
Synthesis of meta-Aminophenol Derivatives via Cu-Catalyzed [1,3]-Rearrangement—Oxa-Michael Addition Cascade Reactions

Abstract
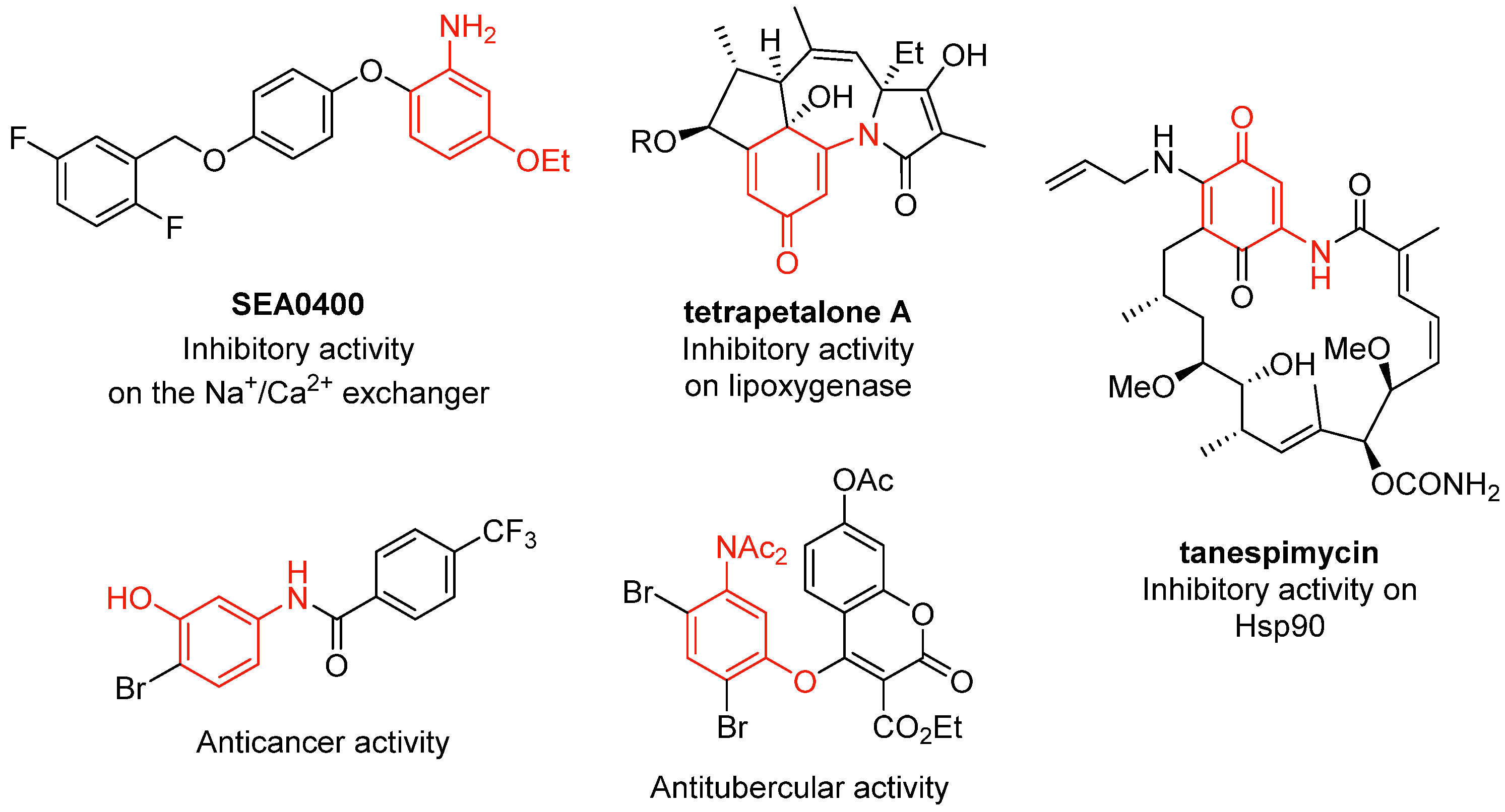
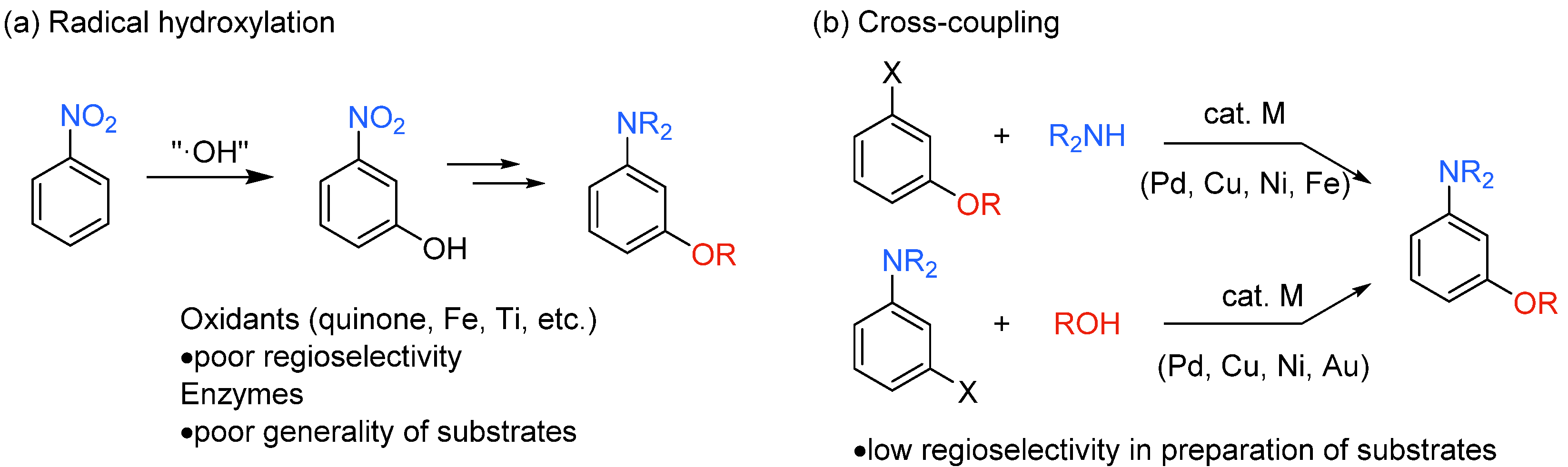
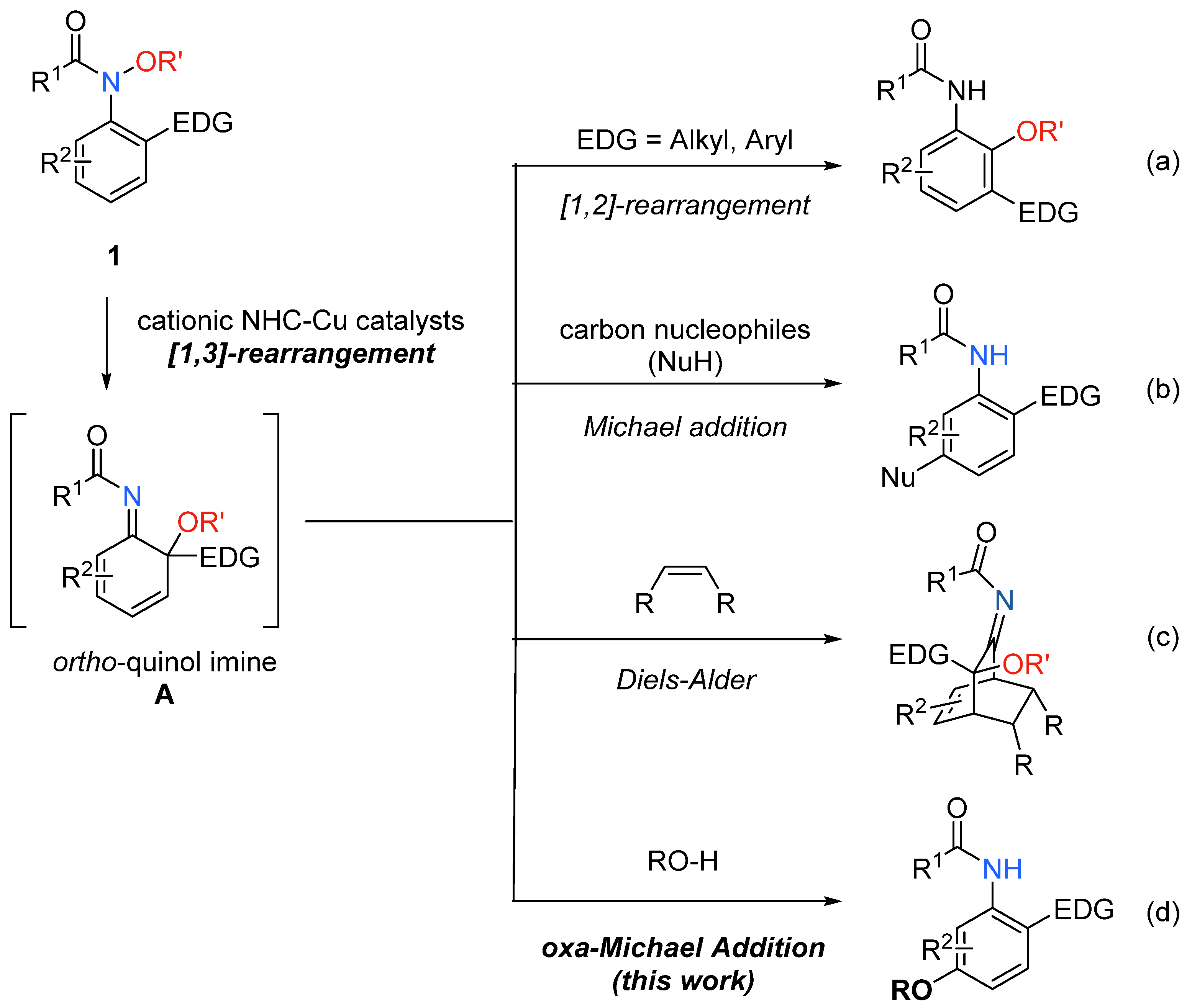
1. Introduction
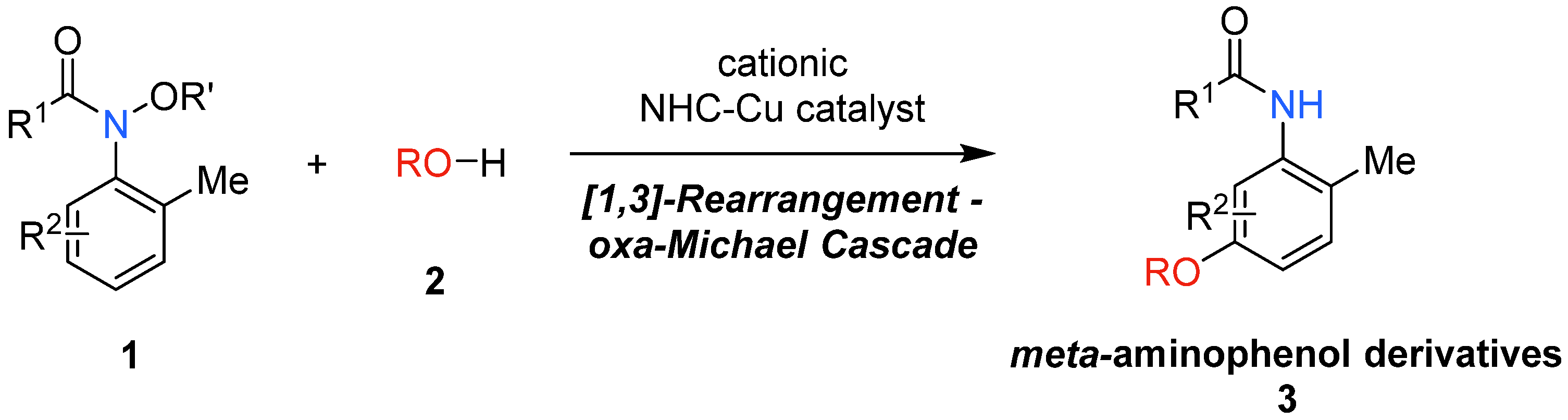
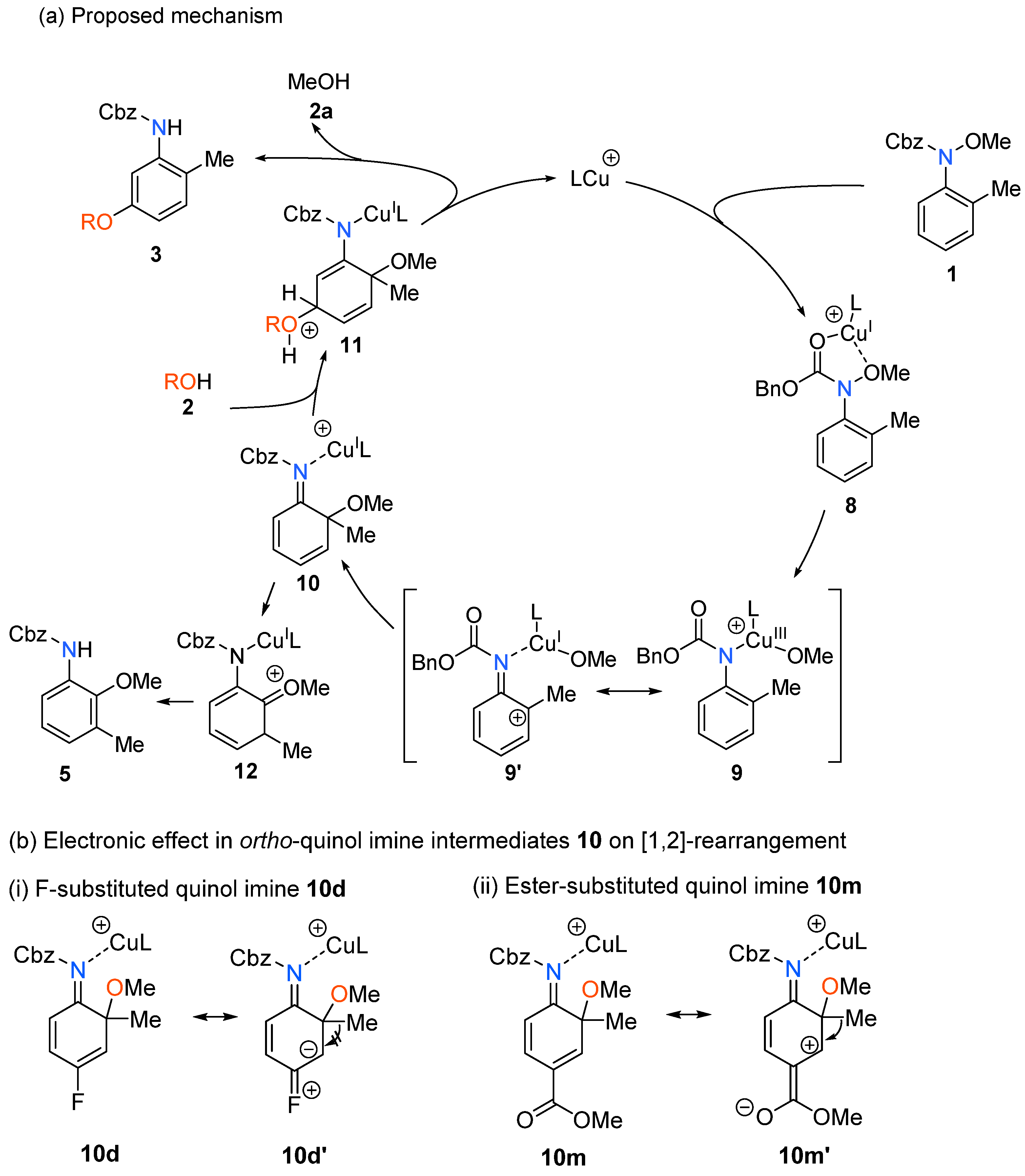
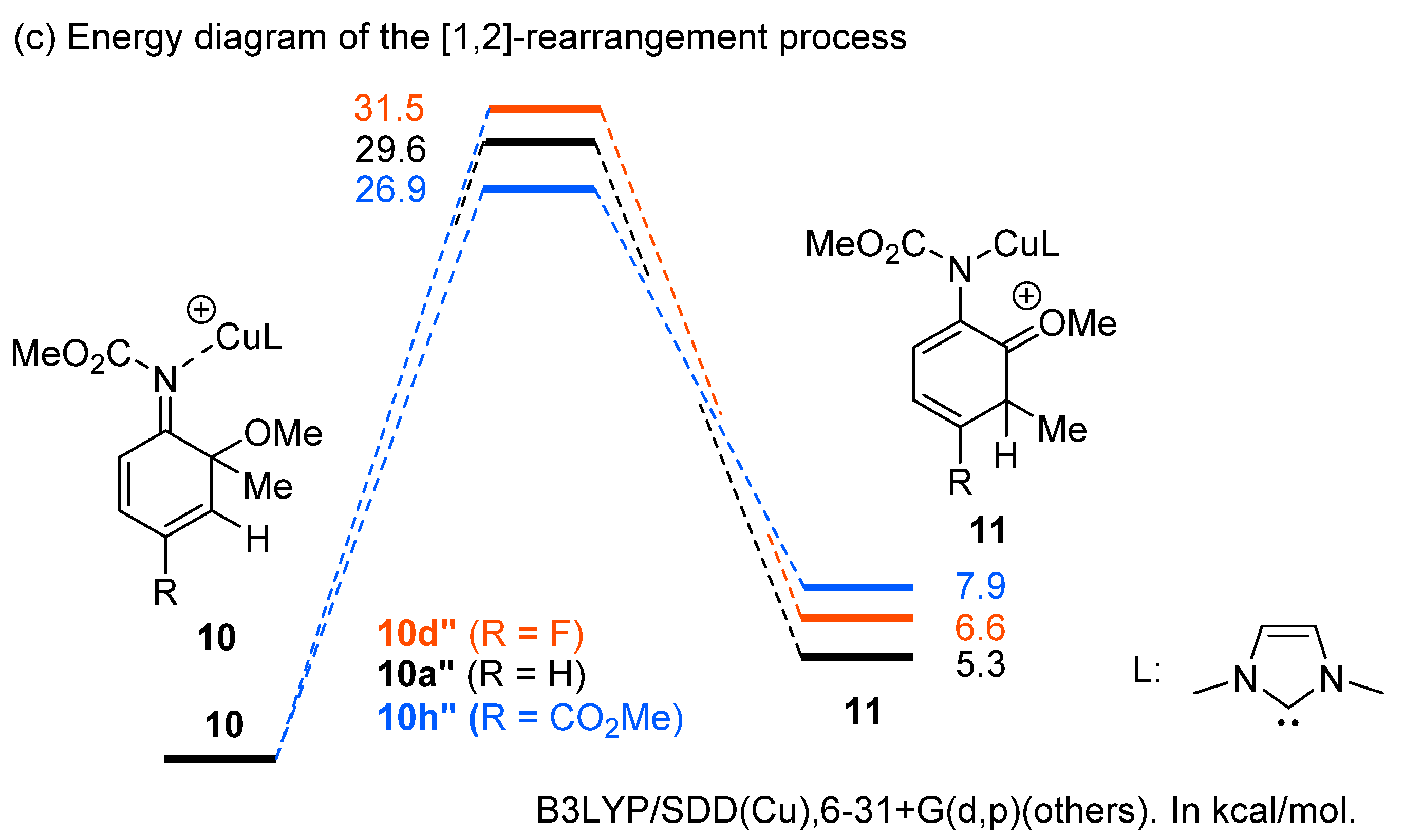
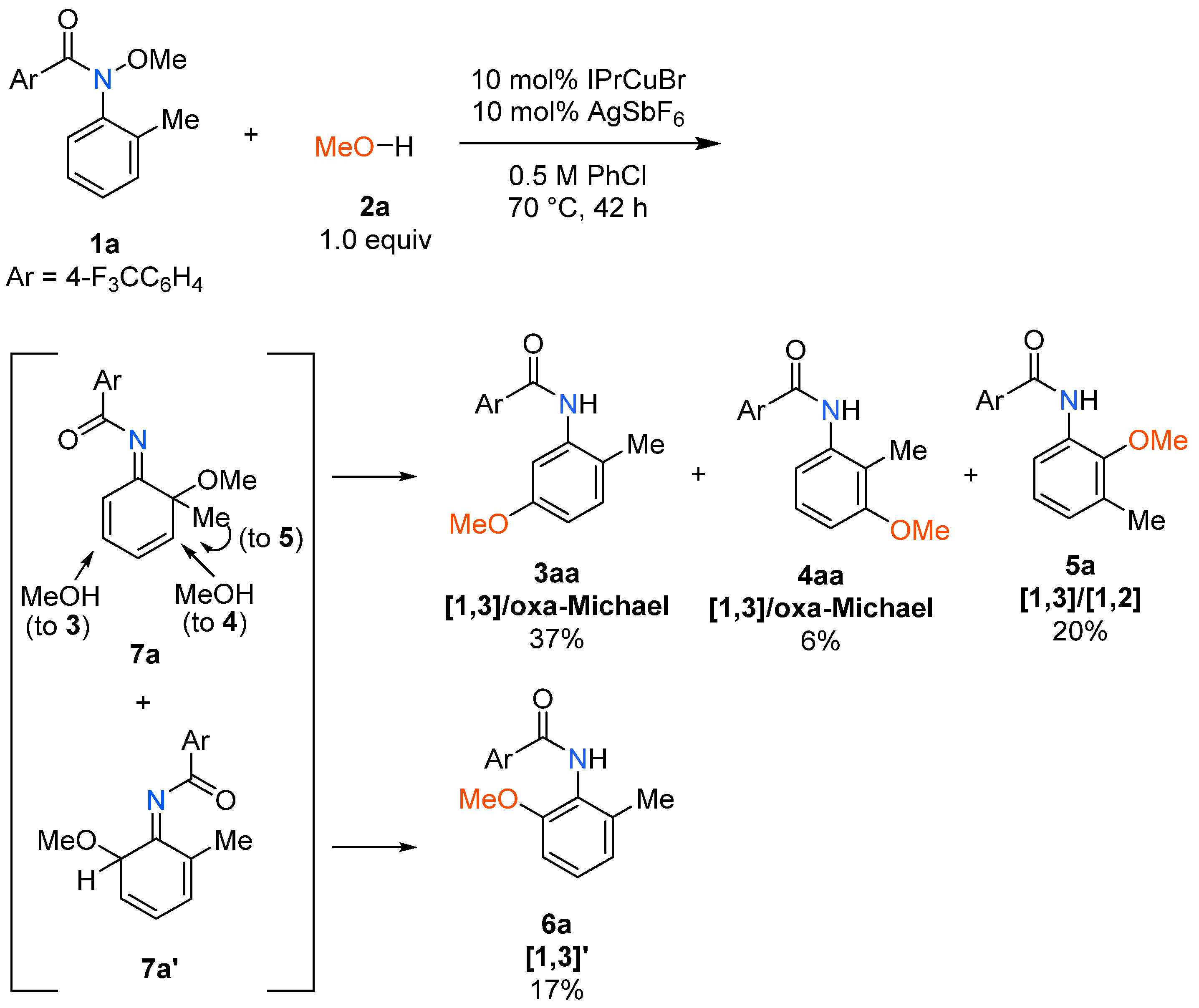
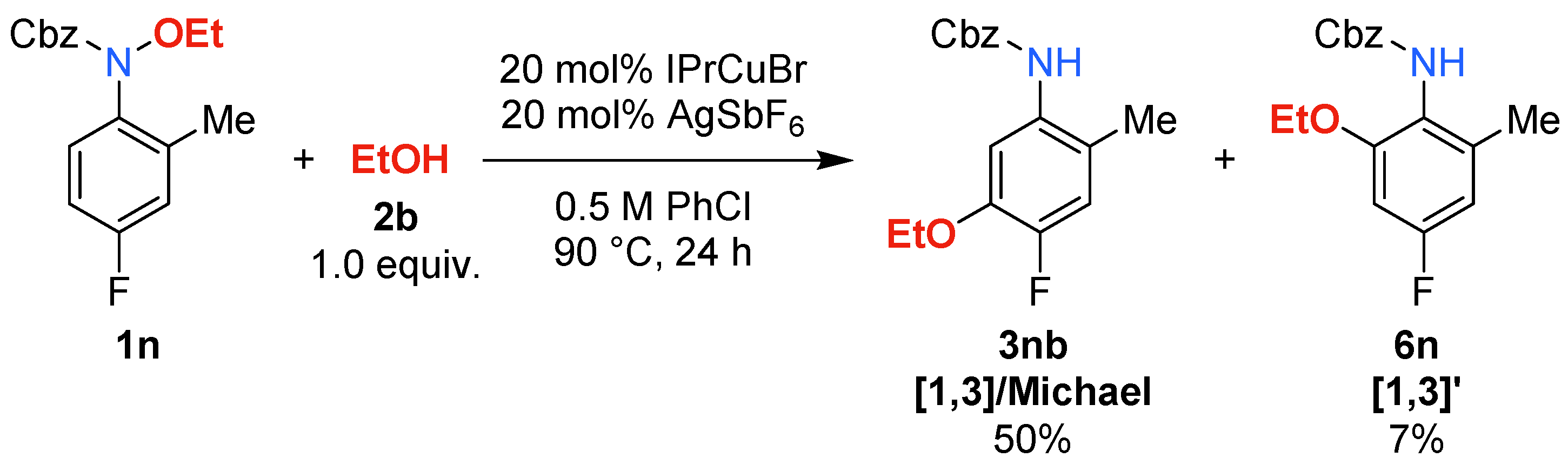
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A
References
- Matsuda, T.; Arakawa, N.; Takuma, K.; Kishida, Y.; Kawasaki, Y.; Sakaue, M.; Takahashi, K.; Takahashi, T.; Suzuki, T.; Ota, T.; et al. SEA0400, a Novel and Selective Inhibitor of the Na+-Ca2+ Exchanger, Attenuates Reperfusion Injury in the In Vitro and In Vivo Cerebral Ischemic Models. J. Pharmacol. Exp. Ther. 2001, 298, 249–256. [Google Scholar] [PubMed]
- Komoda, T.; Sugiyama, Y.; Abe, N.; Imachi, M.; Hirota, H.; Hirota, A. Tetrapetalone A, a novel lipoxygenase inhibitor from Streptomyces sp. Tetrahedron Lett. 2003, 44, 1659–1661. [Google Scholar] [CrossRef]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef]
- Liang, J.; Tang, Y.-X.; Tang, X.-Z.; Liang, H.-J.; Gao, Y.; Fang, C.; Zhang, T.-Y.; Yan, M. Discovery of meta-Amido Bromophenols as New Antitubercular Agents. Chem. Pharm. Bull. 2019, 67, 372–381. [Google Scholar] [CrossRef]
- Radwan, E.M.; Elsayed, E.H.; El-Moneim, M.A.; Moustafa, A.M.Y. Synthesis and Cytotoxicity against Human Breast Carcinoma Cell Evaluation of Some New 3,4-Disubstituted Coumarin Derivatives. Pharm. Chem. J. 2022, 55, 1040–1049. [Google Scholar] [CrossRef]
- Marusawa, H.; Ichikawa, K.; Narita, N.; Murakami, H.; Ito, K.; Tezuka, T. Hydroxyl Radical as a Strong Electrophilic Species. Bioorg. Med. Chem. 2002, 10, 2283–2290. [Google Scholar] [CrossRef]
- Bianchi, D.; Bertoli, M.; Tassinari, R.; Ricchi, M.; Vignola, R. Direct synthesis of phenols by iron-catalyzed biphasic oxidation of aromatic hydrocarbons with hydrogen peroxide. J. Mol. Catal. A Chem. 2003, 200, 111–116. [Google Scholar] [CrossRef]
- Palmisano, G.; Addamo, M.; Augugliaro, V.; Caronna, T.; García-López, E.; Loddo, V.; Palmisano, L. Influence of the substituent on selective photocatalytic oxidation of aromatic compounds in aqueous TiO2 suspensions. Chem. Commun. 2006, 1012–1014. [Google Scholar] [CrossRef]
- Fishman, A.; Tao, Y.; Rui, L.; Wood, T.K. Controlling the Regiospecific Oxidation of Aromatics via Active Site Engineering of Toluene para-Monooxygenase of Ralstonia pickettii PKO1. J. Biol. Chem. 2005, 280, 506–514. [Google Scholar] [CrossRef]
- Michael, S.; Driver, M.S.; Hartwig, J.F. A Second-Generation Catalyst for Aryl Halide Amination: Mixed Secondary Amines from Aryl Halides and Primary Amines Catalyzed by (DPPF)PdCl2. J. Am. Chem. Soc. 1996, 118, 7217–7218. [Google Scholar]
- Wagaw, S.; Yang, B.H.; Buchwald, S.L. A Palladium-Catalyzed Strategy for the Preparation of Indoles: A Novel Entry into the Fischer Indole Synthesis. J. Am. Chem. Soc. 1998, 120, 6621–6622. [Google Scholar] [CrossRef]
- Browning, R.G.; Mahmud, H.; Badarinarayana, V.; Lovely, C.J. Synthesis of chiral N-aryl pyrrolidinones via a palladium-catalyzed cross-coupling reaction. Tetrahedron Lett. 2001, 42, 7155–7157. [Google Scholar] [CrossRef]
- Kim, B.R.; Cho, S.-D.; Kim, E.J.; Lee, I.-H.; Sung, G.H.; Kim, J.-J.; Lee, S.-G.; Yoon, Y.-J. Efficient palladium-catalyzed amination of aryl chlorides using di(dicyclohexylamino)phenylphosphine as a PN2 ligand. Tetrahedron 2012, 68, 287–293. [Google Scholar] [CrossRef]
- Ma, D.; Zhang, Y.; Yao, J.; Wu, S.; Tao, F. Accelerating Effect Induced by the Structure of α-Amino Acid in the Copper-Catalyzed Coupling Reaction of Aryl Halides with α-Amino Acids. Synthesis of Benzolactam-V8. J. Am. Chem. Soc. 1998, 120, 12459–12467. [Google Scholar] [CrossRef]
- Wolter, M.; Klapars, A.; Buchwald, S.L. Synthesis of N-Aryl Hydrazides by Copper-Catalyzed Coupling of Hydrazides with Aryl Iodides. Org. Lett. 2001, 3, 3803–3805. [Google Scholar] [CrossRef]
- Torraca, K.E.; Huang, X.; Parrish, C.A.; Buchwald, S.L. An Efficient Intermolecular Palladium-Catalyzed Synthesis of Aryl Ethers. J. Am. Chem. Soc. 2001, 123, 10770–10771. [Google Scholar] [CrossRef]
- Cheung, C.W.; Stephen, L.; Buchwald, S.L. Mild and General Palladium-Catalyzed Synthesis of Methyl Aryl Ethers Enabled by the Use of a Palladacycle Precatalyst. Org. Lett. 2013, 15, 3998–4001. [Google Scholar] [CrossRef]
- Wolter, M.; Nordmann, G.; Job, G.E.; Buchwald, S.L. Copper-Catalyzed Coupling of Aryl Iodides with Aliphatic Alcohols. Org. Lett. 2002, 4, 973–976. [Google Scholar] [CrossRef]
- Tlili, A.; Xia, N.; Monnier, F.; Taillefer, M. A Very Simple Copper-Catalyzed Synthesis of Phenols Employing Hydroxide Salts. Angew. Chem. Int. Ed. 2009, 48, 8725–8728. [Google Scholar] [CrossRef]
- Yang, L.; Yan, Y.; Cao, N.; Hao, J.; Li, G.; Zhang, W.; Cao, R.; Wang, C.; Xiao, J.; Xue, D. Ni(I)-Catalyzed Hydroxylation of Aryl Halides with Water under Thermal Catalysis. Org. Lett. 2022, 24, 9431–9435. [Google Scholar] [CrossRef]
- Das, A.; Nitin, T.; Patil, N.T. Ligand-Enabled Gold-Catalyzed C(sp2)–O Cross-Coupling Reactions. ACS Catal. 2023, 13, 3847–3853. [Google Scholar] [CrossRef]
- Weaver, M.G.; Bai, W.-J.K.; Jackson, S.K.; Pettus, T.R.R. Diels–Alder Construction of Regiodifferentiated meta-Amino Phenols and Derivatives. Org. Lett. 2014, 16, 1294–1297. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Jo, T.; Ishida, Y.; Tashiro, H.; Terada, M. Cationic N-Heterocyclic Carbene Copper-Catalyzed [1,3]-Alkoxy Rearrangement of N-Alkoxyanilines. Org. Lett. 2017, 19, 3059–3062. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Nozawa, S.; Ishida, Y.; Muranushi, I.; Mayerweg, A.; Terada, M. Copper-catalyzed [1,3]-alkoxy rearrangement for the selective synthesis of polycyclic ortho-aminoarenol derivatives. Org. Chem. Front. 2021, 8, 6390–6394. [Google Scholar] [CrossRef]
- Ishida, Y.; Nakamura, I.; Terada, M. Copper-catalyzed Domino [1,3]/[1,2] Rearrangement for the Efficient Synthesis of Multisubstituted ortho-Anisidines. J. Am. Chem. Soc. 2018, 140, 8629–8633. [Google Scholar] [CrossRef]
- Nakamura, I.; Tashiro, H.; Ishida, Y.; Terada, M. Synthesis of meta-Substituted Anilines via Copper-Catalyzed [1,3]-Methoxy Rearrangement. Org. Lett. 2020, 22, 3794–3798. [Google Scholar] [CrossRef]
- Nakamura, I.; Masukawa, K.; Ishida, Y.; Terada, M. Cu-Catalyzed [1,3]-Alkoxy Rearrangement/Diels–Alder Cascade Reactions via in Situ Generation of Functionalized ortho-Quinol Imines. Org. Lett. 2021, 23, 4127–4132. [Google Scholar] [CrossRef]
- Fang, Y.-Q.; Dang, L. Mechanistic Study of Domino Rearrangement-Promoted Meta C–H Activation in 2-Methyl-N-methoxyaniline via Cu(NHC)+: Motivation and Selectivity. Org. Lett. 2020, 22, 9178–9183. [Google Scholar] [CrossRef]
- Suzuki, M.; Terada, M.; Nakamura, I. Copper-Catalyzed [1,3]-Nitrogen Rearrangement of O-Aryl Ketoximes via Oxidative Addition of N-O Bond in Inverse Electron Flow. Chem. Sci. 2023; accepted manuscript. [Google Scholar] [CrossRef]
- Zhou, S.; Li, Y.; Liu, X.; Hu, W.; Ke, Z.; Xu, X. Enantioselective Oxidative Multi-Functionalization of Terminal Alkynes with Nitrones and Alcohols for Expeditious Assembly of Chiral α-Alkoxy-β-amino-ketones. J. Am. Chem. Soc. 2021, 143, 14703–14711. [Google Scholar] [CrossRef]
- Liu, X.; Tian, X.; Huang, J.; Qian, Y.; Xu, X.; Kang, Z.; Hu, W. Enantioselective Propargylation of Oxonium Ylide with α-Propargylic-3-Indolymethanol: Access to Chiral Propargylic Indoles. Org. Lett. 2022, 24, 1027–1032. [Google Scholar] [CrossRef]
- Yang, X.; Hong, K.; Zhang, S.; Zhang, Z.; Zhou, S.; Huang, J.; Xu, X.; Hu, W. Asymmetric Three-Component Reaction of Two Diazo Compounds and Hyrdroxylamine Derivatives for the Access to Chiral α-Alkoxy-β-amino-carboxylates. ACS Catal. 2022, 12, 12302–12309. [Google Scholar] [CrossRef]
- Fishbein, J.C.; McClelland, R.A. Cyclohexadienes from the rearrangement of O-aroyl-N-acetyl-N-(2,6-dimethylphenyl)hydroxylamines. Reaction in aqueous solution to meta- and para-substituted 2,6-dimethylacetanilides. J. Chem. Soc. Perkin Trans. 2 1995, 653–662. [Google Scholar] [CrossRef]
- Quideau, S.; Pouységu, L.; Ozanne, A.; Gagnepain, J. Oxidative Dearomatization of Phenols and Anilines via λ3- and λ5-Iodane-Mediated Phenylation and Oxygenation. Molecules 2005, 10, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Bian, M.; Ding, H. Recent application of oxa-Michael reaction in complex natural product synthesis. Tetrahedron Lett. 2016, 57, 5519–5539. [Google Scholar] [CrossRef]
- Nising, C.F.; Bräse, S. Recent developments in the field of oxa-Michael reactions. Chem. Soc. Rev. 2012, 41, 988–999. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
| Entry | NHC·CuX | AgX′ | Solvent | Temp. (°C) | 3ba (%) a | 6b (%) a | 1b (%) a |
| 1 | IPrCuBr | AgSbF6 | PhCl | 70 | 70 | 28 | <1 |
| 2 | IMesCuCl | AgSbF6 | PhCl | 70 | 21 | 3 | 76 |
| 3 | SIPrCuCl | AgSbF6 | PhCl | 70 | 13 | 5 | 47 |
| 4 | IPrCuBr | AgBF4 | PhCl | 70 | 43 | 13 | 37 |
| 5 | IPrCuBr | AgNTf2 | PhCl | 70 | <10 | <15 | - b |
| 6 | IPrCuBr | AgSbF6 | DCE | 70 c | 76 | 22 | <2 |
| 7 | IPrCuBr | AgSbF6 | toluene | 70 | 63 | 18 | 11 |
| 8 | IPrCuBr | AgSbF6 | 1,4-dioxane | 70 | 59 | 19 | 7 |
| 9 | IPrCuBr | AgSbF6 | EtOAc | 70 | 29 | 12 | 38 |
| 10 | IPrCuBr | AgSbF6 | DCE | 60 d | <78 | 18 | 3 |
| 11 | IPrCuBr | AgSbF6 | DCE | 80 e | 75 | 22 | 6 |
| 12 | IPrCuBr | - | DCE | 70 | <1 | <1 | 99 |
| 13 | - | AgSbF6 | DCE | 70 | <1 | <1 | 92 |
 | ||||||
| Entry | 1 | R | 2a (Equiv) | 3 (%) a | 6 (%) a | 1 (%) a |
| 1 b | 1c | Bz | 1 | 3ca (73) | 6c (12) | <1 |
| 2 | 1d | Cbz | 1 | 3da (72) | <1 | <1 |
| 3 | 1e | Troc | 1 | 3ea (54) | <1 | 27 |
| 4 | 1f | Alloc | 1 | 3fa (26) | 6f (6) | 32 |
| 5 | 1d | Cbz | 2 | 3da (48) | 6d (4) | 46 |
| 6 | 1d | Cbz | 5 | 3da (18) | 6d (3) | 29 |
| 7 | 1d | Cbz | 0.5 | 3da (50) | 6d (3) | <31 |
| 8 d | 1d | Cbz | 0.5 | 3da (71) | 6d (9) | <11 |
| 9 e | 1d | Cbz | 1 | 3da (85) c | <1 | <1 |
 | ||||||
| Entry | 1 | R | Temp (°C) | 3 (%) b | 1 (%) c | Byproduct (%) c |
| 1 d,e | 1g | Me | 70 | 3ga (64) | <1 | 5g (<3) |
| 2 e | 1h | Ph | 70 | 3ha (45) | <1 | 5h (28), 6h (27) |
| 3 | 1i | Cl | 70 | 3ia (23) | 36 | |
| 4 f | 1i | Cl | 70 | 3ia (30) | 19 | |
| 5 e,f,g | 1i | Cl | 90 c | 3ia (61) | <1 | |
| 6 | 1j | Br | 70 | 3ja (40) | 32 | |
| 7 f | 1j | Br | 70 | 3ja (60) | <1 | |
| 8 f | 1k | I | 70 | 3ka (42) | 6 | 5k (8) |
| 9 g,h | 1l | -CºCPh | 90 | 3la (28) | <1 | |
| 10 g,i | 1m | CO2Me | 90 | <1 | 9 | 5m (80) |
 | ||||
| Entry | 2 | ROH | 3 (%) a | 3da (%) a |
| 1 | 2c | PhCH2CH2OH | 3dc (51) | 14 |
| 2 | 2c b | PhCH2CH2OH | 3dc (51) | 7 |
| 3 | 2d | H2C=CHCH2OH | 3dd (35) | 23 |
| 4 | 2e | tBuOH | - c | - c |
| 5 | 2f | PhOH | - c | - c |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, I.; Tachibana, M.; Konta, R.; Tashiro, H.; Terada, M. Synthesis of meta-Aminophenol Derivatives via Cu-Catalyzed [1,3]-Rearrangement—Oxa-Michael Addition Cascade Reactions. Molecules 2023, 28, 4251. https://doi.org/10.3390/molecules28104251
Nakamura I, Tachibana M, Konta R, Tashiro H, Terada M. Synthesis of meta-Aminophenol Derivatives via Cu-Catalyzed [1,3]-Rearrangement—Oxa-Michael Addition Cascade Reactions. Molecules. 2023; 28(10):4251. https://doi.org/10.3390/molecules28104251
Chicago/Turabian StyleNakamura, Itaru, Mai Tachibana, Riku Konta, Hiroki Tashiro, and Masahiro Terada. 2023. "Synthesis of meta-Aminophenol Derivatives via Cu-Catalyzed [1,3]-Rearrangement—Oxa-Michael Addition Cascade Reactions" Molecules 28, no. 10: 4251. https://doi.org/10.3390/molecules28104251
APA StyleNakamura, I., Tachibana, M., Konta, R., Tashiro, H., & Terada, M. (2023). Synthesis of meta-Aminophenol Derivatives via Cu-Catalyzed [1,3]-Rearrangement—Oxa-Michael Addition Cascade Reactions. Molecules, 28(10), 4251. https://doi.org/10.3390/molecules28104251