
Pentafuhalol-B, a Phlorotannin from Brown Algae, Strongly Inhibits the PLK-1 Overexpression in Cancer Cells as Revealed by Computational Analysis

 ,
, 

 ,
, 
Abstract
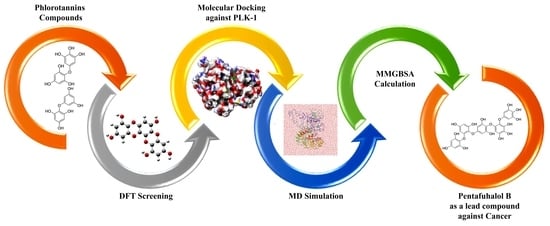
1. Introduction
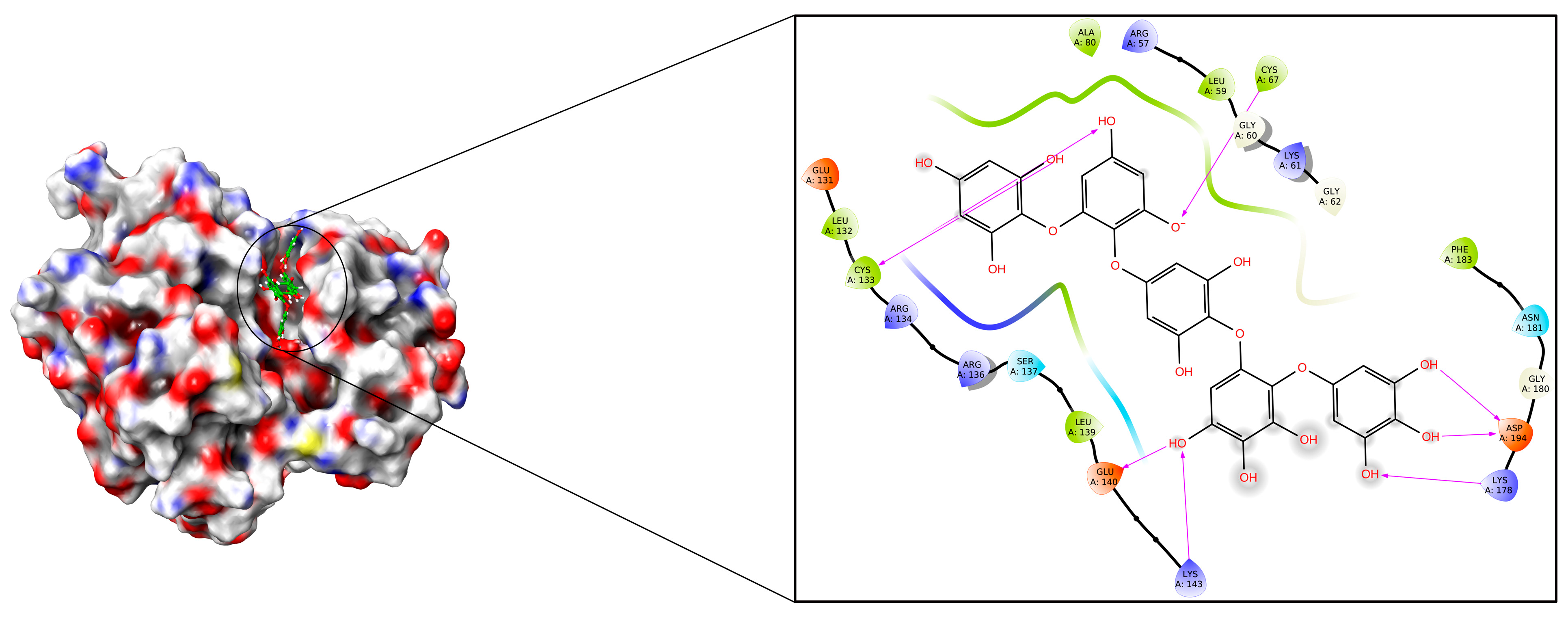
2. Results and Discussions
2.1. Structure Optimisation by DFT Study
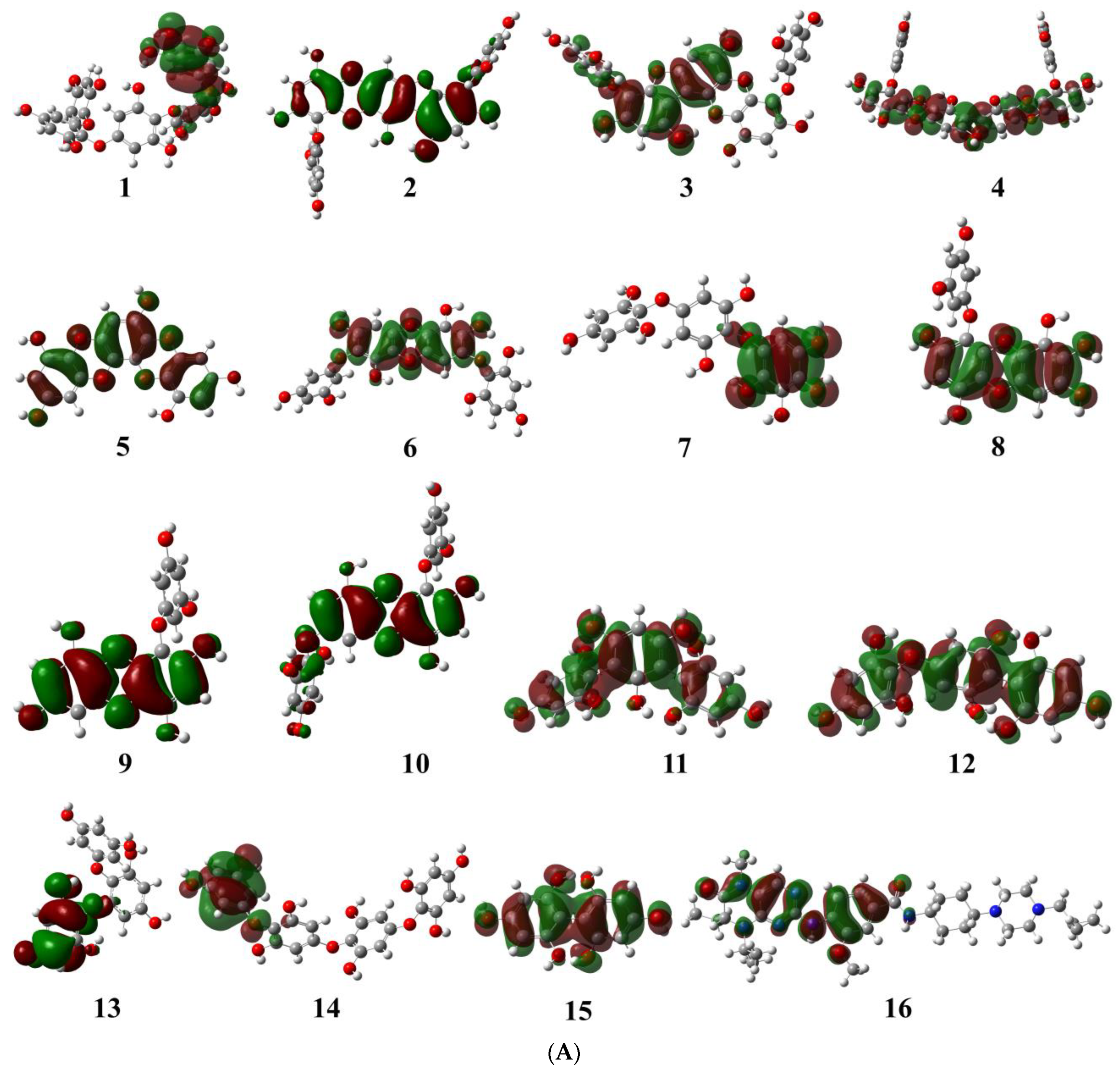
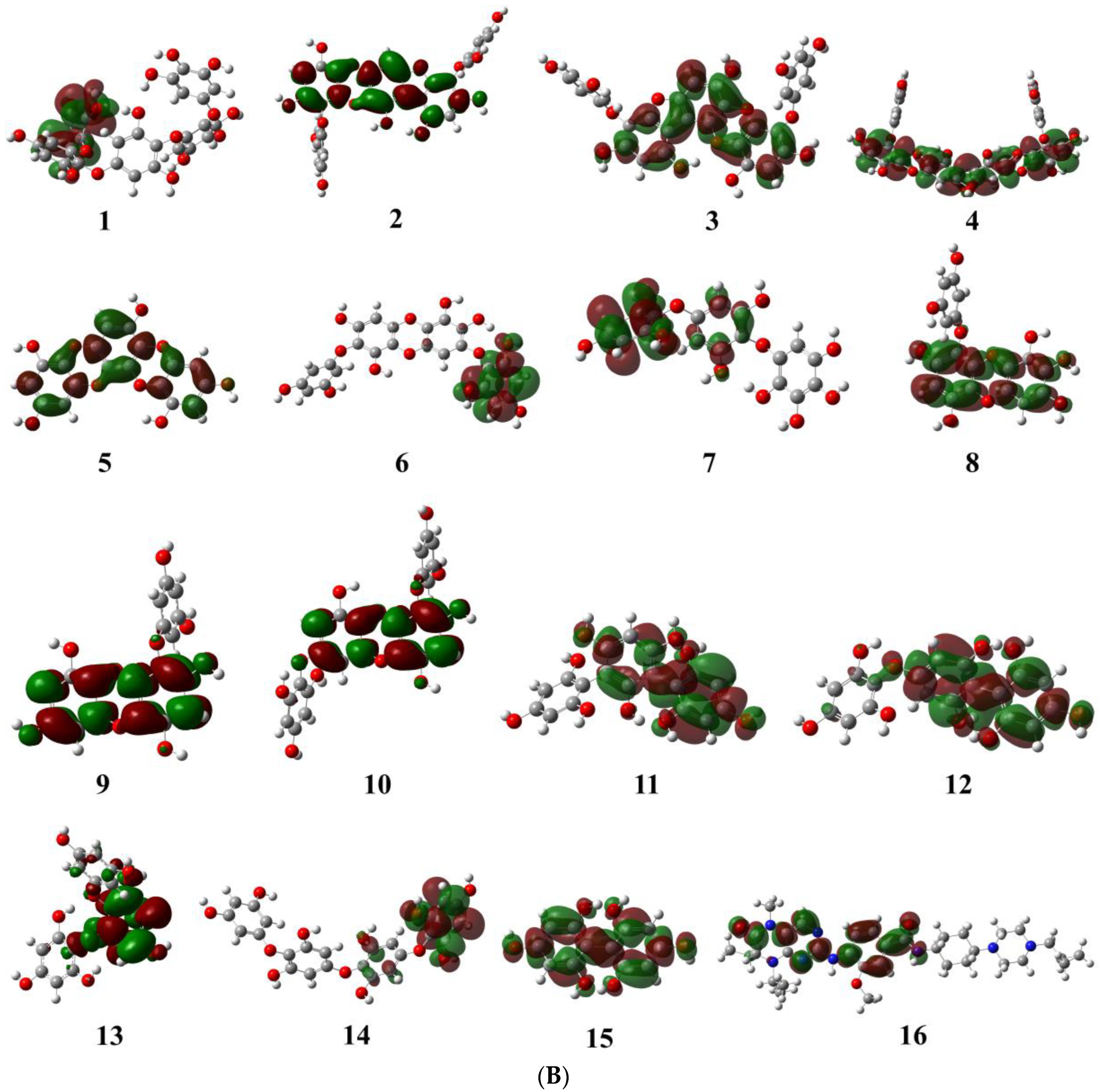
2.1.1. Evaluation and Analysis of Frontier Molecular Orbitals



2.1.2. Evaluation and Analysis of Chemical Descriptor
2.1.3. Evaluation and Analysis of Molecular Electrostatic Potential

2.2. Evaluation and Analysis of Molecular Docking
2.3. Evaluation and Analysis of Molecular Dynamic (MD) Simulation
2.3.1. RMSD and RMSF Studies
2.3.2. Ligand Properties Analysis
Radius of Gyration (Rg) Study
Solvent-Accessible Surface Area (SASA) Study
Molecular Surface Area (MolSA) and Polar Surface Area (PSA) Studies
Ligand–Protein Interactions Studies
2.4. Evaluation and Analysis of MMGBSA Study
3. Materials and Methods
3.1. Density Functional Theory (DFT) Calculations
3.2. Ligand Preparation
3.3. Protein Preparation and Grid Generation
3.4. Molecular Docking and Pose Visualisation
3.5. Molecular Dynamic Simulations
3.6. MMGBSA Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gümüş Yılmaz, G.; Gómez Pinchetti, J.L.; Cifuentes, A.; Herrero, M.; Ibáñez, E. Comparison of Extraction Techniques and Surfactants for the Isolation of Total Polyphenols and Phlorotannins from the Brown Algae Lobophora variegate. Anal. Lett. 2019, 52, 2724–2740. [Google Scholar] [CrossRef]
- Shrestha, S.; Zhang, W.; Smid, S.D. Phlorotannins: A review on biosynthesis, chemistry, and bioactivity. Food. Biosci. 2021, 39, 100832. [Google Scholar] [CrossRef]
- Venkatesan, J.; Keekan, K.K.; Anil, S.; Bhatnagar, I.; Kim, S.K. Phlorotannins. Encycl. Food Chem. 2018, 3, 515–527. [Google Scholar]
- Jung, H.A.; Oh, S.H.; Choi, J.S. Molecular docking studies of phlorotannins from Eisenia bicyclis with BACE1 inhibitory activity. Bioorg. Med. Chem. Lett. 2010, 20, 3211–3215. [Google Scholar] [CrossRef]
- Kang, S.M.; Heo, S.J.; Kim, K.N.; Lee, S.H.; Jeon, Y.J. Isolation and identification of new compound, 2,7′-phloroglucinol-6,6′-bieckol from brown algae, Ecklonia cava and its antioxidant effect. J. Funct. Foods. 2012, 4, 158–166. [Google Scholar] [CrossRef]
- Hakim, M.M.; Patel, I.C. A review on phytoconstituents of marine brown algae. Future J. Pharm. Sci. 2020, 6, 129. [Google Scholar] [CrossRef]
- Heo, S.J.; Hwang, J.Y.; Choi, J.I.; Han, J.S.; Kim, H.J.; Jeon, Y.J. Diphlorethohydroxycarmalol isolated from Ishige okamurae, a brown algae, a potent α-glucosidase and α-amylase inhibitor, alleviates postprandial hyperglycemia in diabetic mice. Eur. J. Pharmacol. 2009, 615, 252–256. [Google Scholar] [CrossRef]
- Yegdaneh, A.; Ghannadi, A.; Dayani, L. Chemical constituents and biological activities of two Iranian Cystoseira species. Res. Pharm. Sci. 2016, 11, 311–317. [Google Scholar] [CrossRef]
- Gunaseelan, S.; Arunkumar, M.; Aravind, M.K.; Gayathri, S.; Rajkeerthana, S.; MohanKumar, V.; Kumar, B.; Varalakshmi, P. Probing marine brown macroalgal phlorotannins as antiviral candidate against SARS-CoV-2: Molecular docking and dynamics simulation approach. Mol. Divers. 2022, 26, 3205–3224. [Google Scholar] [CrossRef]
- He, Z.; Chen, Y.; Chen, Y.; Liu, H.; Yuan, G.; Fan, Y.; Chen, K. Optimization of the microwave-assisted extraction of phlorotannins from Saccharina japonica Aresch and evaluation of the inhibitory effects of phlorotannin-containing extracts on HepG2 cancer cells. Chin. J. Oceanol. Limnol. 2012, 31, 1045–1054. [Google Scholar] [CrossRef]
- Bakunina, I.; Imbs, T.; Likhatskaya, G.; Grigorchuk, V.; Zueva, A.; Malyarenko, O.; Ermakova, S. Effect of Phlorotannins from Brown Algae Costaria costata on α-N-Acetylgalactosaminidase Produced by Duodenal Adenocarcinoma and Melanoma Cells. Mar. Drugs 2023, 21, 33. [Google Scholar] [CrossRef]
- Catarino, M.D.; Fernandes, I.; Oliveira, H.; Carrascal, M.; Ferreira, R.; Silva, A.M.S.; Cruz, M.T.; Mateus, N.; Cardoso, S.M. Antitumor Activity of Fucus vesiculosus-Derived Phlorotannins through Activation of Apoptotic Signals in Gastric and Colorectal Tumor Cell Lines. Int. J. Mol. Sci. 2021, 22, 7604. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A potential target for cancer therapy. Translat. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef]
- Gheghiani, L.; Wang, L.; Zhang, Y.; Moore, X.; Zhang, J.; Smith, S.; Tian, Y.; Wang, L.; Turner, K.; Jackson-Cook, C.; et al. PLK1 induces chromosomal instability and overrides cell-cycle checkpoints to drive tumorigenesis. Cancer Res. 2021, 81, 1293–1307. [Google Scholar] [CrossRef]
- De Cárcer, G.; Venkateswaran, S.V.; Salgueiro, L.; El Bakkali, A.; Somogyi, K.; Rowald, K.; Montañés, P.; Sanclemente, M.; Escobar, B.; de Martino, A.; et al. Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat. Commun. 2018, 9, 3012. [Google Scholar] [CrossRef]
- Kalous, J.; Aleshkina, D. Multiple Roles of PLK1 in Mitosis and Meiosis. Cells 2023, 12, 187. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Ou-Yang, S.; Lu, J.; Kong, X.; Liang, Z.; Luo, C.; Jiang, H. Computational drug discovery. Acta Pharmacol. Sin. 2012, 33, 1131–1140. [Google Scholar] [CrossRef]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Strebhardt, K. Multifaceted polo-like kinases: Drug targets and anti-targets for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 643–660. [Google Scholar] [CrossRef]
- Tariq, K.; Shaheen, I.; Shaheena, R.; Khalil, A. Role of DFT in Drug Design: A mini Review. Drug Des. 2022, 11, 216. [Google Scholar]
- Üstün, E.; Şahin, N. Molecular Docking and DFT Analysis of Methallyl Substituted N-Heterocyclic Carbene Salts for Potential Anticancer Activity. Ordu Üniversitesi Bilim Teknol. Dergisi 2021, 11, 186–192. [Google Scholar] [CrossRef]
- Dhifaoui, S.; Azaid, A.; Bourass, M.; Hassen, L.B.H.; Nasri, H.; Bouachrine, M. DFT Studies on Molecular Structure, Absorption properties, NBO Analysis, and Hirshfeld Surface Analysis of Iron(III) Porphyrin Complex. Phys. Chem. Res. 2021, 9, 701–713. [Google Scholar]
- Abuelizz, H.A.; Taie, H.A.A.; Bakheit, A.H.; Mostafa, G.A.E.; Marzouk, M.; Rashid, H.; Al-Salahi, R. Investigation of 4-Hydrazinobenzoic Acid Derivatives for Their Antioxidant Activity: In Vitro Screening and DFT Study. ACS Omega 2021, 6, 31993–32004. [Google Scholar] [CrossRef] [PubMed]
- Saji, R.S.; Prasana, J.C.; Muthu, S.; George, J. Experimental and theoretical spectroscopic (FT-IR, FT-Raman, UV-VIS) analysis, natural bonding orbitals and molecular docking studies on 2-bromo-6-methoxynaphthalene: A potential anti-cancer drug. Heliyon 2021, 7, e07213. [Google Scholar] [CrossRef] [PubMed]
- Aziz, H.A.; Santoso, G.A.; Mulya, F.; Pranowo, H.D. Molecular and electronic structure of some symmetrically meso-substituted Hg(II)-porphyrin complexes. Asian J. Chem. 2017, 29, 2224–2226. [Google Scholar] [CrossRef]
- Luo, J.; Xue, Z.Q.; Liu, W.M.; Wu, J.L.; Yang, Z.Q. Koopmans’ theorem for large molecular systems within density functional theory. J. Phys. Chem. A 2006, 110, 12005–12009. [Google Scholar] [CrossRef]
- Amati, M.; Stoia, S.; Baerends, E.J. The Electron Affinity as the Highest Occupied Anion Orbital Energy with a Sufficiently Accurate Approximation of the Exact Kohn-Sham Potential. J. Chem. Theory Comput. 2020, 16, 443–452. [Google Scholar] [CrossRef]
- Bakheit, A.H.; Attwa, M.W.; Kadi, A.A.; Ghabbour, H.A.; Alkahtani, H.M. Exploring the Chemical Reactivity, Molecular Docking, Molecular Dynamic Simulation and ADMET Properties of a Tetrahydrothienopyridine Derivative Using Computational Methods. Crystals 2023, 13, 1020. [Google Scholar] [CrossRef]
- Vidhya, V.; Austine, A.; Arivazhagan, M. Molecular structure, aromaticity, vibrational investigation and dual descriptor for chemical reactivity on 1- chloroisoquinoline using quantum chemical studies. Results Mater. 2020, 6, 100097. [Google Scholar] [CrossRef]
- Thanikaivelan, P.; Subramanian, V.; Rao, J.R.; Nair, B.U. Application of quantum chemical descriptor in quantitative structure activity and structure property relationship. Chem. Phys. Lett. 2000, 323, 59–70. [Google Scholar] [CrossRef]
- Alias, A.; Zabidi, Z.; Zakaria, N.; Mahmud, Z.; Ali, R. Biological Activity Relationship of Cyclic and Noncyclic Alkanes Using Quantum Molecular Descriptors. Open J. Appl. Sci. 2021, 11, 966–984. [Google Scholar] [CrossRef]
- Balabin, R. Enthalpy difference between conformations of normal alkanes: Intramolecular basis set superposition error (BSSE) in the case of n-butane and n-hexane. J. Chem. Phys. 2008, 129, 164101. [Google Scholar] [CrossRef]
- Haynes, P.; Skylaris, C.; Mostofi, A.; Payne, M. Elimination of basis set superposition error in linear-scaling density-functional calculations with local orbitals optimised in situ. Chem. Phys. Lett. 2006, 422, 345–349. [Google Scholar] [CrossRef][Green Version]
- Uzzaman, M. Thermodynamic, HOMO-LUMO, MEP, and ADMET Studies of Metronidazole and its Modified Derivatives Based on DFT. J. Biomed. Eng. Biosci. 2019, 3, 262–264. [Google Scholar]
- Grillo, I.; Urquiza-Carvalho, G.; Rocha, G. Quantum chemical descriptors based on semiempirical methods for large biomolecules. J. Chem. Phys. 2023, 158, 201001. [Google Scholar] [CrossRef]
- Jeevitha, D.; Sadasivam, K.; Praveena, R.; Jayaprakasam, R. DFT study of glycosyl group reactivity in quercetin derivatives. J. Mol. Struct. 2016, 1120, 15–24. [Google Scholar] [CrossRef]
- Adekoya, O.C.; Adekoya, G.J.; Sadiku, E.R.; Hamam, Y.; Ray, S.S. Application of DFT Calculations in Designing Polymer-Based Drug Delivery Systems: An Overview. Pharmaceutics 2022, 14, 1972. [Google Scholar] [CrossRef]
- Beria, I.; Bossi, R.T.; Brasca, M.G.; Caruso, M.; Ceccarelli, W.; Fachin, G.; Fasolini, M.; Forte, B.; Fiorentini, F.; Pesenti, E.; et al. NMS-P937, a 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivative as potent and selective Polo-like kinase 1 inhibitor. Bioorg. Med. Chem. Lett. 2011, 21, 2969–2974. [Google Scholar] [CrossRef]
- Schöffski, P. Polo-Like Kinase (PLK) Inhibitors in Preclinical and Early Clinical Development in Oncology. Oncologist 2019, 14, 559–570. [Google Scholar] [CrossRef]
- Ellis, P.; Leighl, N.; Hirsh, V.; Reaume, M.; Blais, N.; Wierzbicki, R.; Sadrolhefazi, B.; Gu, Y.; Liu, D.; Pilz, K.; et al. A Randomized, Open-Label Phase II Trial of Volasertib as Monotherapy and in Combination with Standard-Dose Pemetrexed Compared with Pemetrexed Monotherapy in Second-Line Treatment for Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2015, 16, 457–465. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Salsbury, F.R. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr. Opin. Pharmacol. 2010, 10, 738–744. [Google Scholar] [CrossRef]
- Halder, D.; Das, S.; Aiswarya, R.; Jeyaprakash, R.S. Molecular docking and dynamics based approach for the identification of kinase inhibitors targeting PI3Kα against non-small cell lung cancer: A computational study. RSC Adv. 2022, 12, 21452–21467. [Google Scholar] [CrossRef]
- Ghosh, A.; Mukerjee, N.; Sharma, B.; Pant, A.; Kishore Mohanta, Y.; Jawarkar, R.D.; Bakal, R.L.; Terefe, E.M.; Batiha, G.E.I.; Mostafa-Hedeab, G.; et al. Target Specific Inhibition of Protein Tyrosine Kinase in Conjunction with Cancer and SARS-CoV-2 by Olive Nutraceuticals. Front. Pharmacol. 2022, 12, 812565. [Google Scholar] [CrossRef]
- Nipun, T.S.; Ema, T.I.; Mia, M.A.R.; Hossen, M.S.; Arshe, F.A.; Ahmed, S.Z.; Masud, A.; Taheya, F.F.; Khan, A.A.; Haque, F.; et al. Active site-specific quantum tunneling of hACE2 receptor to assess its complexing poses with selective bioactive compounds in co-suppressing SARS-CoV-2 influx and subsequent cardiac injury. J. Adv. Vet. Anim. Res. 2021, 8, 540–556. [Google Scholar] [CrossRef]
- Bhrdwaj, A.; Abdalla, M.; Pande, A.; Madhavi, M.; Chopra, I.; Soni, L.; Vijayakumar, N.; Panwar, U.; Aqueel, M.; Prajapati, L.; et al. Structure-Based Virtual Screening, Molecular Docking, Molecular Dynamics Simulation of EGFR for the clinical treatment of Glioblastoma. App. Biochem. Biotechnol. 2023, 195, 5094–5119. [Google Scholar] [CrossRef]
- Brylinski, M. Aromatic interactions at the ligand-protein interface: Implications for the development of docking scoring functions. Chem. Biol. Drug Des. 2017, 91, 380–390. [Google Scholar] [CrossRef]
- Sheikh, S.Y.; Ansari, W.A.; Hassan, F.; Faruqui, T.; Khan, M.F.; Akhter, Y.; Khan, A.R.; Siddiqui, M.A.; Al-Khedhairy, A.A.; Nasibullah, M. Drug repositioning to discover novel Ornithine Decarboxylase inhibitors against Visceral Leishmaniasis. J. Mol. Recog. 2023, 36, e3021. [Google Scholar] [CrossRef]
- Gopinath, P.; Kathiravan, M.K. Docking studies and molecular dynamics simulation of triazole benzene sulfonamide derivatives with human carbonic anhydrase IX inhibition activity. RSC Adv. 2021, 11, 38079–38093. [Google Scholar]
- Saini, M.; Sangwan, R.; Khan, M.F.; Kumar, A.; Verma, R.; Jain, S. Specioside (SS) & verminoside (VS) (Iridoid glycosides): Isolation, characterization and comparable quantum chemical studies using density functional theory (DFT). Heliyon 2019, 5, e01118. [Google Scholar]
- Rasool, N.; Yasmin, F.; Sahai, S.; Hussain, W.; Inam, H.; Arshad, A. Biological perspective of thiazolide derivatives against Mpro and MTase of SARS-CoV-2: Molecular docking, DFT and MD simulation investigations. Chem. Phys. Lett. 2021, 771, 138463. [Google Scholar] [CrossRef]
- Hashem, H.E. In Silico Approach of Some Selected Honey Constituents as SARS-CoV-2 Main Protease (COVID-19) Inhibitors Eurasian. J. Med. Oncol. 2020, 4, 196–200. [Google Scholar]
- Kumar, S.; Kim, J. PLK-1 Targeted Inhibitors and their Potential against Tumorigenesis. BioMed Res. Int. 2015, 2015, 1–21. [Google Scholar] [CrossRef]
- Garuti, L.; Roberti, M.; Bottegoni, G. Polo-Like Kinases Inhibitors. Curr. Med. Chem. 2012, 19, 3937–3948. [Google Scholar] [CrossRef]
- Puranik, N.V.; Srivastava, P.; Swami, S.; Choudhari, A.; Sarkar, D. Molecular modeling studies and: In vitro screening of dihydrorugosaflavonoid and its derivatives against Mycobacterium tuberculosis. RSC Adv. 2018, 8, 10634–10643. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Thai, N.Q.; Truong, D.T.; Li, M.S. Remdesivir strongly binds to both RNA-dependent RNA polymerase and main protease of SARS-CoV-2: Evidence from molecular simulations. J. Phys. Chem. B 2020, 124, 11337–11348. [Google Scholar] [CrossRef]
- Ansari, W.A.; Ahamad, T.; Khan, M.A.; Khan, Z.A.; Khan, M.F. Exploration of Luteolin as Potential Anti-COVID-19 Agent: Molecular Docking, Molecular Dynamic Simulation, ADMET and DFT Analysis. Lett. Drug Des. Discov. 2021, 19, 741–756. [Google Scholar] [CrossRef]
- Shah, B.; Modi, P.; Sagar, S.R. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020, 252, 117652. [Google Scholar] [CrossRef]
- Ansari, W.A.; Khan, M.A.; Rizvi, F.; Ali, K.; Hussain, M.K.; Saquib, M.; Khan, M.F. Computational Screening of Plant-Derived Natural Products against SARS-CoV-2 Variants. Future Pharmacol. 2022, 2, 558–578. [Google Scholar] [CrossRef]
- De Campos, L.J.; Palermo, N.Y.; Conda-Sheridan, M. Targeting SARS-CoV-2 Receptor Binding Domain with Stapled Peptides: An In Silico Study. J. Phys. Chem. B 2021, 125, 6572–6586. [Google Scholar] [CrossRef]
- Sheikh, S.Y.; Hassan, F.; Khan, M.F.; Ahmad, T.; Ansari, W.A.; Akhter, Y.; Khafagy, E.; Khan, A.R.; Nasibullah, M. Drug Repurposing to Discover Novel Anti-Inflammatory Agents Inhibiting JAK3/STAT Signaling. Russ. J. Bioorg. Chem. 2022, 48, 958–975. [Google Scholar] [CrossRef]
- Khan, M.F.; Ansari, W.A.; Ahamad, T.; Khan, M.A.; Khan, Z.A.; Sarfraz, A.; Khan, M.A. Bioactive components of different nasal spray solutions may defeat SARS-Cov2: Repurposing and in silico studies. J. Mol. Model. 2022, 28, 212. [Google Scholar] [CrossRef]
- Ansari, W.A.; Rizvi, F.; Khan, M.A.; Khan, Z.A.; Khan, M.F. Computational Study Reveals the Inhibitory Effects of Chemical Constituents from Azadirachta indica (Indian Neem) Against Delta and Omicron Variants of SARS-CoV-2. Coronaviruses 2022, 3, 62–72. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | EHOMO (eV) | ELUMO (eV) | Egap (eV) | IE (eV) | EA (eV) | η (eV) | σ (eV) | MEP (au) | BSSE (Kcal) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | −4.513 | −0.810 | 3.703 | 4.513 | 0.810 | 1.851 | 0.540 | −0.104 × 10−2 to +0.104 × 10−2 | 5.404 |
| 2 | −5.315 | −0.910 | 4.405 | 5.315 | 0.910 | 2.202 | 0.454 | −8.301 × 10−2 to +8.301 × 10−2 | 3.396 |
| 3 | −5.501 | −1.012 | 4.489 | 5.501 | 1.012 | 2.244 | 0.445 | −7.982 × 10−2 to +7.982 × 10−2 | 4.225 |
| 4 | −5.081 | −0.452 | 4.629 | 5.081 | 0.452 | 2.314 | 0.432 | −7.656 × 10−2 to +7.656 × 10−2 | 2.684 |
| 5 | −5.316 | −0.647 | 4.669 | 5.316 | 0.647 | 2.334 | 0.428 | −8.071 × 10−2 to +8.071 × 10−2 | 2.630 |
| 6 | −5.292 | −0.530 | 4.762 | 5.292 | 0.530 | 2.381 | 0.419 | −9.166 × 10−2 to +9.166 × 10−2 | 4.878 |
| 7 | −5.094 | −0.308 | 4.786 | 5.094 | 0.308 | 2.393 | 0.417 | −9.690 × 10−2 to +9.690 × 10−2 | 4.144 |
| 8 | −5.279 | −0.469 | 4.810 | 5.279 | 0.469 | 2.405 | 0.415 | −8.195 × 10−2 to +8.195 × 10−2 | 4.482 |
| 9 | −5.272 | −0.423 | 4.853 | 5.272 | 0.423 | 2.425 | 0.412 | −7.643 × 10−2 to +7.643 × 10−2 | 3.279 |
| 10 | −5.496 | −0.603 | 4.876 | 5.496 | 0.603 | 2.432 | 0.411 | −8.170 × 10−2 to +8.170 × 10−2 | 1.068 |
| 11 | −4.987 | 0.097 | 5.084 | 4.987 | −0.097 | 2.542 | 0.393 | −7.903 × 10−2 to +7.903 × 10−2 | 4.021 |
| 12 | −5.699 | −0.602 | 5.097 | 5.699 | 0.602 | 2.548 | 0.392 | −8.353 × 10−2 to +8.353 × 10−2 | 3.745 |
| 13 | −5.681 | −0.520 | 5.166 | 5.681 | 0.520 | 2.583 | 0.387 | −8.430 × 10−2 to +8.430 × 10−2 | 4.298 |
| 14 | −5.514 | −0.276 | 5.238 | 5.514 | 0.276 | 2.619 | 0.381 | −8.594 × 10−2 to +8.594 × 10−2 | 1.516 |
| 15 | −5.570 | −0.307 | 5.263 | 5.570 | 0.307 | 2.631 | 0.380 | −7.782 × 10−2 to +7.782 × 10−2 | 2.557 |
| 16 | −5.192 | −0.970 | 4.222 | 5.192 | 0.970 | 2.111 | 0.473 | −7.462 × 10−2 to +7.462 × 10−2 | 7.918 |
| Compounds | * Binding Energy | H-Bonds Interaction | Hydrophobic Interaction | Other Interaction |
|---|---|---|---|---|
| 1 | −7.665 | Cys67, Cys133, Glu140, Lys143, Lys178, Asp194 | Arg57, Leu59, Gly60, Lys61, Gly62, Ala80, Glu131, Leu132, Arg134, Arg136, Ser137, Leu139, Gly180, Asn181, Phe183 | - |
| 2 | −7.635 | Arg57, Arg134, Cys133, Glu140, Lys178, Asn181 | Leu59, Gly60, Lys61, Gly62, Cys67, Ala80, Leu132, Arg135, Arg136, Leu139, Lys143, Gly180, Phe183, Asp194 | - |
| 3 | −6.846 | Cys133, Glu140, Lys178, Asp194 | Leu59, Gly60, Gly62, Cys67, Ala80, Lys82, Val114, Leu130, Glu131, Leu132, Arg136, Leu139, Lys143, Gly180, Asn181, | Π-Π-Phe183 |
| 4 | −6.729 | Cys67, Cys133, Glu140, Asp176, | Leu59, Gly60, Lys61, Gly62, Gly63, Ala80, Lys82,Hie105, Val114, Leu130, Glu131, Leu132, Arg136, Ser137, leu139, Asn181, Phe183, Gly193, Gly196, Leu197 | Salt-Lys178 |
| 5 | −6.511 | Leu59, Cys133, Glu131, Asp194 | Arg57, Gly60, Gly62, Cys67, Ala80, Val114, Leu130, Leu132, Arg134, Arg136, Phe183 | Salt-Lys82 |
| 6 | −5.897 | Cys67, Arg136, Lys143, Asp194 | Leu59, Gly60, Gly62, Ala80, Lys82, Hie105, Val114, Leu130, Glu131, Leu132, Cys133, Leu139, Glu140, Asn181, Gly193 | Π-Π-Phe183 |
| 7 | −5.767 | Cys67, Cys133, Asp194 | Arg57, Leu59, Gly60, Lys61, Gly62,Leu132, Arg134, Arg136, Glu140Lys178, Asn181, Phe183 | - |
| 8 | −5.708 | Leu59, Cys133, Asp194 | Gly60, Gly62, Cys67, Ala80, Hie105, Val114, Leu130, Glu131, Leu132, Arg134, Arg136, Lys178, Gly180, Asn181,Gly193 | Salt-Lys82, Π-Π-Phe183 |
| 9 | −5.656 | Cys67, Cys133, Glu131, Asp194 | Leu59, Gly60, Lys61, Gly62, Ala80, Lys82, Hie105, Val114, Leu130, Leu132, Arg136, Glu140, Phe183, Gly193, Phe195 | - |
| 10 | −5.489 | Leu59, Lys61, Cys67, Cys133, Asp194 | Gly60, Gly62, Ala80, Leu130, Leu132, Arg134, Arg136, Glu140, Lys178, Asn181, Phe183 | Salt-Arg57 |
| 11 | −5.348 | Leu59, Lys133, Asp194 | Arg57, Gly60, Lys61, Gly62, Ala65, Cys67, Lys82, Leu132, Arg134, Arg136, Ser137, Leu139, Glu140, Gly180, Asn181, Phe183 | - |
| 12 | −5.142 | Cys67, Cys133, Asp194, Lys178, Gly180 | Leu59, Gly60, Lys61, Gly62, Ala80, Glu131, Leu132, Arg136, Ser137, Asn181 | Π-Π-Phe183 |
| 13 | −4.976 | Cys133, Glu140, Asp194 | Leu59, Gly60, Lys61, Gly62, Ala80, Lys82, Leu130, Glu131, Leu132, Arg136, Ser137, Leu139, Lys178, Gly180, Asn181 | Π-Π-Phe183 |
| 14 | −4.953 | Leu59, Cys133, Arg134, Asp194 | Arg57, Gly60, Lys61, Gly62, Cys67, Lys82, Leu132, Arg135, Arg136, Ser137, glu140, Lys178, Asn181 | Π-Π-Phe183 |
| 15 | −3.087 | Glu140, Asp194 | Gly60, Lys61, Gly62, Ala65, Cys67, Lys82, Ser137, Leu139, Lys178, Gly180, Asn181, Phe183 | - |
| 16 | −5.172 | Cys67, Cys133, Arg136 | Arg57, Phe58, Leu59, Gly60, Lys61, Gly62, Gly63, Ala80, Glu131, Leu132, Arg134, Arg135, Glu140, Lys178, Gly180, Asn181, Phe183, Gly196, Leu197, Thr214 | Π-cation Arg136, Salt-Asp176, Asp194 |
| Parameters | * PtB-2YAC Complex |
|---|---|
| RMSD of Protein (Å) | 1.167–2.956 |
| RMSD of PtB (Å) | 1.394–4.630 |
| RMSF of Protein (Å) | 0.454–6.985 |
| rGyr (Å) | 4.588–5.316 |
| MolSA (Å2) | 457.037–503.076 |
| SASA (Å2) | 105.693–293.441 |
| PSA (Å2) | 508.501–597.879 |
| Complex | ΔGbind | ΔGcoul | ΔGH-bond | ΔGlipo | ΔGGB | ΔGvdW |
|---|---|---|---|---|---|---|
| PtB-2YAC | −67.915 | −191.533 | −5.742 | −11.187 | −7.178 | −62.129 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ansari, W.A.; Rab, S.O.; Saquib, M.; Sarfraz, A.; Hussain, M.K.; Akhtar, M.S.; Ahmad, I.; Khan, M.F. Pentafuhalol-B, a Phlorotannin from Brown Algae, Strongly Inhibits the PLK-1 Overexpression in Cancer Cells as Revealed by Computational Analysis. Molecules 2023, 28, 5853. https://doi.org/10.3390/molecules28155853
Ansari WA, Rab SO, Saquib M, Sarfraz A, Hussain MK, Akhtar MS, Ahmad I, Khan MF. Pentafuhalol-B, a Phlorotannin from Brown Algae, Strongly Inhibits the PLK-1 Overexpression in Cancer Cells as Revealed by Computational Analysis. Molecules. 2023; 28(15):5853. https://doi.org/10.3390/molecules28155853
Chicago/Turabian StyleAnsari, Waseem Ahmad, Safia Obaidur Rab, Mohammad Saquib, Aqib Sarfraz, Mohd Kamil Hussain, Mohd Sayeed Akhtar, Irfan Ahmad, and Mohammad Faheem Khan. 2023. "Pentafuhalol-B, a Phlorotannin from Brown Algae, Strongly Inhibits the PLK-1 Overexpression in Cancer Cells as Revealed by Computational Analysis" Molecules 28, no. 15: 5853. https://doi.org/10.3390/molecules28155853
APA StyleAnsari, W. A., Rab, S. O., Saquib, M., Sarfraz, A., Hussain, M. K., Akhtar, M. S., Ahmad, I., & Khan, M. F. (2023). Pentafuhalol-B, a Phlorotannin from Brown Algae, Strongly Inhibits the PLK-1 Overexpression in Cancer Cells as Revealed by Computational Analysis. Molecules, 28(15), 5853. https://doi.org/10.3390/molecules28155853