
7-Aminoalkoxy-Quinazolines from Epigenetic Focused Libraries Are Potent and Selective Inhibitors of DNA Methyltransferase 1




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Compounds for Experimental Screening
- (1)
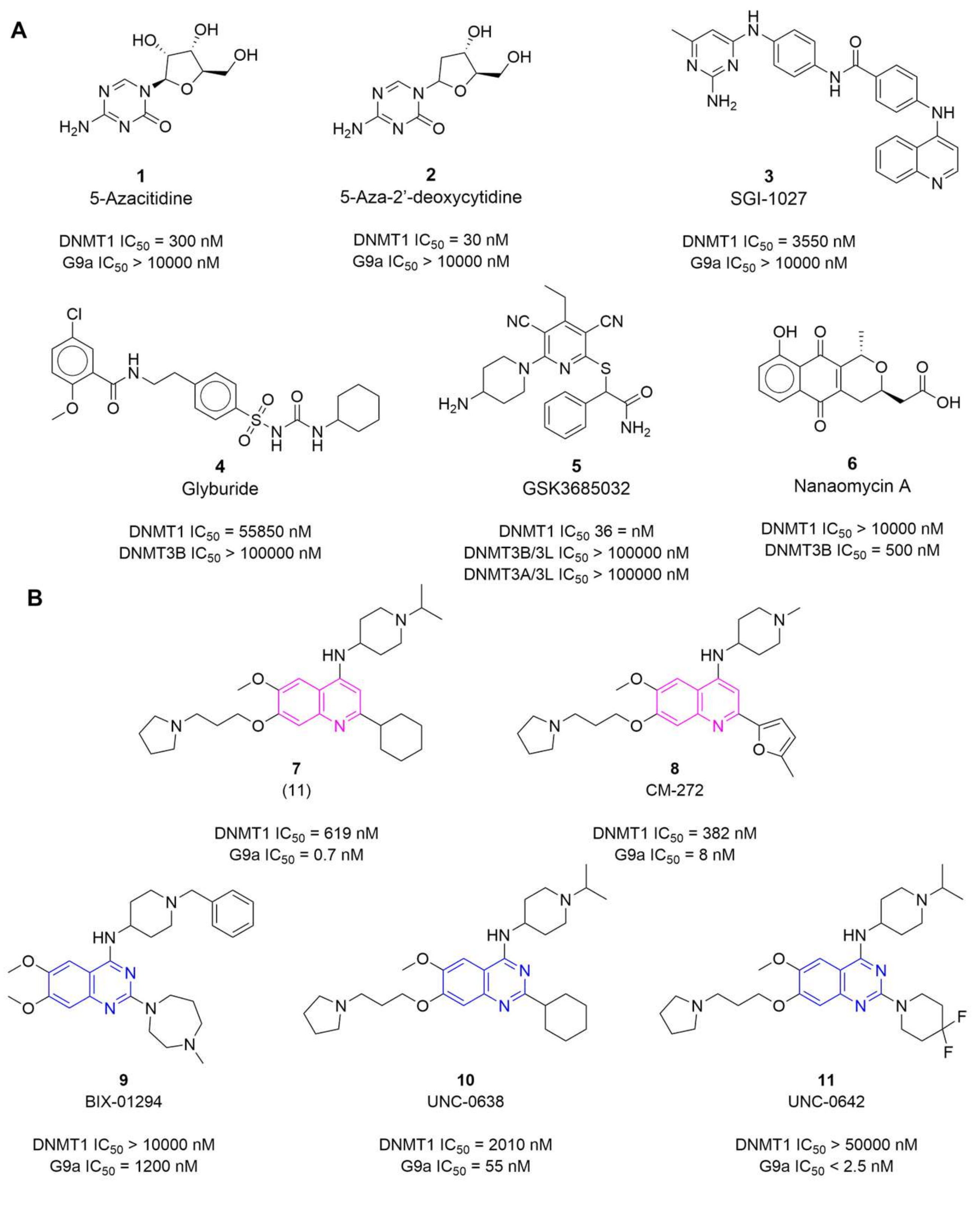
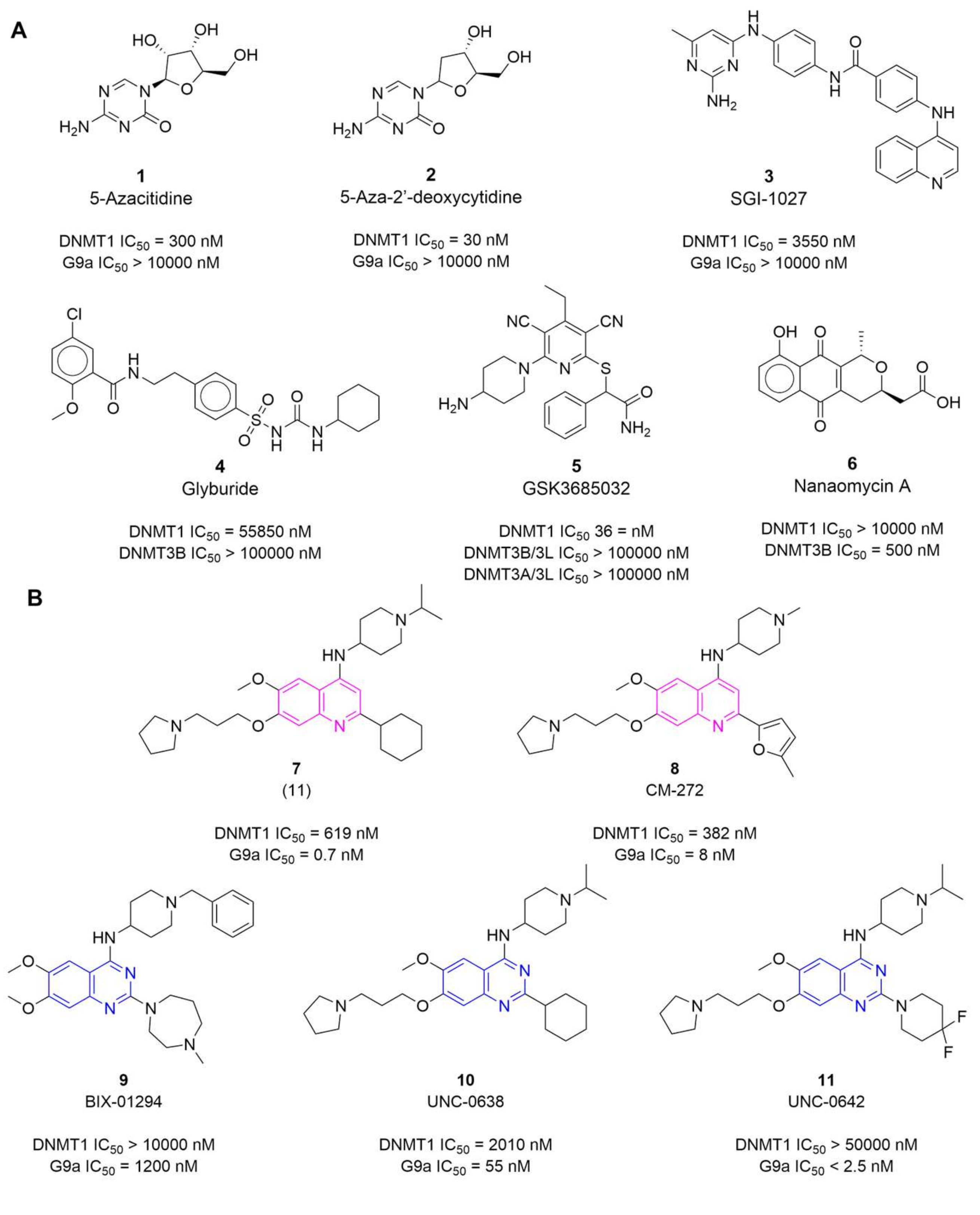
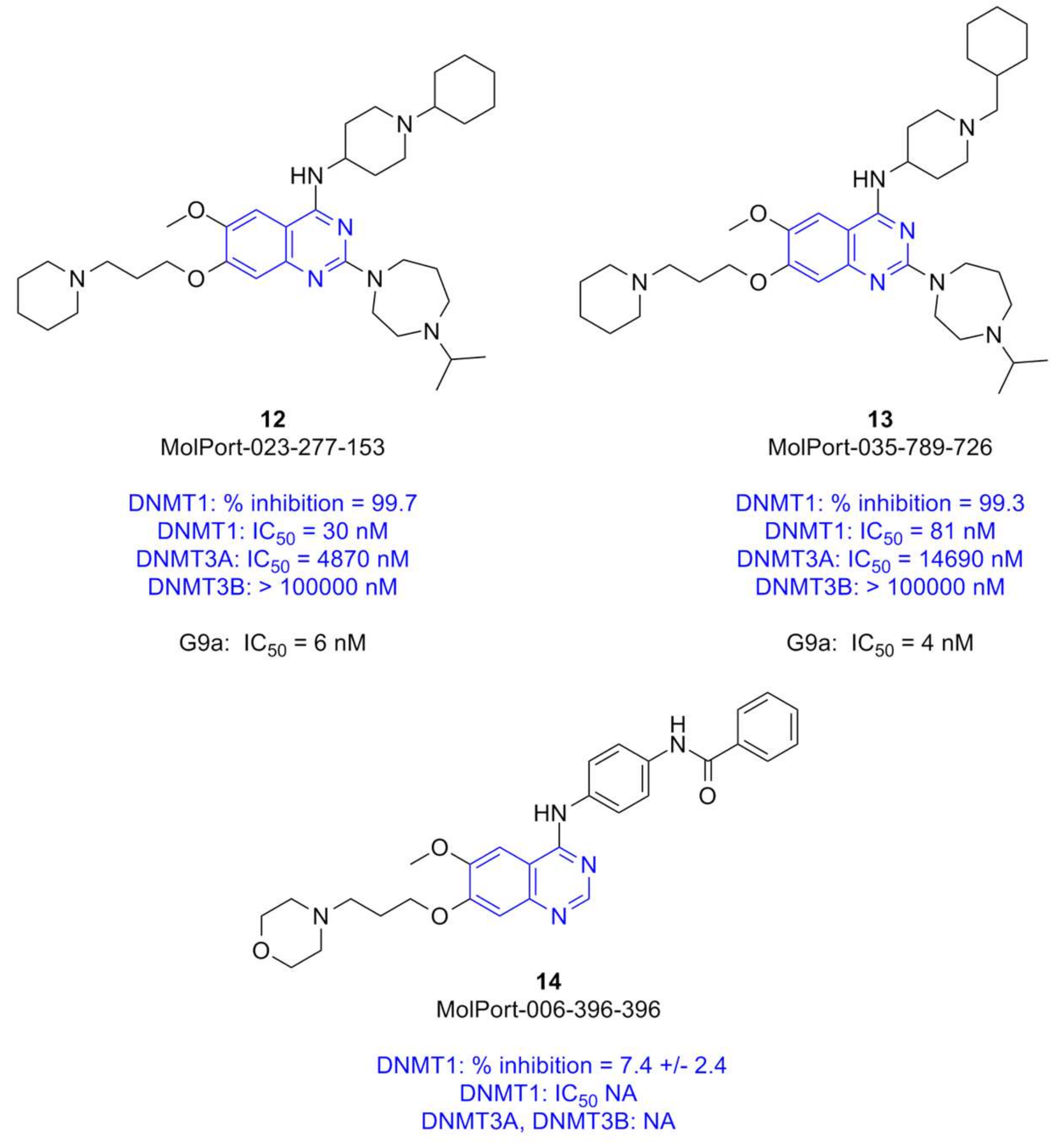
- Knowledge of the promising activity profile of quinoline-based derivatives as dual inhibitors of DNMTs/G9a (vide supra, Figure 1B) in enzymatic and epigenetic functional cellular response;
- (2)
- High structural similarity of the quinoline-based compounds to the selected quinazoline-based derivatives 12 and 13, that are known to be potent G9a inhibitors in enzymatic and cell-based assays (vide supra, Figure 1B).
- (3)
- Commercial availability of the physical samples from the chemical vendors (Table S3 in the Supplementary Materials).
2.2. Biochemical DNMT Inhibition Assays
2.3. Computational Methods
2.3.1. Protein and Ligands Preparation
2.3.2. Molecular Docking
2.3.3. Molecular Dynamics
3. Results and Discussion
3.1. Biochemical Inhibition Assays
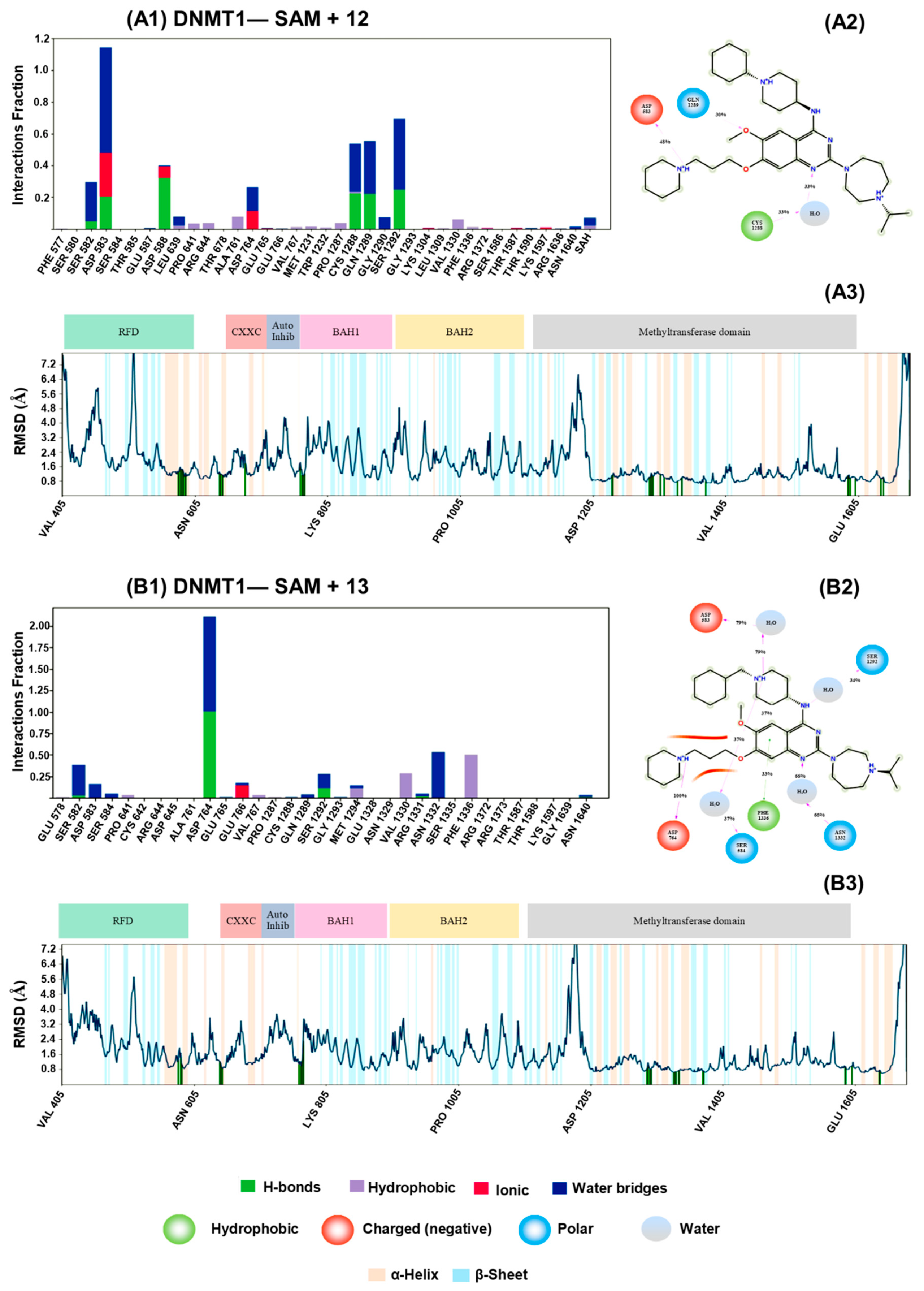
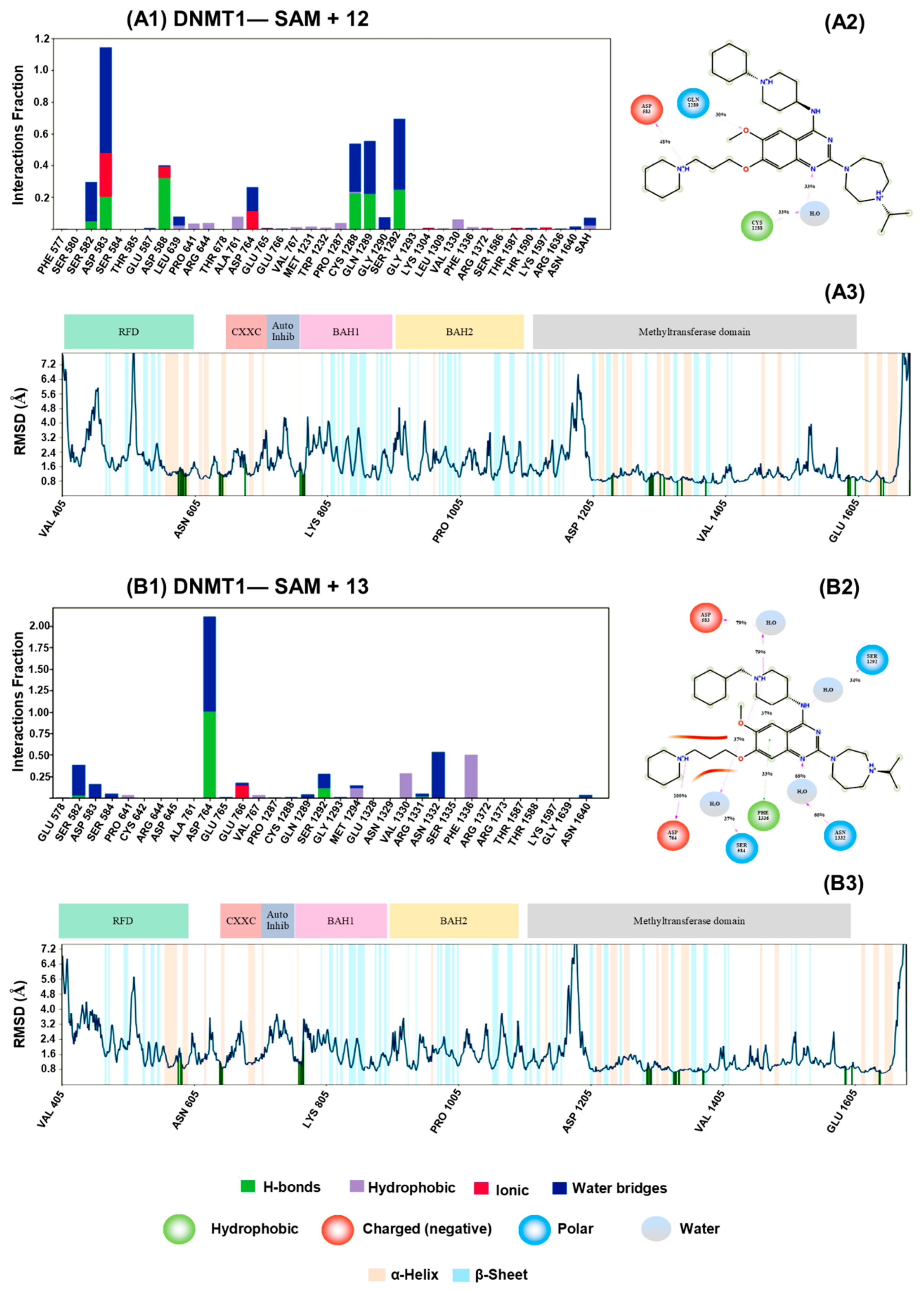
3.2. Molecular Docking and Dynamics Simulations with DNMTs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Holdgate, G.A.; Bardelle, C.; Lanne, A.; Read, J.; O’Donovan, D.; Smith, J.M.; Selmi, N.; Sheppard, R. Drug discovery for epigenetics targets. Drug Discov. Today 2021, 27, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kumar, V.; Sehrawat, N.; Yadav, M.; Chaudhary, M.; Upadhyay, S.K.; Kumar, S.; Sharma, V.; Kumar, S.; Dilbaghi, N.; et al. Current paradigms in epigenetic anticancer therapeutics and future challenges. Semin. Cancer Biol. 2021, 13, 3364. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenetics 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Xie, T.; Wang, Z.; Wang, X.; Zeng, S.; Kang, Y.; Hou, T. DNA methyltransferases: Emerging targets for the discovery of inhibitors as potent anticancer drugs. Drug Discov. Today 2019, 24, 2323–2331. [Google Scholar] [CrossRef]
- Conery, A.R.; Rocnik, J.L.; Trojer, P. Small molecule targeting of chromatin writers in cancer. Nat. Chem. Biol. 2022, 18, 124–133. [Google Scholar] [CrossRef]
- Vougiouklakis, T.; Bernard, B.J.; Nigam, N.; Burkitt, K.; Nakamura, Y.; Saloura, V. Clinicopathologic significance of protein lysine methyltransferases in cancer. Clin. Epigenetics 2020, 12, 146. [Google Scholar] [CrossRef]
- Nepali, K.; Liou, J.P. Recent developments in epigenetic cancer therapeutics: Clinical advancement and emerging trends. J. Biomed. Sci. 2021, 28, 27. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, C.; Wu, C.; Cui, W.; Wang, L. DNA methyltransferases in cancer: Biology, paradox, aberrations, and targeted therapy. Cancers 2020, 12, 2123. [Google Scholar] [CrossRef]
- Weng, Y.-L.; An, R.; Shin, J.; Song, H.; Ming, G.-l. DNA modifications and neurological disorders. Neurotherapeutics 2013, 10, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Bayraktar, G.; Kreutz, M.R. Neuronal DNA methyltransferases: Epigenetic mediators between synaptic activity and gene expression? Neurosci. A Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2018, 24, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, R.; Zwergel, C.; Artico, M.; Taurone, S.; Ralli, M.; Greco, A.; Mai, A. The emerging role of epigenetics in human autoimmune disorders. Clin. Epigenetics 2019, 11, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zouali, M. DNA methylation signatures of autoimmune diseases in human b lymphocytes. Clin. Immunol. 2021, 222, 108622. [Google Scholar] [CrossRef] [PubMed]
- Arguelles, A.O.; Meruvu, S.; Bowman, J.D.; Choudhury, M. Are epigenetic drugs for diabetes and obesity at our door step? Drug Discov. Today 2016, 21, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, A.; Hayashi, K.; Yoshimoto, N.; Nakamichi, R.; Homma, K.; Itoh, H. DNA damage and expression of DNA methylation modulators in urine-derived cells of patients with hypertension and diabetes. Sci. Rep. 2020, 10, 3377. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Liu, Y. DNA methylation in human diseases. Genes Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef]
- Hauser, B.M.; Lau, A.; Gupta, S.; Bi, W.L.; Dunn, I.F. The epigenomics of pituitary adenoma. Front. Endocrinol. 2019, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark Res. 2017, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Ou, T.T.; Wu, C.C.; Li, R.N.; Lin, Y.C.; Lin, C.H.; Tsai, W.C.; Liu, H.W.; Yen, J.H. Global DNA methylation, dnmt1, and mbd2 in patients with systemic lupus erythematosus. Lupus 2011, 20, 131–136. [Google Scholar] [CrossRef]
- Klein, C.J.; Botuyan, M.V.; Wu, Y.; Ward, C.J.; Nicholson, G.A.; Hammans, S.; Hojo, K.; Yamanishi, H.; Karpf, A.R.; Wallace, D.C.; et al. Mutations in dnmt1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 2011, 43, 595–600. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños María, P.; Mosquera Juan, F.; Mutowo, P.; Nowotka, M.; et al. Chembl: Towards direct deposition of bioassay data. Nucl. Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Kuck, D.; Caulfield, T.; Lyko, F.; Medina-Franco, J.L. Nanaomycin a selectively inhibits dnmt3b and reactivates silenced tumor suppressor genes in human cancer cells. Mol. Cancer Ther. 2010, 9, 3015–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappalardi, M.B.; Keenan, K.; Cockerill, M.; Kellner, W.A.; Stowell, A.; Sherk, C.; Wong, K.; Pathuri, S.; Briand, J.; Steidel, M.; et al. Discovery of a first-in-class reversible dnmt1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nat. Cancer 2021, 2, 1002–1017. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, A.G.; Groy, A.; Gore, E.R.; Atkins, C.; Long, E.R.; Montoute, M.N.; Wu, Z.; Halsey, W.; McNulty, D.E.; Ennulat, D.; et al. In vitro and in vivo induction of fetal hemoglobin with a reversible and selective dnmt1 inhibitor. Haematologica 2021, 106, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Naveja, J.J.; Medina-Franco, J.L. Insights from pharmacological similarity of epigenetic targets in epipolypharmacology. Drug Discov. Today 2018, 23, 141–150. [Google Scholar] [CrossRef] [PubMed]
- de Lera, A.R.; Ganesan, A. Two-hit wonders: The expanding universe of multitargeting epigenetic agents. Curr. Opin. Chem. Biol. 2020, 57, 135–154. [Google Scholar] [CrossRef]
- Tomaselli, D.; Lucidi, A.; Rotili, D.; Mai, A. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med. Res. Rev. 2020, 40, 190–244. [Google Scholar] [CrossRef]
- Rabal, O.; San José-Enériz, E.; Agirre, X.; Sánchez-Arias, J.A.; Vilas-Zornoza, A.; Ugarte, A.; de Miguel, I.; Miranda, E.; Garate, L.; Fraga, M.; et al. Discovery of reversible DNA methyltransferase and lysine methyltransferase g9a inhibitors with antitumoral in vivo efficacy. J. Med. Chem. 2018, 61, 6518–6545. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Méndez-Lucio, O.; Yoo, J.; Dueñas, A. Discovery and development of DNA methyltransferase inhibitors using in silico approaches. Drug Discov. Today 2015, 20, 569–577. [Google Scholar] [CrossRef]
- Saldívar-González, F.I.; Gómez-García, A.; Chávez-Ponce de León, D.E.; Sánchez-Cruz, N.; Ruiz-Rios, J.; Pilón-Jiménez, B.A.; Medina-Franco, J.L. Inhibitors of DNA methyltransferases from natural sources: A computational perspective. Front. Pharmacol. 2018, 9, 1144. [Google Scholar] [CrossRef] [Green Version]
- Sessions, Z.; Sánchez-Cruz, N.; Prieto-Martínez, F.D.; Alves, V.M.; Santos, H.P., Jr.; Muratov, E.; Tropsha, A.; Medina-Franco, J.L. Recent progress on cheminformatics approaches to epigenetic drug discovery. Drug Discov. Today 2020, 25, 2268–2276. [Google Scholar] [CrossRef]
- Prado-Romero, D.L.; Medina-Franco, J.L. Advances in the exploration of the epigenetic relevant chemical space. ACS Omega 2021, 6, 22478–22486. [Google Scholar] [CrossRef] [PubMed]
- Flores-Padilla, E.A.; Juárez-Mercado, K.E.; Naveja, J.J.; Kim, T.D.; Alain Miranda-Quintana, R.; Medina-Franco, J.L. Chemoinformatic characterization of synthetic screening libraries focused on epigenetic targets. Mol. Inf. 2021, 40, e2100285. [Google Scholar] [CrossRef] [PubMed]
- Gros, C.; Fleury, L.; Nahoum, V.; Faux, C.; Valente, S.; Labella, D.; Cantagrel, F.; Rilova, E.; Bouhlel, M.A.; David-Cordonnier, M.-H.; et al. New insights on the mechanism of quinoline-based DNA methyltransferase inhibitors. J. Biol. Chem. 2015, 290, 6293–6302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-López, E.; Rabal, O.; Oyarzabal, J.; Medina-Franco, J.L. Towards the understanding of the activity of g9a inhibitors: An activity landscape and molecular modeling approach. J. Comp.-Aided Mol. Des. 2020, 34, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Barsyte-Lovejoy, D.; Allali-Hassani, A.; He, Y.; Herold, J.M.; Chen, X.; Yates, C.M.; Frye, S.V.; Brown, P.J.; Huang, J.; et al. Optimization of cellular activity of G9a inhibitors 7-aminoalkoxy-quinazolines. J. Med. Chem. 2011, 54, 6139–6150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Vázquez, F.S.; Hernández-Luis, F.; Medina Franco, J.L. Quinazolines as inhibitors of chromatin-associated proteins in histones. Med. Chem. Res. 2019, 28, 395–416. [Google Scholar] [CrossRef]
- Juárez-Mercado, K.E.; Prieto-Martínez, F.D.; Sánchez-Cruz, N.; Peña-Castillo, A.; Prada-Gracia, D.; Medina-Franco, J.L. Expanding the structural diversity of DNA methyltransferase inhibitors. Pharmaceuticals 2020, 14, 17. [Google Scholar] [CrossRef]
- Reaction Biology Corporation. Available online: http://www.reactionbiology.com (accessed on 5 March 2022).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in casp8. Proteins 2009, 77 (Suppl. 9), 114–122. [Google Scholar] [CrossRef] [Green Version]
- Cho, A.E.; Guallar, V.; Berne, B.J.; Friesner, R. Importance of accurate charges in molecular docking: Quantum mechanical/molecular mechanical (qm/mm) approach. J. Comput Chem 2005, 26, 915–931. [Google Scholar] [CrossRef] [Green Version]
- Molecular Operating Environment (MOE). Chemical Computing Group Inc.: Montreal, QC, Canada, 2021. Available online: http://www.chemcomp.com (accessed on 5 March 2022).
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on cpu and gpu architectures with namd. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Guianvarc’h, D.; Arimondo, P.B. Challenges in developing novel DNA methyltransferases inhibitors for cancer therapy. Fut. Med. Chem. 2014, 6, 1237–1240. [Google Scholar] [CrossRef] [PubMed]
- López-López, E.; Prieto-Martínez, F.D.; Medina-Franco, J.L. Activity landscape and molecular modeling to explore the SAR of dual epigenetic inhibitors: A focus on G9a and DNMT1. Molecules 2018, 23, 3282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeltsch, A.; Jurkowska, R.Z. Allosteric control of mammalian DNA methyltransferases—A new regulatory paradigm. Nucleic Acids Res. 2016, 44, 8556–8575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, M.; Estève, P.O.; Chin, H.G.; Samaranayke, M.; Kim, G.D.; Pradhan, S. CXXC domain of human DNMT1 is essential for enzymatic activity. Biochemistry 2008, 47, 10000–10009. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Cruz, N.; Medina-Franco, J.L. Epigenetic target profiler: A web server to predict epigenetic targets of small molecules. J. Chem. Inf. Model. 2021, 61, 1550–1554. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medina-Franco, J.L.; López-López, E.; Martínez-Fernández, L.P. 7-Aminoalkoxy-Quinazolines from Epigenetic Focused Libraries Are Potent and Selective Inhibitors of DNA Methyltransferase 1. Molecules 2022, 27, 2892. https://doi.org/10.3390/molecules27092892
Medina-Franco JL, López-López E, Martínez-Fernández LP. 7-Aminoalkoxy-Quinazolines from Epigenetic Focused Libraries Are Potent and Selective Inhibitors of DNA Methyltransferase 1. Molecules. 2022; 27(9):2892. https://doi.org/10.3390/molecules27092892
Chicago/Turabian StyleMedina-Franco, José L., Edgar López-López, and Liliam P. Martínez-Fernández. 2022. "7-Aminoalkoxy-Quinazolines from Epigenetic Focused Libraries Are Potent and Selective Inhibitors of DNA Methyltransferase 1" Molecules 27, no. 9: 2892. https://doi.org/10.3390/molecules27092892
APA StyleMedina-Franco, J. L., López-López, E., & Martínez-Fernández, L. P. (2022). 7-Aminoalkoxy-Quinazolines from Epigenetic Focused Libraries Are Potent and Selective Inhibitors of DNA Methyltransferase 1. Molecules, 27(9), 2892. https://doi.org/10.3390/molecules27092892