 123.8, 121.5, 121.2, 117.2, 116.8, 44.1, 23.6. HRMS (ESI) calcd. for C21H17NNaO2+ ([M + Na]+): 338.1151, found: 338.1147.
- N-(2-(Trifluoromethyl)-9H-xanthen-9-yl)acetamide (4e) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4e (51.6 mg, 56% yield). White solid; m.p.: 244~245 °C. 1H NMR (600 MHz, CDCl3) δ 7.75 (s, 1H), 7.55 (dd, J = 8.4, 1.2 Hz, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.35–7.31 (m, 1H), 7.20 (d, J = 8.4 Hz, 1H), 7.15 (t, J = 7.8 Hz, 1H), 7.13 (d, J = 8.4 Hz, 1H), 6.52 (d, J = 9.0 Hz, 1H), 6.10 (br, s, 1H), 2.04 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.4, 153.5, 150.7, 129.8, 129.64, 127.4 (q, J = 3.6), 126.5 (q, J = 3.2), 125.9 (q, J = 271.5), 124.5, 123.1, 121.8, 121.3, 120.7, 117.4, 116.9, 43.7, 23.5. 19F NMR (565 MHz, CDCl3) δ − 77.30. HRMS (ESI) calcd. for C16H12F3NNaO2+ ([M + Na]+): 330.0172, found: 330.0175.
- N-(4-Methyl-9H-xanthen-9-yl)acetamide (4f) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4f (52.5 mg, 69% yield). White solid; m.p.: 239~240 °C. 1H NMR (600 MHz, CDCl3) 7.50 (d, J = 7.2 Hz, 1H), 7.31 (t, J = 8.4 Hz, 2H), 7.16 (d, J = 8.4 Hz, 2H), 7.13–7.10 (m, 1H), 7.02 (t, J = 7.8 Hz, 1H), 6.50 (d, J = 9.0 Hz, 1H), 5.92 (br, d, J = 8.4 Hz, 1H), 2.40 (s, 3H), 2.00 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.0, 151.2, 151.0, 139.6, 129.7, 129.4, 129.2, 124.6, 123.5, 121.3, 118.1, 116.9, 116.6, 43.8, 23.4, 21.2. HRMS (ESI) calcd. for C16H15NNaO2+ ([M + Na]+): 276.0995, found: 276.0997.
- N-(4-Phenyl-9H-xanthen-9-yl)acetamide (4g) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4g (54.7 mg, 58% yield). White solid; m.p.: 251~152 °C. 1H NMR (600 MHz, CDCl3) δ 7.60 (d, J = 7.0 Hz, 2H), 7.51–7.46 (m, 4H), 7.42–7.38 (m, 1H), 7.36 (dd, J = 7.8, 1.6 Hz, 1H), 7.29–7.26 (m, 1H), 7.19 (t, J = 7.8 Hz, 1H), 7.12 (td, J = 7.8, 1.2 Hz, 1H), 7.03 (d, J = 8.4 Hz, 1H), 6.56 (d, J = 9.0 Hz, 1H), 6.06 (br, J = 8.4 Hz, 1H), 2.02 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.2, 151.3, 148.3, 137.5, 130.8, 130.2, 129.8, 129.6, 129.3, 129.1, 128.3, 127.5, 123.9, 123.7, 122.0, 121.3, 116.9, 44.5, 23.6. HRMS (ESI) calcd. for C21H17NNaO2+ ([M + Na]+): 338.1151, found: 338.1151.
- N-(2,3-Dimethyl-9,10-dihydroanthracen-9-yl)acetamide (4h) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4h (57.2 mg, 72% yield). White solid; m.p.: 247~248 °C. 1H NMR (600 MHz, CDCl3) δ 7.66 (dd, J = 7.8, 0.6 Hz, 1H), 7.30–7.27 (m, 1H), 7.12 (d, J = 8.4 Hz, 1H), 7.10–7.07 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.55 (d, J = 9.0 Hz, 1H), 5.85 (br, d, J = 7.8 Hz, 1H), 2.27 (s, 3H), 2.26 (s, 3H), 1.91 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 168.4, 151.1, 150.8, 136.8, 131.9, 130.7, 130.2, 129.2, 123.6, 122.8, 119.0, 116.3, 114.1, 42.9, 23.3, 20.2, 15.2. HRMS (ESI) calcd. for C17H17NNaO2+ ([M + Na]+): 290.1151, found: 290.1152.
- N-(1,3-Dimethyl-9,10-dihydroanthracen-9-yl)acetamide (4i) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4i (51.6 mg, 67% yield). White solid; m.p.: 244~245 °C. 1H NMR (600 MHz, CDCl3) δ 7.66–7.63 (m, 1H), 7.29 (td, J = 7.8, 1.8 Hz, 1H), 7.11–7.08 (m, 2H), 6.82 (d, J = 4.8 Hz, 2H), 6.47 (d, J = 9.6 Hz, 1H), 5.78 (br, d, J = 9.0 Hz, 1H), 2.34 (s, 3H), 2.32 (s, 3H), 1.91 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 168.4, 152.4, 151.3, 139.4, 138.6, 130.2, 129.2, 126.4, 123.7, 122.9, 116.5, 116.2, 115.1, 42.4, 23.4, 21.2, 18.7. HRMS (ESI) calcd. for C17H17NNaO2+ ([M + Na]+): 290.1151, found: 290.1155.
- N-(1,2,3,10-Tetrahydrocyclopenta[b]xanthen-10-yl)acetamide (4j) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4j (51.9 mg, 70% yield). White solid; m.p.: 236~237 °C. 1H NMR (600 MHz, CDCl3) δ 7.58 (dd, J = 8.4, 1.8 Hz, 1H), 7.30–7.27 (m, 1H), 7.18 (d, J = 8.4 Hz, 1H), 7.11–7.07 (m, 2H), 6.94 (d, J = 8.4 Hz, 1H), 6.48 (d, J = 9.6 Hz, 1H), 5.81 (br, d, J = 9.6 Hz, 1H), 2.99–2.94 (m, 1H), 2.90 (t, J = 8.4 Hz, 2H), 2.87–2.81 (m, 1H), 2.17–2.08 (m, 2H), 1.96 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 168.5, 151.327, 150.6, 144.8, 139.4, 130.2, 129.3, 125.1, 123.6, 122.2, 116.6, 116.4, 114.9, 42.8, 32.5, 31.4, 25.7, 23.4. HRMS (ESI) calcd. for C18H17NNaO2+ ([M + Na]+): 302.1151, found 302.1154.
- N-(10-Methoxy-12H-benzo[a]xanthen-12-yl)acetamide (4k) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 3:1) to afford the product 4k (61.3 mg, 64% yield). White solid; m.p.: 258~259 °C. 1H NMR (600 MHz, CDCl3) δ 8.07 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 6.6 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.60 (t, J = 6.6 Hz, 1H), 7.46 (t, J = 7.2 Hz, 1H), 7.30 (d, J = 9.0 Hz, 1H), 7.25 (d, J = 3.0 Hz, 1H), 7.13 (d, J = 9.0 Hz, 1H), 7.08 (d, J = 9.4 Hz, 1H), 6.92 (dd, J = 8.4, 3.0 Hz, 1H), 5.85 (br, d, J = 9.6 Hz, 1H), 3.83 (s, 3H), 1.92 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 168.7, 156.2, 150.3, 144.8, 131.8, 130. 5, 130.4, 128.7, 127.8, 124.8, 123.0, 122.5, 118.0, 117.6, 116.8, 112.6, 111.5, 56.0, 42.2, 23.4. HRMS (ESI) calcd. for C20H17NNaO2+ ([M + Na]+): 342.1101, found 342.1102.
- N-(7H-benzo[c]xanthen-7-yl)acetamide (4l) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4l (47.6 mg, 55% yield). White solid; m.p.: 250~251 °C. 1H NMR (600 MHz, CDCl3)δ 8.43 (d, J = 8.4 Hz, 1H), 7.83 (d, J = 7.2 Hz, 1H), 7.60 –7.54 (m, 4H), 7.51–7.48 (m, 1H), 7.38–7.35 (m, 1H), 7.30 (dd, J = 8.4, 1.2 Hz, 1H), 7.19–7.16 (m, 1H), 6.66 (d, J = 9.0 Hz, 1H), 6.04 (br, d, J = 9.0 Hz, 1H), 2.02 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.1, 151.0, 146.3, 134.0, 129.9, 129.3, 127.6, 127.0, 126.3, 126.3, 124.0, 123.9, 123.3, 121.8, 121.2, 116.8, 114.8, 44.2, 23.5. HRMS (ESI) calcd. for C19H15
- N-(12H-benzo[a]xanthen-12-yl)acetamide (4m) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4m (50.3 mg, 58% yield). White solid; m.p.: 249~250 °C. 1H NMR (600 MHz, CDCl3) δ 8.43 (d, J = 8.4 Hz, 1H), 7.83 (d, J = 7.8 Hz, 1H), 7.60–7.54 (m, 4H), 7.50 (d, J = 8.4 Hz, 1H), 7.37 (t, J = 7.8 Hz, 1H), 7.30 (d, J = 7.8 Hz, 1H), 7.17 (t, J = 7.2 Hz, 1H), 6.67 (d, J = 9.6 Hz, 1H), 6.01 (br, J = 9.0 Hz, 1H), 2.02 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.1, 151.1, 146.4, 134.1, 130.0, 129.4, 127.8, 127.1, 126.4, 126.4, 124.1, 123.4, 121.9, 121.4, 116.9, 114.9, 44.4, 23.6. HRMS (ESI) calcd. for C19H15NNaO2+ ([M + Na]+): 312.0095, found 312.0098.
- N-(9H-xanthen-9-yl)butyramide (4n) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 9:1) to afford the product 4n (40.1 mg, 50% yield). White solid; m.p.: 262~263 °C. 1H NMR (600 MHz, CDCl3) δ 7.45 (d, J = 7.8 Hz, 2H), 7.30 (t, J = 7.2 Hz, 2H), 7.12–7.09 (m, 4H), 6.52 (d, J = 9.0 Hz, 1H), 6.05 (br, d, J = 8.4 Hz, 1H), 2.17 (t, J = 7.8 Hz, 2H), 1.72–1.67 (m, 2H), 0.94 (t, J = 7.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 172.2, 151.2, 129.8, 129.4, 123.7, 121.4, 116.8, 43.8, 38.9, 19.3, 13.9. HRMS (ESI) calcd. for C18H19NO2+ ([M + H]+): 267.1379, found: 267.1377.
- N-(9H-thioxanthen-9-yl)acetamide (4o) was prepared following general procedure, and the reaction mixture was purified by flash column chromatography with petroleum ether and ethylacetate (PE/EA = 5:1) to afford the product 4o (48.9 mg, 64% yield). White solid; m.p.: 237~238 °C. 1H NMR (600 MHz, CDCl3) δ 7.58 (dd, J = 6.0, 2.4 Hz, 2H), 7.47–7.45 (m, 2H), 7.28–7.25 (m, 4H), 6.30 (d, J = 9.0 Hz, 1H), 6.23 (br, d, J = 7.2 Hz, 1H), 1.88 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 169.0, 134.9, 133.0, 129.6, 128.0, 127.2, 127.2, 53.4, 23.5. HRMS (ESI) calcd. for C15H13NNaOS+ ([M + Na]+): 278.0610, found: 278.0613.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Lee, B.J.; DeGlopper, K.S.; Yoon, T.P. Site-Selective Alkoxylation of Benzylic C−H Bonds by Photoredox Catalysis. Angew. Chem. Int. Ed. 2019, 59, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.Y.; Lai, X.L.; Wang, Y.; Hu, H.H.; Song, J.; Yang, Y.; Wang, C.; Xu, H.C. Photoelectrochemical Asymmetric Catalysis Enables Site- and Enantioselective Cyanation of Benzylic C−H Bonds. Nat. Catal. 2022, 5, 943–951. [Google Scholar] [CrossRef]
- Wu, F.P.; Yang, Y.; Fuentes, D.P.; Wu, X.F. Copper-catalyzed Carbonylative Catenation of Olefins: Direct Synthesis of γ-boryl Esters. Chem 2022, 8, 1982–1992. [Google Scholar] [CrossRef]
- Guo, S.; AbuSalim, D.I.; Cook, S.P. Aqueous Benzylic C−H Trifluoromethylation for Late-Stage Functionalization. J. Am. Chem. Soc. 2018, 140, 12378–12382. [Google Scholar] [CrossRef]
- Dewanji, A.; Krach, P.E.; Rueping, M. The Dual Role of Benzophenone in Visible-Light/Nickel Photoredox-Catalyzed C−H Arylations: Hydrogen-Atom Transfer and Energy Transfer. Angew. Chem. Int. Ed. 2019, 58, 3566–3570. [Google Scholar] [CrossRef]
- Ishida, N.; Masuda, Y.; Imamura, Y.; Yamazaki, K.; Murakami, M. Carboxylation of Benzylic and Aliphatic C−H Bonds with CO2 Induced by Light/Ketone/Nickel. J. Am. Chem. Soc. 2019, 141, 19611–19615. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Lambert, T.H. Electrophotocatalytic Diamination of Vicinal C−H Bonds. Science 2021, 371, 620–626. [Google Scholar] [CrossRef]
- Oliva, M.; Coppola, G.A.; Van der Eycken, E.V.; Sharma, U.K. Photochemical and Electrochemical Strategies Towards Benzylic C−H Functionalization: A Recent Update. Adv. Synth. Catal. 2021, 363, 1810–1834. [Google Scholar] [CrossRef]
- Khatua, H.; Das, S.; Patra, S.; Das, S.K.; Roy, S.; Chattopadhyay, B. Iron-Catalyzed Intermolecular Amination of Benzylic C(sp3)−H Bonds. J. Am. Chem. Soc. 2022, 144, 21858–21866. [Google Scholar] [CrossRef]
- Das, M.; Zamani, L.; Bratcher, C.; Musacchio, P.Z. Azolation of Benzylic C−H Bonds via Photoredox-Catalyzed Carbocation Generation. J. Am. Chem. Soc. 2023, 145, 3861–3868. [Google Scholar] [CrossRef]
- Lewandowska-Andralojc, A.; Grills, D.C.; Zhang, J.; Bullock, R.M.; Miyazawa, A.; Kawanishi, Y.; Fujita, E. Kinetic and Mechanistic Studies of Carbon-to-Metal Hydrogen Atom Transfer Involving Os-Centered Radicals: Evidence for Tunneling. J. Am. Chem. Soc. 2014, 136, 3572–3578. [Google Scholar] [CrossRef]
- Wu, H.; Su, C.; Tandiana, R.; Liu, C.; Qiu, C.; Bao, Y.; Wu, J.; Xu, Y.; Lu, J.; Fan, D.; et al. Graphene-Oxide-Catalyzed Direct CH−CH-Type Cross-Coupling: The Intrinsic Catalytic Activities of Zigzag Edges. Angew. Chem. Int. Ed. 2018, 57, 10848–10853. [Google Scholar] [CrossRef]
- Fenteany, G.; Sharma, G.; Gaur, P.; Borics, A.; Wéber, E.; Kiss, E.; Haracska, L. A Series of Xanthenes Inhibiting Rad6 Function and Rad6-Rad18 Interaction in the PCNA Ubiquitination Cascade. iScience 2022, 25, 104053–104076. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Shi, J.; Zhu, A.; Xu, Z.F.; Liu, S.H.; Wang, Y.S.; Guo, Z.K.; Jiao, R.H.; Tan, R.X.; Ge, H.M. Total Biosynthesis of Mutaxanthene Unveils a Flavoprotein Monooxygenase Catalyzing Xanthene Ring Formation. Angew. Chem. Int. Ed. 2023, 62, e202218660. [Google Scholar] [CrossRef] [PubMed]
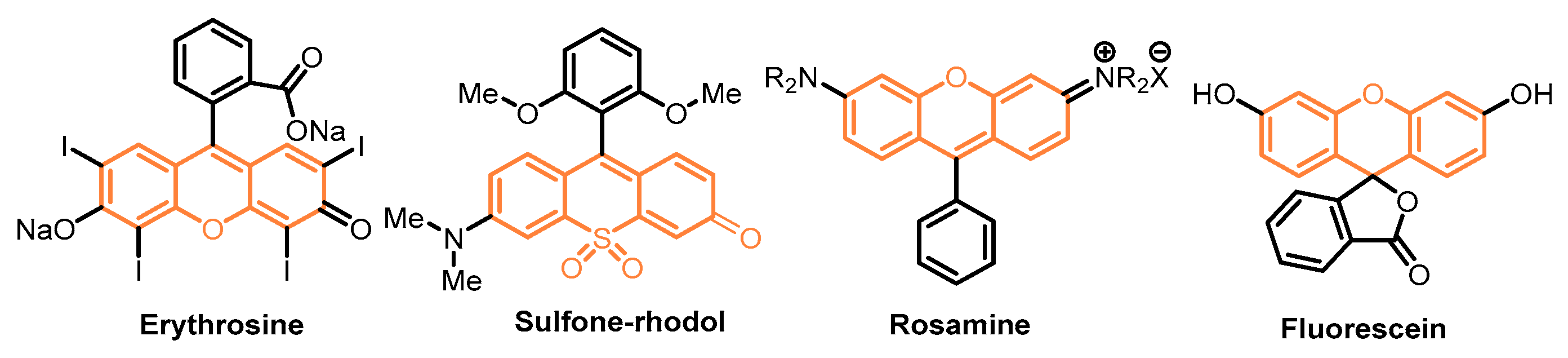
- Maia, M.; Resende, D.I.; Durães, F.; Pinto, M.M.; Sousa, E. Xanthenes in Medicinal Chemistry–Synthetic Strategies and Biological Activities. Eur. J. Med. Chem. 2021, 210, 113085–113114. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Huang, T.; Wu, W.; Liang, F.; Cao, S. LDA-Mediated Synthesis of Triarylmethanes by Arylation of Diarylmethanes with Fluoroarenes at Room Temperature. Org. Lett. 2015, 17, 5096–5099. [Google Scholar] [CrossRef]
- Bhanuchandra, M.; Yorimitsu, H.; Osuka, A. Synthesis of Spirocyclic Diarylfluorenes by One-Pot Twofold SNAr Reactions of Diaryl Sulfones with Diarylmethanes. Org. Lett. 2016, 18, 384–387. [Google Scholar] [CrossRef]
- Murugesan, K.; Senthamarai, T.; Alshammari, A.S.; Altamimi, R.M.; Kreyenschulte, C.; Pohl, M.M.; Lund, H.; Jagadeesh, R.V.; Beller, M. Cobalt-Nanoparticles Catalyzed Efficient and Selective Hydrogenation of Aromatic Hydrocarbons. ACS Catal. 2019, 9, 8581–8591. [Google Scholar] [CrossRef]
- Das, S.; Roy, S.; Bhowmik, A.; Sarkar, W.; Mondal, I.; Mishra, A.; Saha, S.J.; Karmakar, S.; Deb, I. A Radical–radical Cross-coupling Reaction of Xanthene with Sulfonyl Hydrazides: Facile Access to Xanthen-9-sulfone Derivatives. Chem. Commun. 2022, 58, 2902–2905. [Google Scholar] [CrossRef]
- Yang, Y.Z.; Song, R.J.; Li, J.-H. Intermolecular Anodic Oxidative Cross-Dehydrogenative C(sp3)–N Bond-Coupling Reactions of Xanthenes with Azoles. Org. Lett. 2019, 21, 3228–3231. [Google Scholar] [CrossRef]
- Yang, Y.Z.; Wu, Y.C.; Song, R.J.; Li, J.H. Electrochemical Dehydrogenative Cross-coupling of Xanthenes with Ketones. Chem. Commun. 2020, 56, 7585–7588. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.-J.; Zhong, Y.-J.; Feng, Y.-F.; Gao, L.; Tang, H.-T.; Pan, Y.-M.; Ma, X.-L.; Mo, Z.-Y. Electrochemically Mediated Direct C(sp3)−H Sulfonylation of Xanthene Derivatives. Adv. Synth. Catal. 2022, 364, 726–731. [Google Scholar] [CrossRef]
- Tang, S.; Guillot, R.; Grimaud, L.; Vitale, M.R.; Vincent, G. Electrochemical Benzylic C−H Functionalization with Isocyanides. Org. Lett. 2022, 24, 2125–2130. [Google Scholar] [CrossRef]
- Zhang, B.; Xiang, S.K.; Zhang, L.; Cui, Y.; Jiao, N. Organocatalytic Asymmetric Intermolecular Dehydrogenative A-Alkylation of Aldehydes Using Molecular Oxygen as Oxidant. Org. Lett. 2011, 13, 5212–5215. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, W.; Nakano, K. Efficient C(sp3)−H Bond Functionalization of Isochroman by AZADOL Catalysis. Org. Lett. 2015, 17, 1549–1552. [Google Scholar] [CrossRef]
- Zhou, K.; Yu, Y.; Lin, Y.M.; Li, Y.; Gong, L. Copper-catalyzed Aerobic Asymmetric Cross-dehydrogenative Coupling of C(sp3)−H Bonds Driven by Visible Light. Green Chem. 2020, 22, 4597–4603. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, Z.; Zhang, A.H.; Yu, S. Access to Cyanoimines Enabled by Dual Photoredox/Copper-Catalyzed Cyanation of O-Acyl Oximes. Org. Lett. 2020, 22, 7315–7320. [Google Scholar] [CrossRef]
- Sarma, D.; Majumdar, B.; Sarma, T.K. Visible-light Induced Enhancement in the Multi-catalytic Activity of Sulfated Carbon Dots for Aerobic Carbon–carbon Bond Formation. Green Chem. 2019, 21, 6717–6726. [Google Scholar] [CrossRef]
- Gui, J.; Sun, M.; Wu, H.; Li, J.; Yang, J.; Wang, Z. Direct Benzylic C−H Difluoroalkylation with Difluoroenoxysilanes by Transition Metal-free Photoredox Catalysis. Org. Chem. Front. 2022, 9, 4569–4574. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, F.; McCann, S.D.; Wang, D.; Chen, P.; Stahl, S.S.; Liu, G. Enantioselective cyanation of benzylic C−H bonds via copper-catalyzed radical relay. Science 2016, 353, 1014–1018. [Google Scholar] [CrossRef]
- Suh, S.E.; Nkulu, L.E.; Lin, S.; Krska, S.W.; Stahl, S.S. Benzylic C−H Isocyanation/amine Coupling Sequence Enabling High-throughput Synthesis of Pharmaceutically Relevant Ureas. Chem. Sci. 2021, 12, 10380–10387. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ding, R.; Guo, H.Y.; Xia, S.; Shu, L.; Wang, P.L.; Li, H. Catalyst-free Benzylic C(sp3)−H Cross-coupling with Organotrifluoroborates Enabled by Electrochemistry. Green Chem. 2022, 24, 7883–7888. [Google Scholar] [CrossRef]
- Zhong, Q.; Gao, H.; Wang, P.L.; Zhou, C.; Miao, T.; Li, H. Electrochemical Site-Selective Alkylation of Azobenzenes with (Thio)Xanthenes. Molecules 2022, 27, 4967. [Google Scholar] [CrossRef]
- Gao, H.; Chen, X.; Wang, P.L.; Shi, M.M.; Shang, L.L.; Guo, H.Y.; Li, H.; Li, P. Electrochemical Benzylic C−H Arylation of Xanthenes and Thioxanthenes Without a Catalyst and Oxidant. Org. Chem. Front. 2022, 9, 1911–1916. [Google Scholar] [CrossRef]
- Yang, N.; Li, A.; Gao, H.; Liao, L.-M.; Yang, Y.-P.; Wang, P.-L.; Li, H. Electrochemical oxidation-induced benzylic C(sp3)−H functionalization towards the atom-economic synthesis of oxazole heterocycles. Green Chem. 2023, 25, 5128–5133. [Google Scholar] [CrossRef]
- Liu, S.; Klussmann, M. Acid Promoted Radical-chain Difunctionalization of Styrenes with Stabilized Radicals and (N, O)-nucleophiles. Chem. Commun. 2020, 56, 1557–1560. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Tian, L.; Wang, Y. C–N Coupling of Azoles or Imides with Carbocations Generated by Electrochemical Oxidation. Eur. J. Org. Chem. 2019, 2019, 4089–4094. [Google Scholar] [CrossRef]
- Shinohara, R.; Ogawa, N.; Kawashima, H.; Wada, K.; Saito, S.; Yamazaki, T.; Kobayashi, Y. SN2 Reaction of Diarylmethyl Anions at Secondary Alkyl and Cycloalkyl Carbons. Eur. J. Org. Chem. 2019, 2019, 1461–1478. [Google Scholar] [CrossRef]
- Li, K.J.; Jiang, Y.Y.; Xu, K.; Zeng, C.C.; Sun, B.G. Electrochemically Dehydrogenative C–H/P−H Cross-coupling: Effective Synthesis of Phosphonated Quinoxalin-2(1H)-ones and Xanthenes. Green Chem. 2019, 21, 4412–4421. [Google Scholar] [CrossRef]
- Lin, M.-Y.; Xu, K.; Jiang, Y.-Y.; Liu, Y.-G.; Sun, B.-G.; Zeng, C.-C. Intermolecular electrochemical C(sp3)-H/N-H cross-coupling of xanthenes with N-alkoxyamides: Radical pathway mediated by ferrocene as a redox catalyst. Adv. Synth. Catal. 2018, 360, 1665–1672. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Okada, Y.; Yoshimoto, Y.; Nakamura, M. Tuning Chemoselectivity in Iron-Catalyzed Sonogashira-Type Reactions Using a Bisphosphine Ligand with Peripheral Steric Bulk: Selective Alkynylation of Nonactivated Alkyl Halides. Angew. Chem. Int. Ed. 2011, 50, 10973–10976. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.-Y.; Li, Z.-L.; Ye, L.; Tan, B.; Liu, X.-Y. Organic base-catalysed solvent-tuned chemoselective carbotrifluoromethylation and oxytrifluoromethylation of unactivated alkenes. Chem. Commun. 2016, 52, 9052–9055. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, Y.; Hayashi, K.; Carlson, E.; Shaji, S.; Waldmann, D.; Simmons, B.J.; Edwards, J.T.; Zapf, C.W.; Saito, M.; Baran, P.S. Chemoselective Electrosynthesis Using Rapid Alternating Polarity. J. Am. Chem. Soc. 2021, 143, 16580–16588. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zeng, L.; Hu, J.; Ma, R.; Liu, X.; Jiao, Y.; He, H.; Chen, S.; Xu, Z.; Wang, H.; et al. Electrochemical Difunctionalization of Terminal Alkynes: Access to 1,4-Dicarbonyl Compounds. Org. Lett. 2022, 24, 289–292. [Google Scholar] [CrossRef]
- Zhang, L.; Fu, Y.; Shen, Y.; Liu, C.; Sun, M.; Cheng, R.; Zhu, W.; Qian, X.; Ma, Y.; Ye, J. Ritter-type amination of C(sp3)-H bonds enabled by electrochemistry with SO42−. Nat. Commun. 2022, 13, 4138. [Google Scholar] [CrossRef]
- Pintér, Á.; Sud, A.; Sureshkumar, D.; Klussmann, M. Autoxidative Carbon-Carbon Bond Formation from Carbon-Hydrogen Bonds. Angew. Chem. Int. Ed. 2010, 49, 5004–5007. [Google Scholar] [CrossRef]
- Pintér, Á.; Klussmann, M. Sulfonic Acid-Catalyzed Autoxidative Carbon-Carbon Coupling Reaction Under Elevated Partial Pressure of Oxygen. Adv. Synth. Catal. 2012, 354, 701–711. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Electrode | Temp (°C) | Yield (%) | |
| 3a | 4a | |||
| 1 | C(+)|Pt(−) | rt | 32 | 40 |
| 2 | C(+)|Pt(−) | 30 | 38 | 37 |
| 3 | C(+)|Pt(−) | 40 | 45 | 21 |
| 4 | C(+)|Pt(−) | 50 | 71 | 6 |
| 5 | C(+)|Pt(−) | 60 | 65 | trace |
| 6 | C(+)|Pt(−) | 70 | 52 | trace |
| 7 | C(+)|Ni(−) | 50 | 14 | 25 |
| 8 | Pt(+)|C(−) | 50 | 46 | 17 |
| 9 | GF(+)|GF(−) | 50 | n.d. | 43 |
| 10 c | C(+)|Pt(−) | 50 | 55 | 8 |
| 11 d | C(+)|Pt(−) | 50 | 37 | 10 |
| 12 e | C(+)|Pt(−) | 50 | 48 | trace |
| 13 f | C(+)|Pt(−) | 50 | 22 | trace |
| 14 | GF(+)|GF(−) | rt | n.d. | 35 |
| 15 f | GF(+)|GF(−) | rt | n.d. | 50 |
| 16 f,g | GF(+)|GF(−) | rt | n.d. | 77 |
| 17 c,f,g | GF(+)|GF(−) | rt | n.d. | 61 |
| 18 d,f,g | GF(+)|GF(−) | rt | n.d. | 33 |
| 19 e,f,g | GF(+)|GF(−) | rt | n.d. | 58 |
| 20 h | GF(+)|GF(−) | rt | n.d. | n.d. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Yang, N.; Li, C.; He, J.; Li, H. Tuning Benzylic C−H Functionalization of (Thio)xanthenes with Electrochemistry. Molecules 2023, 28, 6139. https://doi.org/10.3390/molecules28166139
Wang C, Yang N, Li C, He J, Li H. Tuning Benzylic C−H Functionalization of (Thio)xanthenes with Electrochemistry. Molecules. 2023; 28(16):6139. https://doi.org/10.3390/molecules28166139
Chicago/Turabian StyleWang, Changji, Na Yang, Chao Li, Jian He, and Hongji Li. 2023. "Tuning Benzylic C−H Functionalization of (Thio)xanthenes with Electrochemistry" Molecules 28, no. 16: 6139. https://doi.org/10.3390/molecules28166139
APA StyleWang, C., Yang, N., Li, C., He, J., & Li, H. (2023). Tuning Benzylic C−H Functionalization of (Thio)xanthenes with Electrochemistry. Molecules, 28(16), 6139. https://doi.org/10.3390/molecules28166139