
Quantum Computational Investigation of (E)-1-(4-methoxyphenyl)-5-methyl-N′-(3-phenoxybenzylidene)-1H-1,2,3-triazole-4-carbohydrazide

 , , ,
, , , 
Abstract
:1. Introduction
2. Results and Discussion
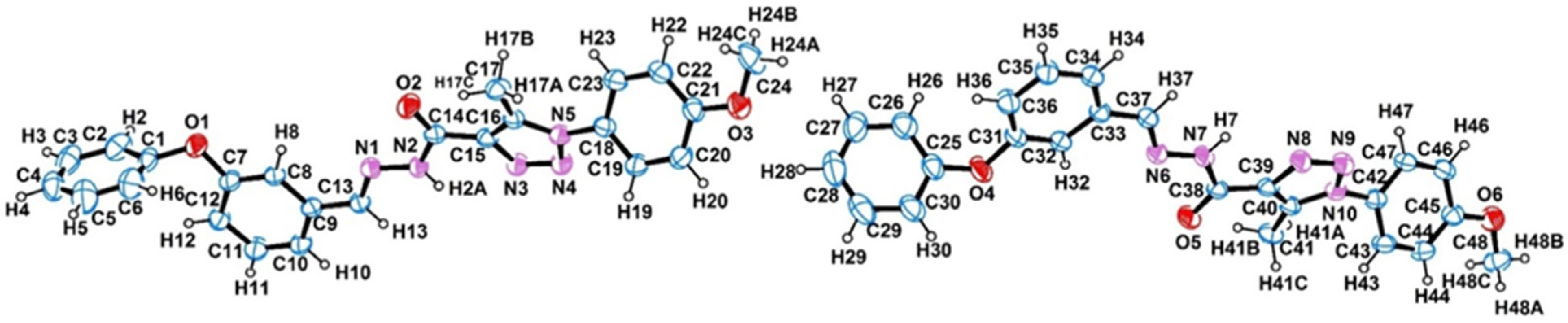
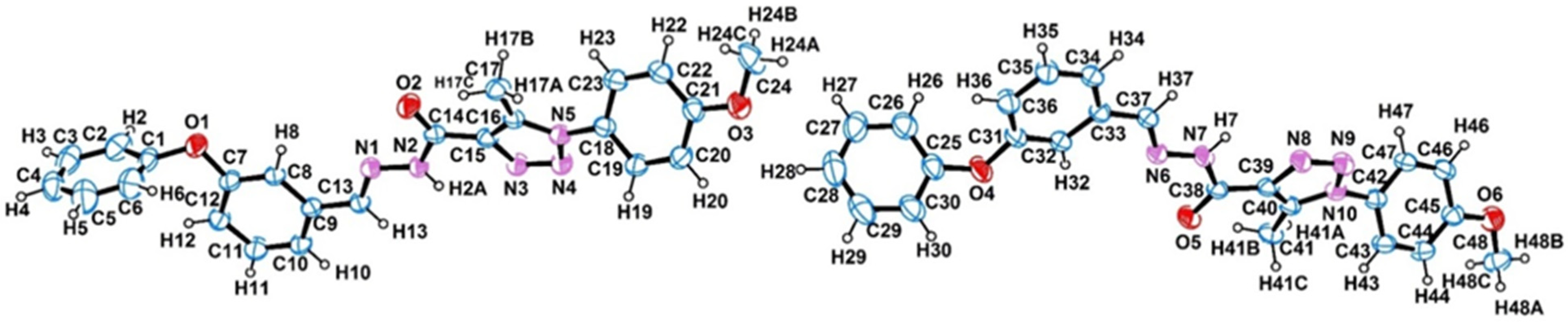
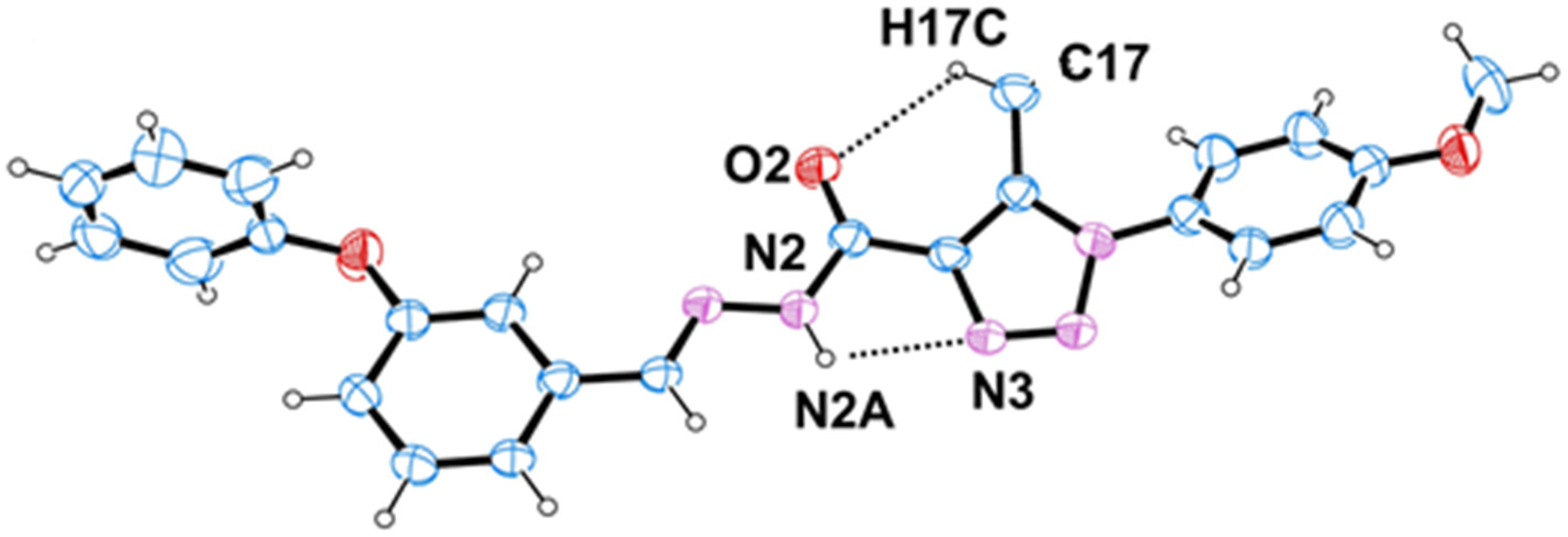
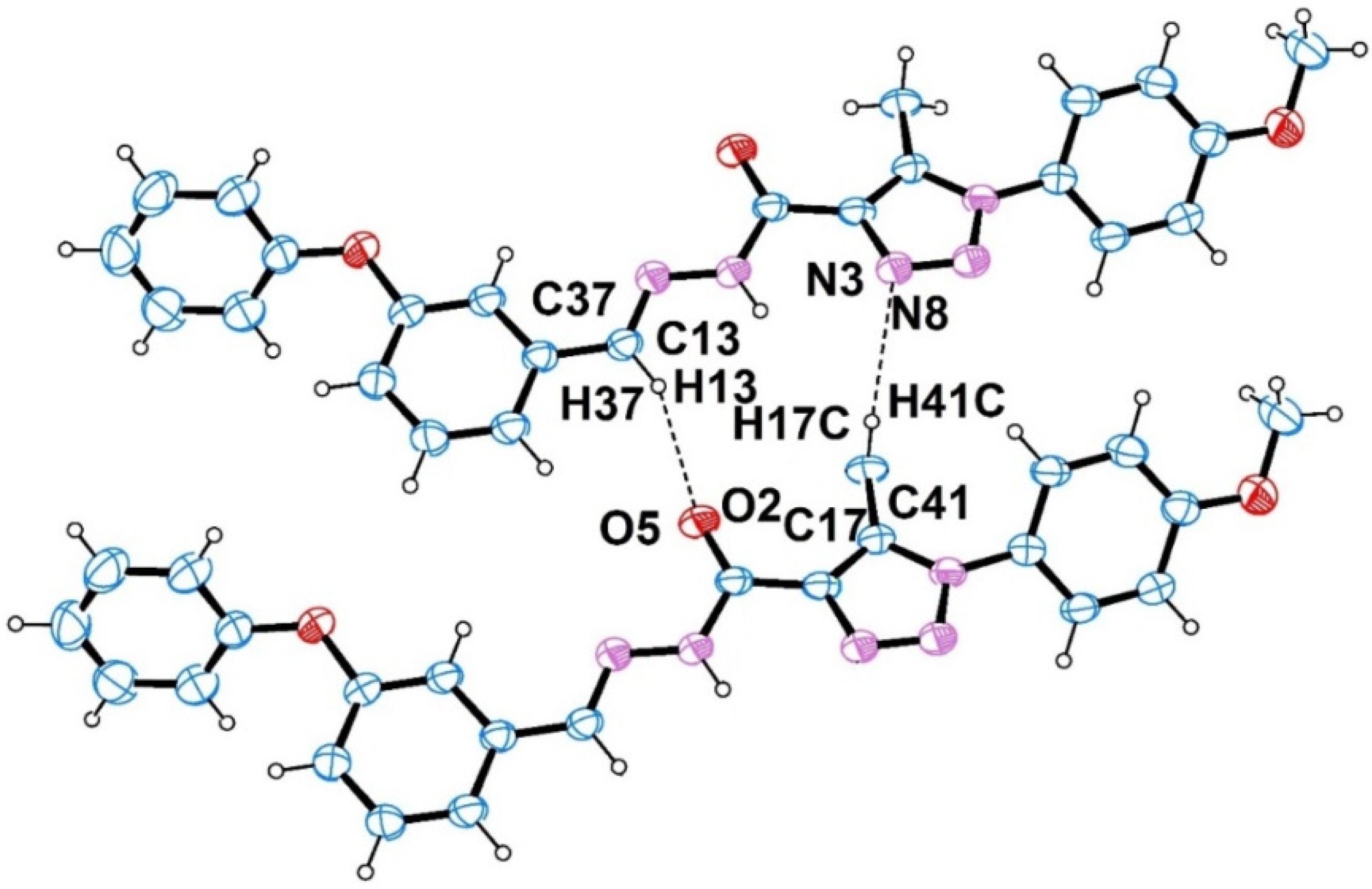
2.1. Crystal Structure
2.2. Surface Studies
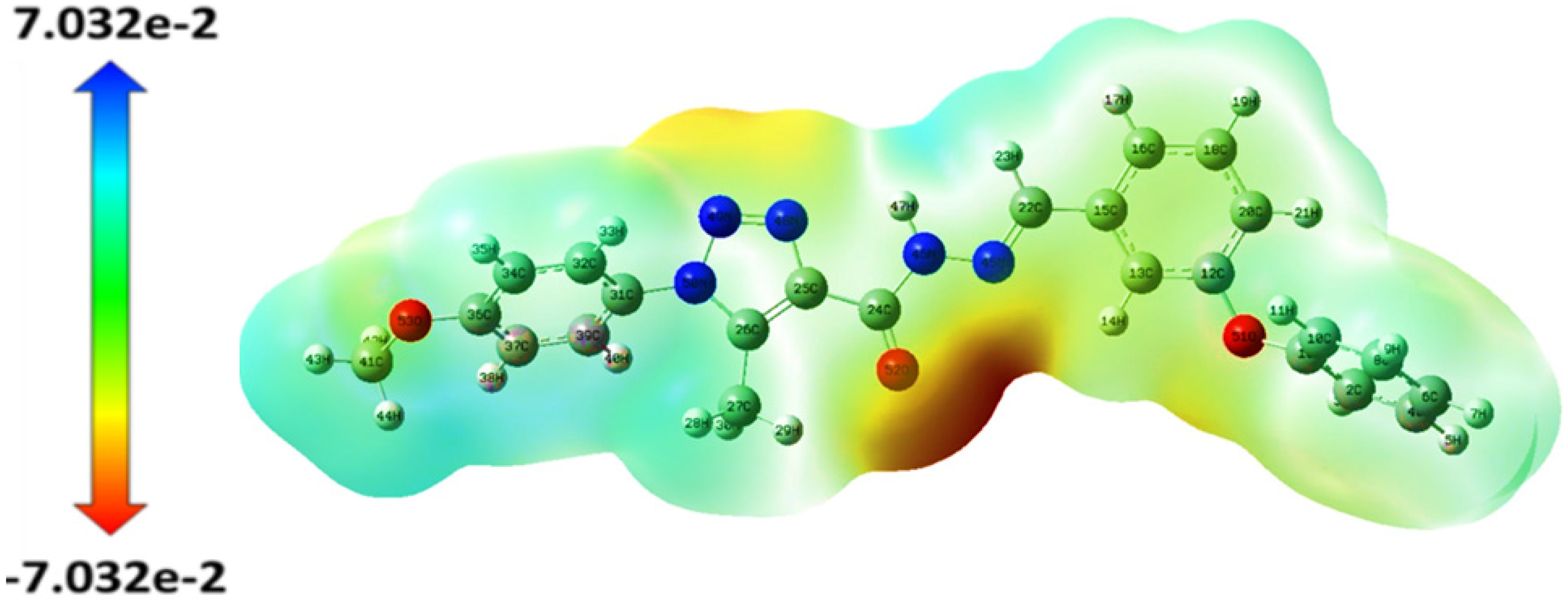
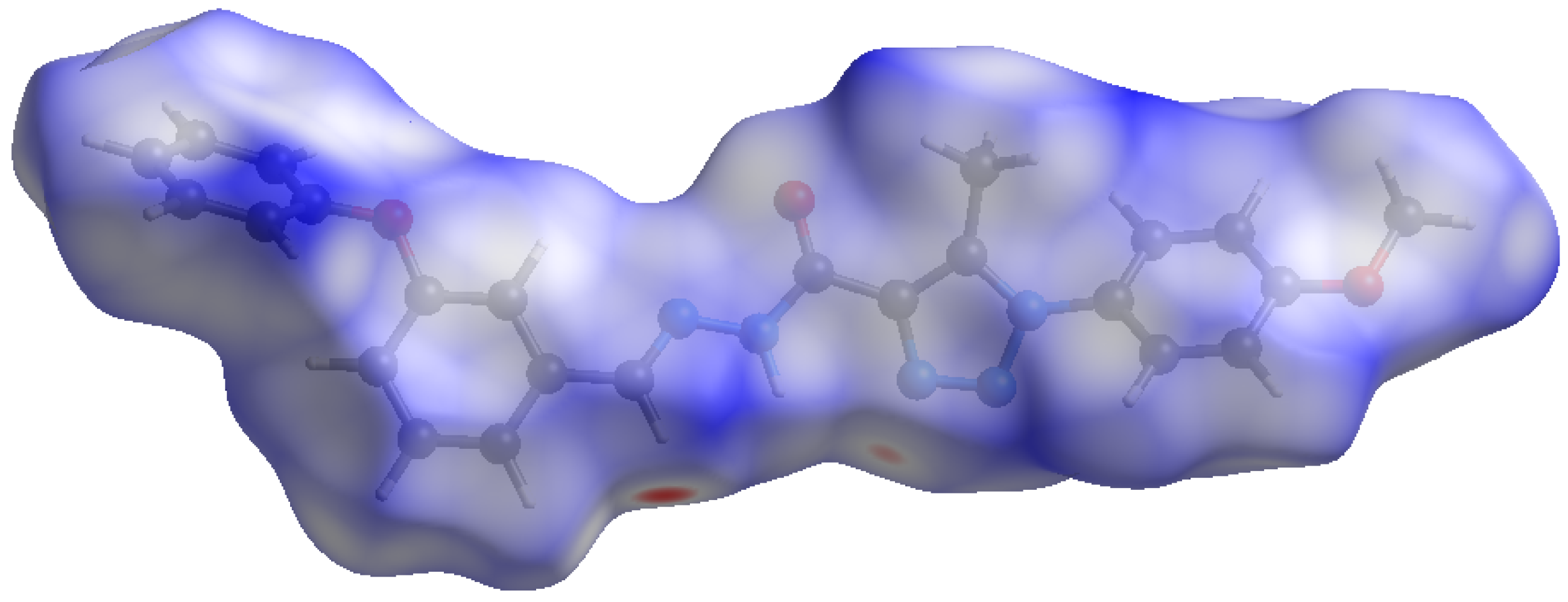
2.2.1. MEP Surface
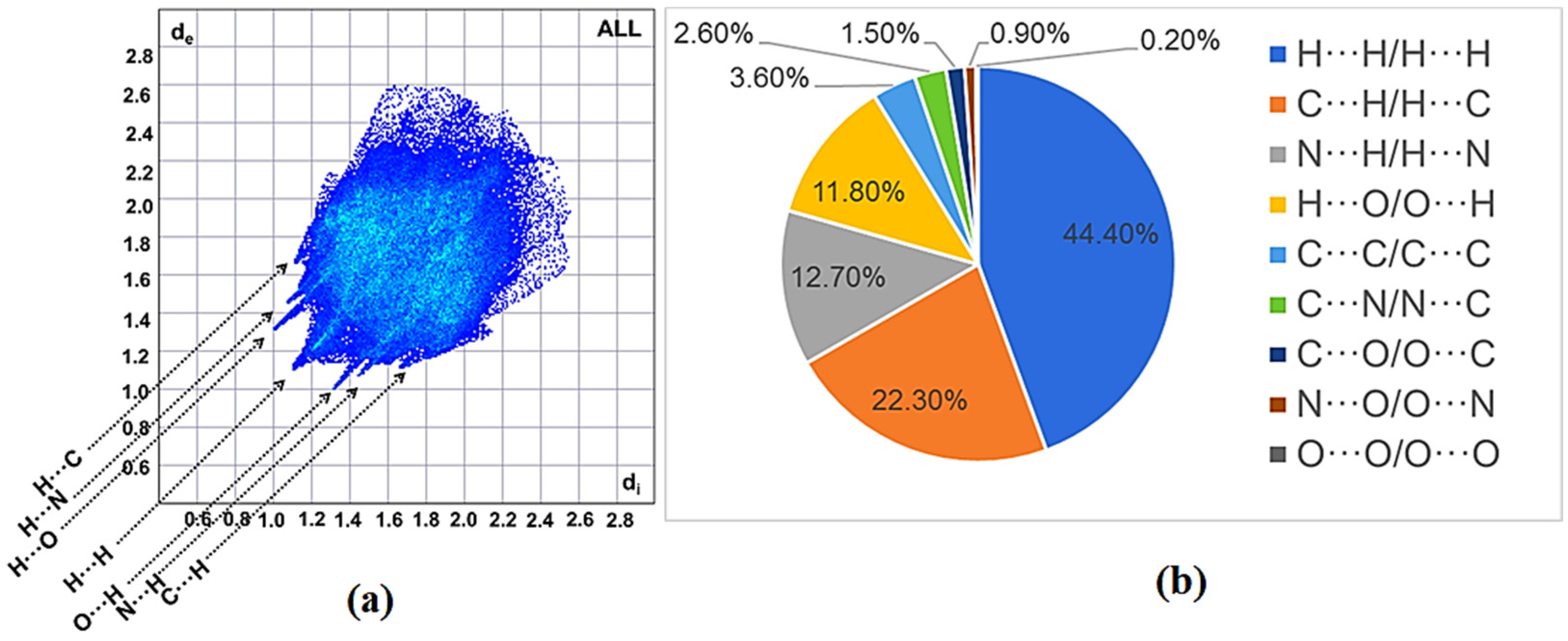
2.2.2. Hirshfeld Surface
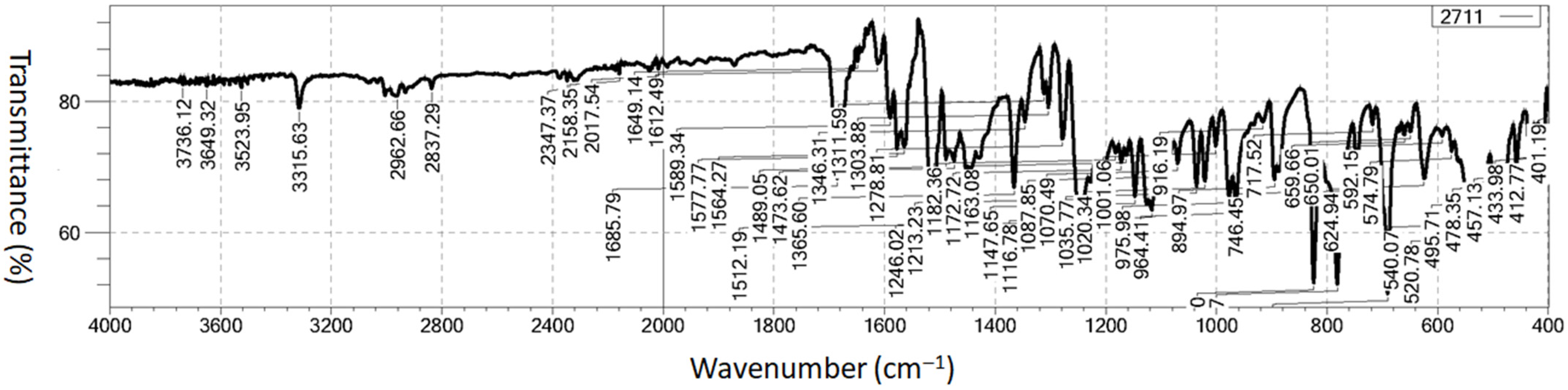
2.3. Vibrational Analysis
2.3.1. Aromatic C–H, Aliphatic (CH3) and Aromatic C–C Vibrations
2.3.2. Carbohydrazide (N–H, N–N, C=N, C=O) and Other C–O Group Vibrations
2.3.3. 1H-1,2,3-Triazole Ring Vibrations
2.4. NMR Chemical Shift Analyses
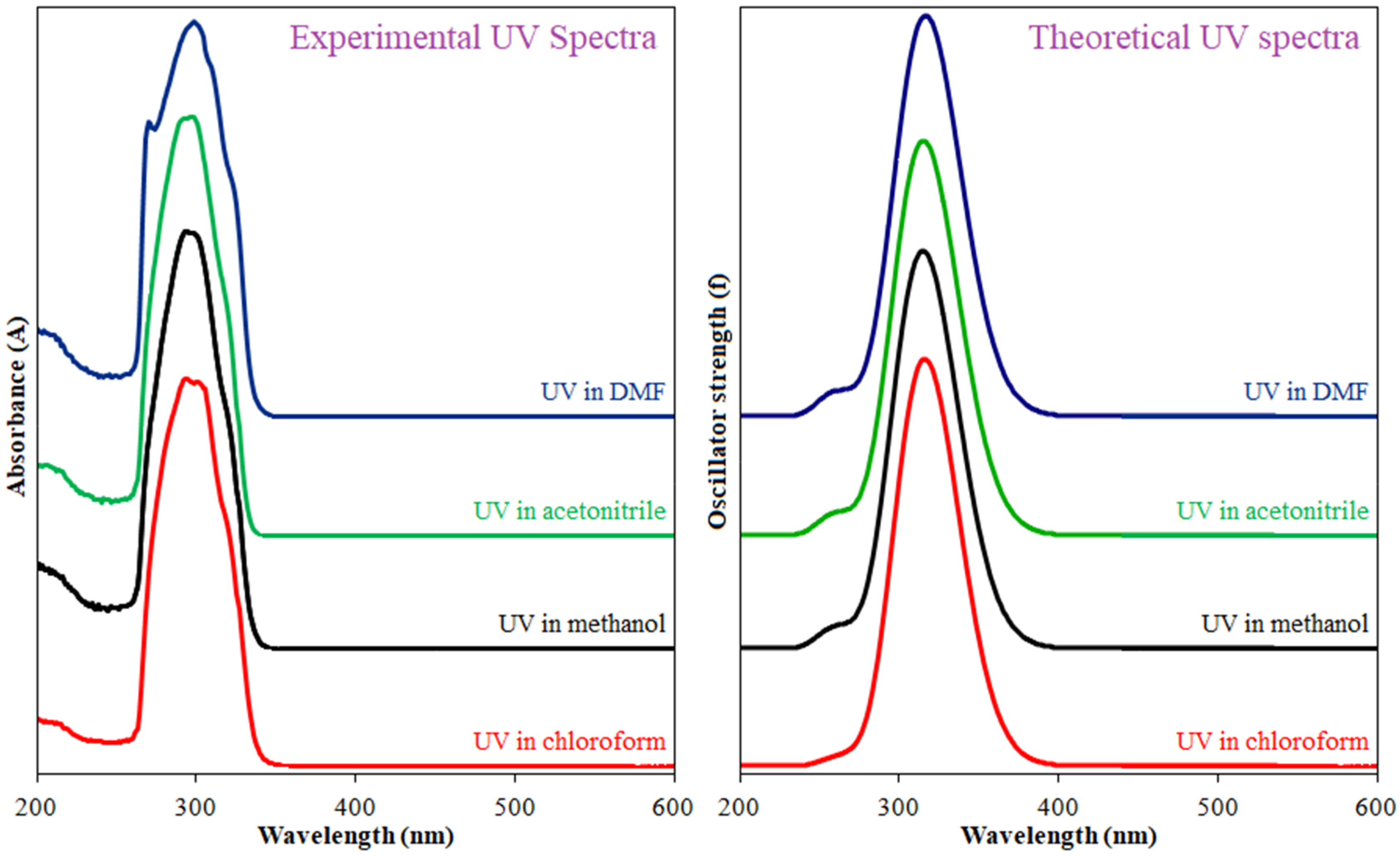
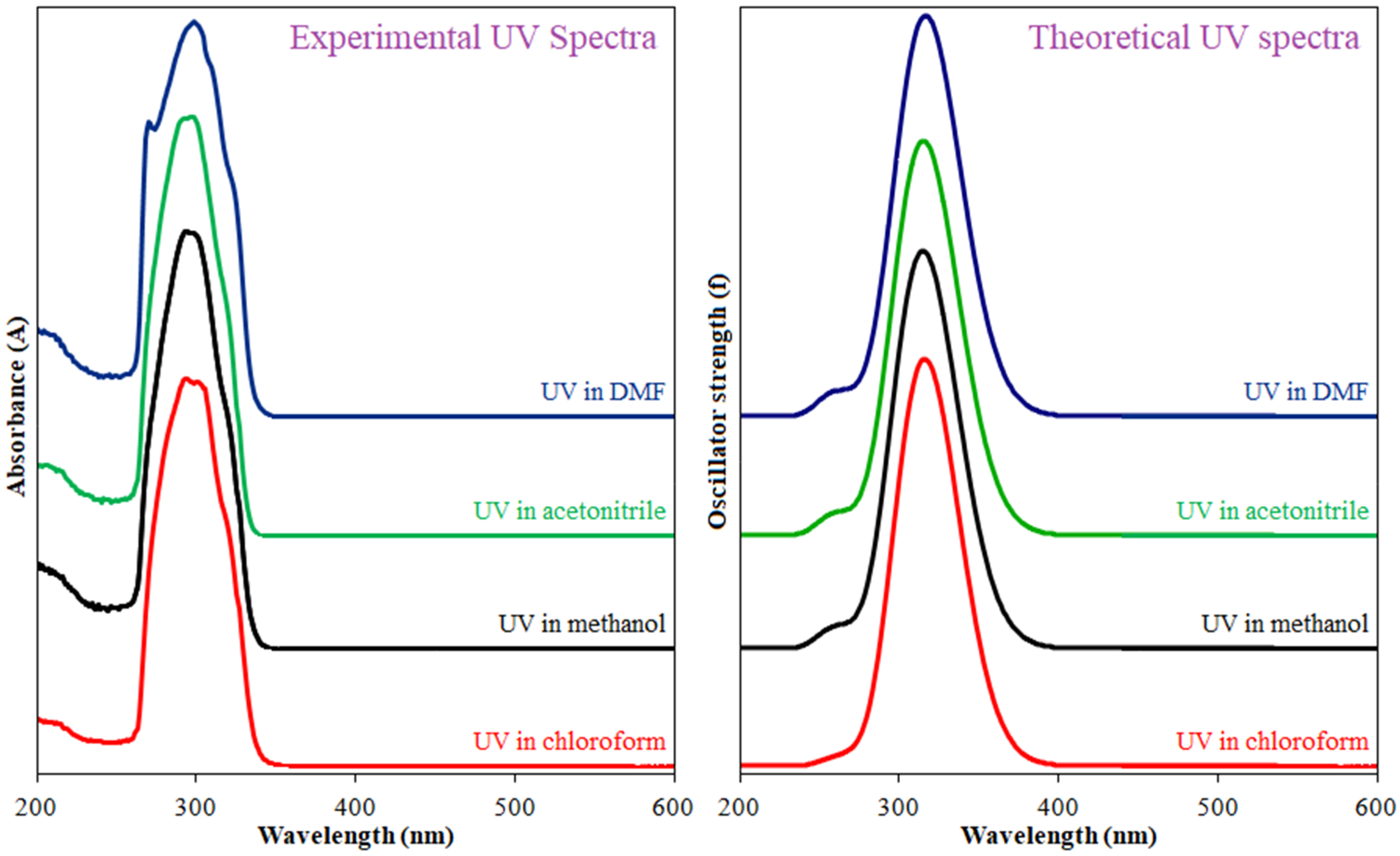
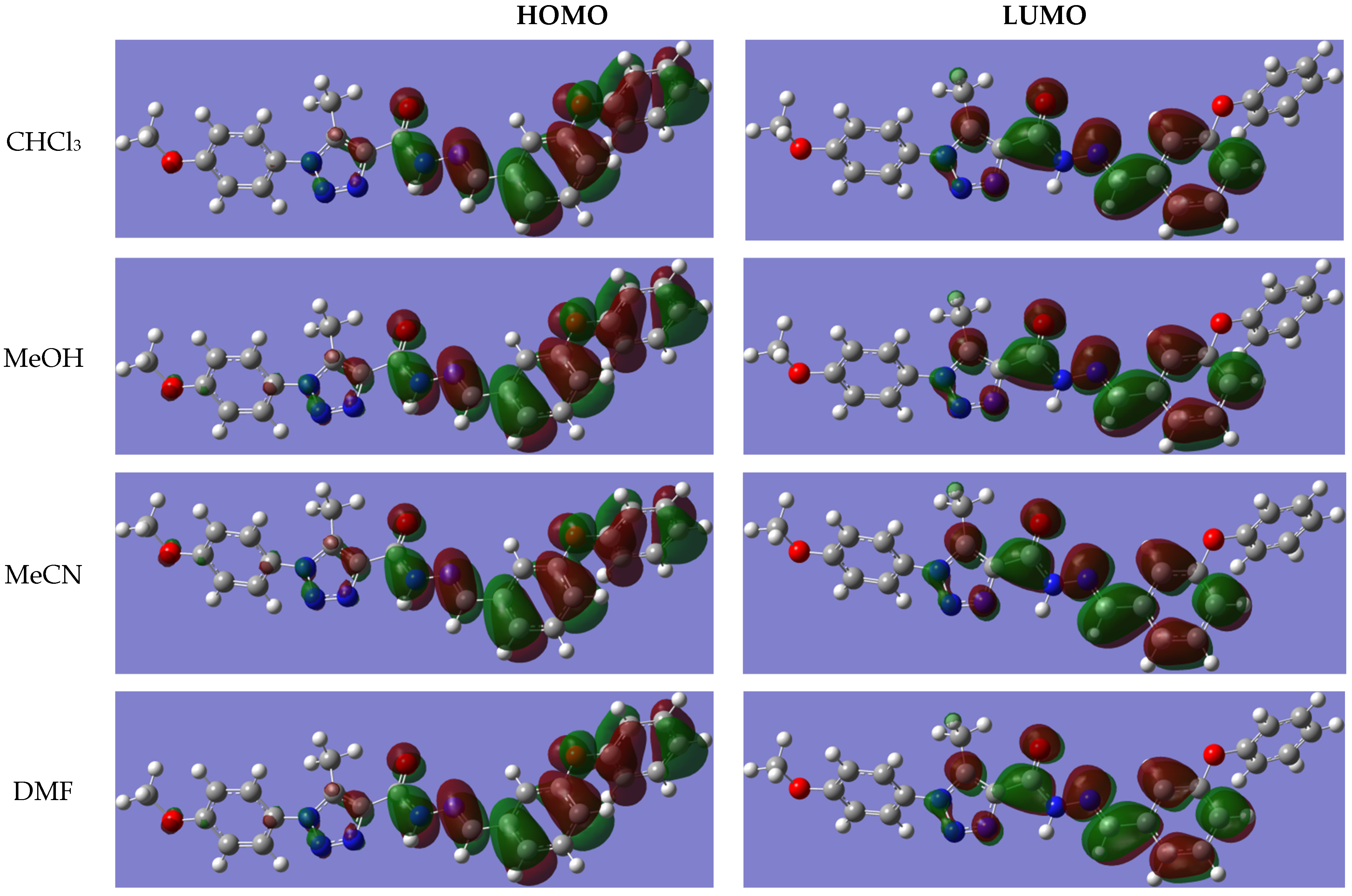
2.5. UV–Visible Spectrum and Frontier Orbital Analysis
2.6. Nonlinear Optical (NLO) Properties
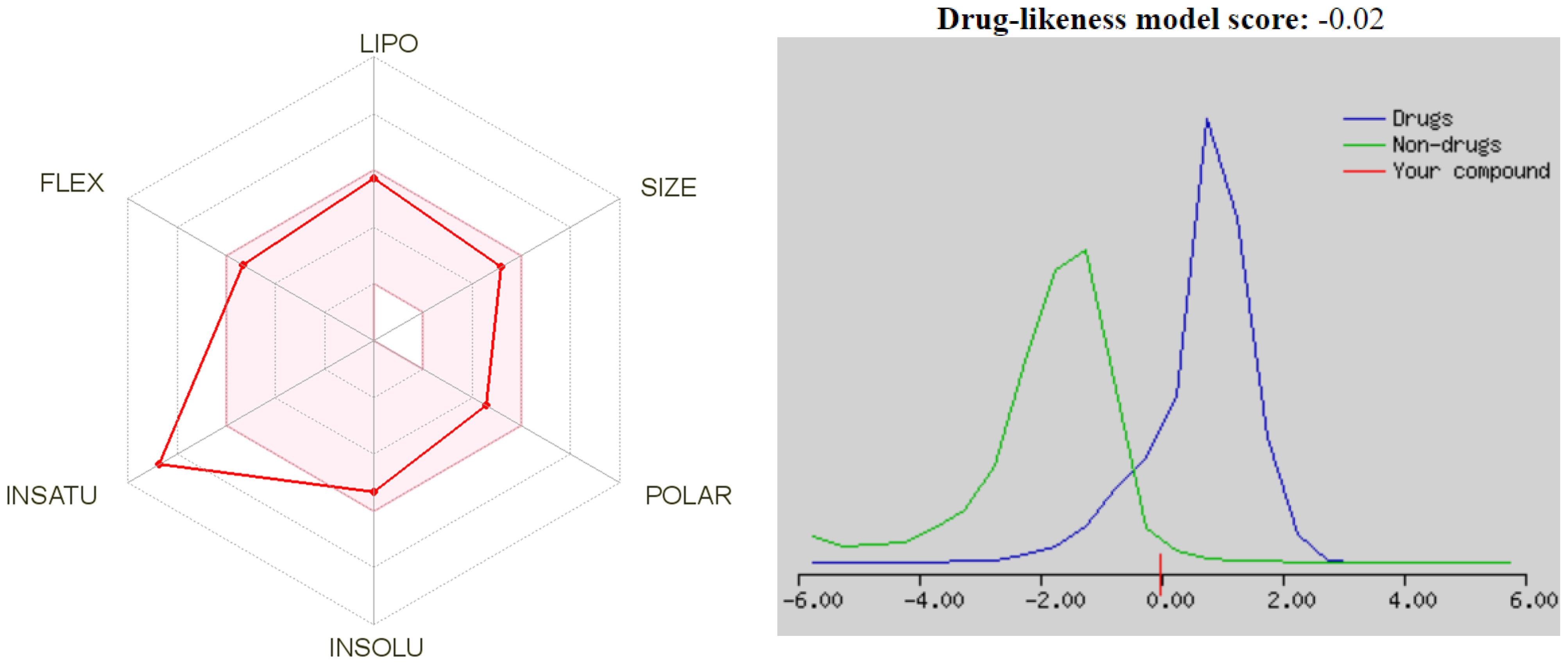
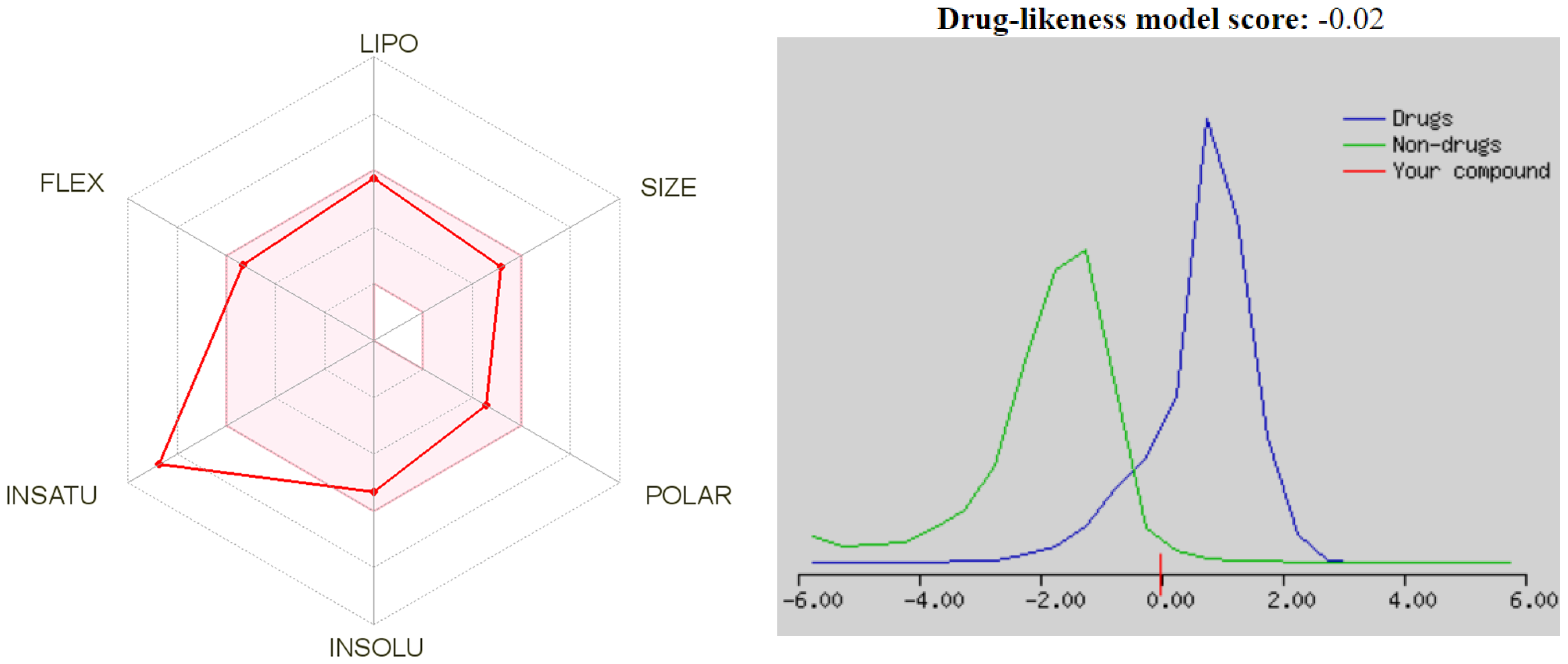
2.7. Drug-Likeness and ADME Studies
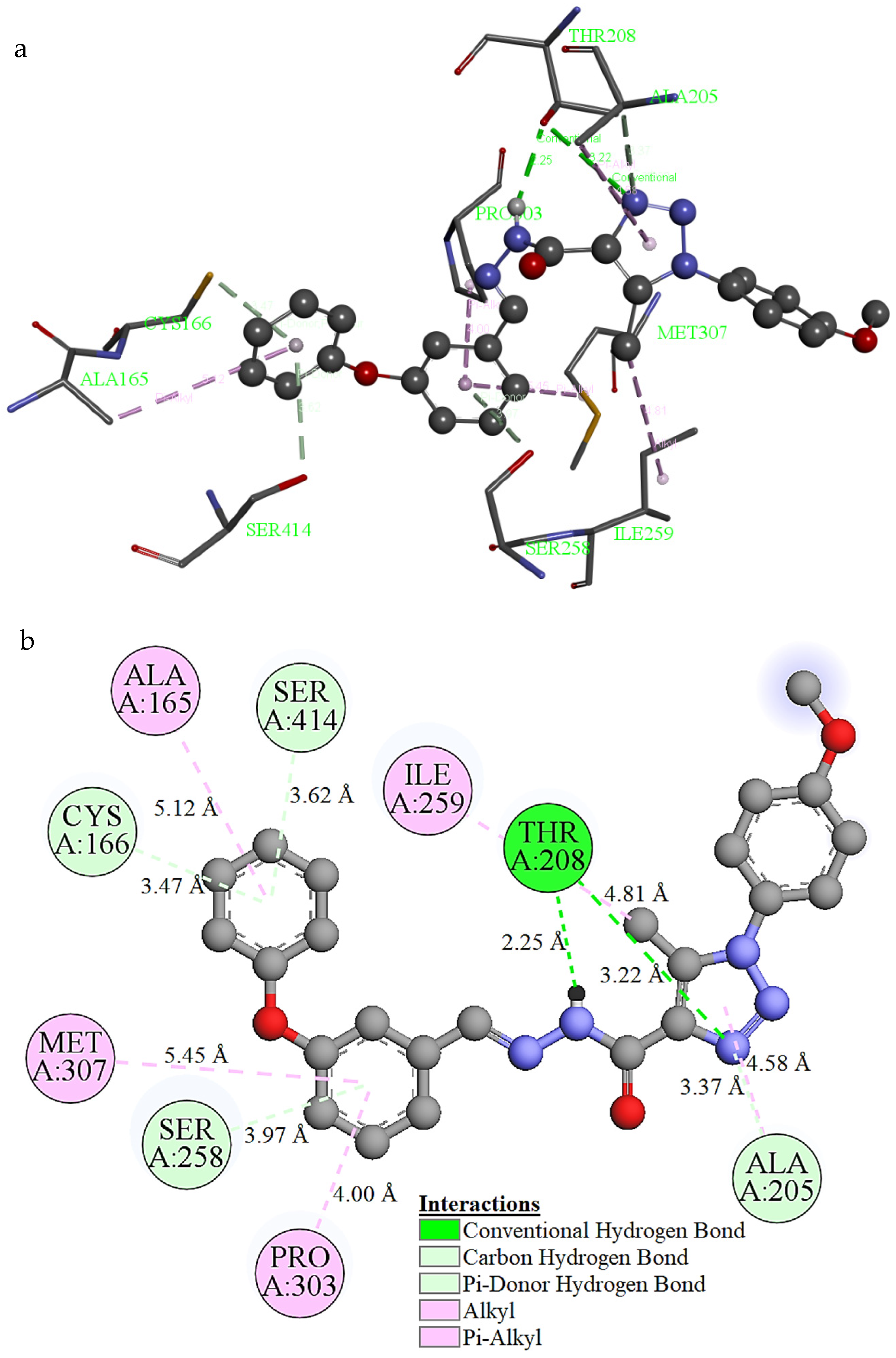
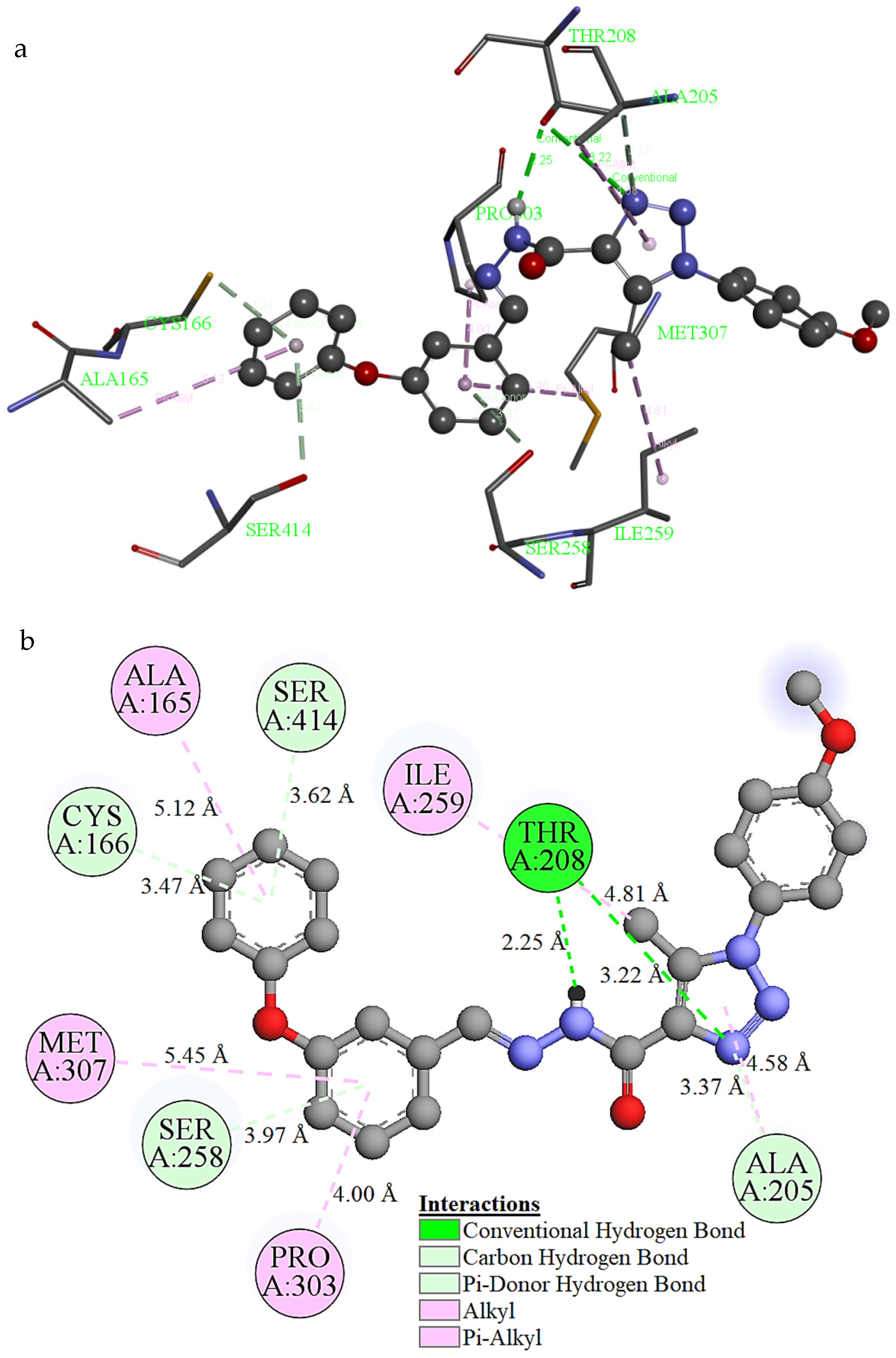
2.8. Molecular Docking Study
3. Materials and Methods
3.1. Instrumentation
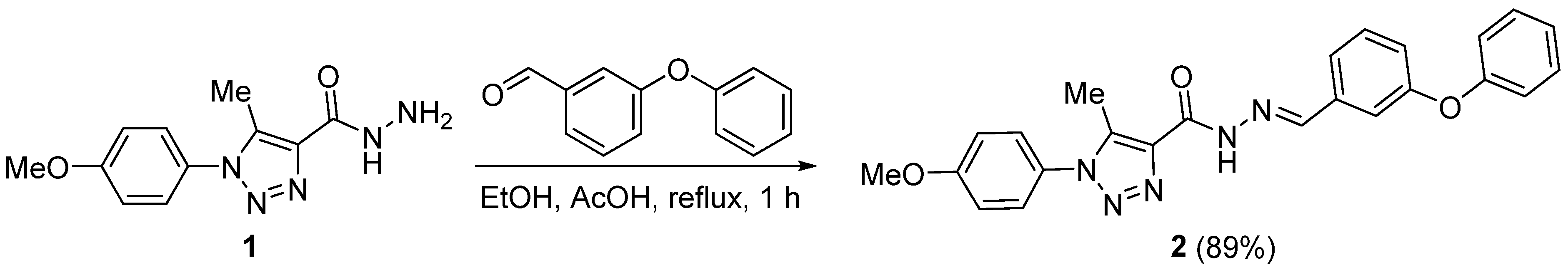
3.2. Synthesis of 2
3.3. Theoretical Details
3.4. Crystal Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- El Azab, I.H.; El-Sheshtawy, H.S.; Bakr, R.B.; Elkanzi, N.A.A. New 1,2,3-triazole-containing hybrids as antitumor candidates: Design, click reaction synthesis, DFT calculations, and molecular docking study. Molecules 2021, 26, 708. [Google Scholar] [CrossRef] [PubMed]
- Celik, F.; Unver, Y.; Barut, B.; Ozel, A.; Sancak, K. Synthesis, characterization and biological activities of new symmetric bis-1,2,3-triazoles with click chemistry. Med. Chem. 2018, 14, 230–241. [Google Scholar] [CrossRef]
- Hussaini, S.M.A.; Yedla, P.; Babu, K.S.; Shaik, T.B.; Chityal, G.K.; Kamal, A. Synthesis and biological evaluation of 1,2,3-triazole tethered pyrazoline and chalcone derivatives. Chem. Biol. Drug Des. 2016, 88, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Gibault, F.; Sturbaut, M.; Coevoet, M.; Pugnière, M.; Burtscher, A.; Allemand, F.; Melnyk, P.; Hong, W.; Rubin, B.P.; Pobbati, A.V.; et al. Design, synthesis and evaluation of a series of 1,5-diaryl-1,2,3-triazole-4-carbohydrazones as inhibitors of the YAP-TAZ/TEAD complex. ChemMedChem 2021, 16, 2823–2844. [Google Scholar] [CrossRef] [PubMed]
- Bokor, E.; Docsa, T.; Gergely, P.; Somsak, L. Synthesis of 1-(D-glucopyranosyl)-1,2,3-triazoles and their evaluation as glycogen phosphorylase inhibitors. Bioorgan. Med. Chem. 2010, 18, 1171–1180. [Google Scholar] [CrossRef]
- Kuntala, N.; Mareddy, J.; Telu, J.R.; Banothu, V.; Pal, S.; Anireddy, J.S. Nonsteroidal anti-inflammatory drugs based new 1,2,3-triazole derivatives: Their design, one-pot synthesis and in vitro evaluation. J. Heterocycl. Chem. 2021, 58, 2018–2032. [Google Scholar] [CrossRef]
- Haroun, M.; Tratrat, C.; Kochkar, H.; Nair, A.B. Recent advances in the development of 1,2,3-triazole-containing derivatives as potential antifungal agents and inhibitors of lanoster ol 14α-demethylase. Curr. Top. Med. Chem. 2021, 21, 462–506. [Google Scholar] [CrossRef]
- Tripathi, R.P.; Yadav, A.K.; Arya, A.; Bisht, S.S.; Chaturvedi, V.; Sinha, S.K. Application of Huisgen (3 + 2) cycloaddition reaction: Synthesis of 1-(2,3-dihydrobenzofuran-2-yl-methyl [1,2,3]-triazoles and their antitubercular evaluations. Eur. J. Med. Chem. 2010, 45, 142–148. [Google Scholar] [CrossRef]
- Zhao, X.; Lu, B.W.; Lu, J.R.; Xin, C.W.; Li, J.F.; Liu, Y. Design, synthesis and antimicrobial activities of 1,2,3-triazole derivatives. Chin. Chem. Lett. 2012, 23, 933–935. [Google Scholar] [CrossRef]
- Zhou, L.; Adel, A.; Korn, M.; Burda, R.; Balzarini, J.; Clercq, E.; Kern, E.R.; Torrence, P.F. Synthesis and antiviral activities of 1,2,3-triazole functionalized thymidines: 1,3-dipolar cycloaddition for efficient regioselective diversity generation. Antivir. Chem. Chemother. 2005, 16, 375–383. [Google Scholar] [CrossRef]
- Humphries, R.M.; Hindler, J.A.; Wong-Beringer, A.; Miller, S.A. Activity of ceftolozane-tazobactam and ceftazidime-avibactam against beta-lactam-resistant Pseudomonas aeruginosa isolates. Antimicrob. Agents Chemother. 2017, 61, e01858-17. [Google Scholar] [CrossRef] [Green Version]
- Corapcioglu, F.; Sarper, N.; Zengin, E. Monotherapy with piperacillin/tazobactam versus cefepime as empirical therapy for febrile neutropenia in pediatric cancer patients: A randomized comparison. Pediatr. Hematol. Oncol. 2006, 23, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Bonnefond, M.-L.; Florent, R.; Lambert, B.; Abeilard, E.; Giffard, F.; Louis, M.-H.; Elie, N.; Briand, M.; Vivien, D.; Poulain, L.; et al. Inhibition of store-operated channels by carboxyamidotriazole sensitizes ovarian carcinoma cells to anti-BclxL strategies through Mcl-1 down-regulation. Oncotarget 2018, 9, 33896–33911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmaja, R.D.; Chanda, K. A short review on synthetic advances toward the synthesis of rufinamide, an antiepileptic drug. Org. Process Res. Dev. 2018, 22, 457–466. [Google Scholar] [CrossRef]
- Jiang, Y.; Kuang, C.; Yang, Q. The use of calcium carbide in the synthesis of 1-monosubstituted aryl 1,2,3-triazole via click chemistry. Synlett 2009, 20, 3163–3166. [Google Scholar] [CrossRef]
- Shu, W.-M.; Zhang, X.-F.; Zhang, X.-X.; Li, M.; Wang, A.-J.; Wu, A.-X. Metal-free cascade [4 + 1] cyclization access to 4-aryl-NH-1,2,3-triazoles from N-tosylhydrazones and sodium azide. J. Org. Chem. 2019, 84, 14919–14925. [Google Scholar] [CrossRef]
- Zhang, H.; Dong, D.-Z.; Wang, Z.-L. Direct synthesis of N-unsubstituted 4-aryl-1,2,3-triazoles mediated by Amberlyst-15. Synthesis 2016, 48, 131–135. [Google Scholar] [CrossRef]
- Barluenga, J.; Valdés, C.; Beltrán, G.; Escribano, M.; Aznar, F. Developments in Pd catalysis: Synthesis of 1H-1,2,3-triazoles from sodium azide and alkenyl bromides. Angew. Chem. Int. Ed. 2006, 45, 6893–6896. [Google Scholar] [CrossRef] [PubMed]
- Banday, A.H.; Hruby, V.J. Regioselective N/C-heterocyclization of allenylindium bromide across aryl azides: One-pot synthesis of 5-methyl-1,2,3-triazoles. Synlett 2014, 25, 1859–1862. [Google Scholar] [CrossRef]
- Sert, Y.; Balakit, A.A.; Öztürk, N.; Ucun, F.; El-Hiti, G.A. Experimental (FT-IR, NMR and UV) and theoretical (M06–2X and DFT) investigation, and frequency estimation analyses on (E)-3-(4-bromo-5-methylthiophen-2-yl)acrylonitrile. Spectrochim. Acta A 2014, 131, 502–511. [Google Scholar] [CrossRef]
- Sert, Y.; Ucun, F.; El-Hiti, G.A.; Smith, K.; Hegazy, A.S. Spectroscopic investigations and DFT calculations on 3-(diacetylamino)-2-ethyl-3H-quinazolin-4-one. J. Spectrosc. 2016, 2016, 5396439. [Google Scholar] [CrossRef] [Green Version]
- Balakit, A.A.; Sert, Y.; Çırak, Ç.; Smith, K.; El-Hiti, G.A. Synthesis, spectroscopic (FT-IR and Laser-Raman) and theoretical DFT simulations of 3-bromo-2-methyl-5-(4-nitrophenyl)thiophene. J. Appl. Spectrosc. 2017, 84, 888–899. [Google Scholar] [CrossRef]
- Sert, Y.; El-Hiti, G.A.; Gökce, H.; Ucun, F.; Abdel-Wahab, B.F.; Kariuki, B.M. DFT, molecular docking and experimental FT-IR, laser-Raman, NMR and UV investigations on a potential anticancer agent containing triazole ring system. J. Mol. Struct. 2020, 1211, 128077. [Google Scholar] [CrossRef]
- Romani, D.; Noureddine, O.; Issaoui, N.; Brandán, S.A. Properties and reactivities of niclosamide in different media, a potential antiviral to treatment of COVID-19 by using DFT calculations and molecular docking. Biointerface Res. Appl. Chem. 2020, 10, 7295–7328. [Google Scholar] [CrossRef]
- Gatfaoui, S.; Sagaama, A.; Issaoui, N.; Roisnel, T.; Marouani, H. Synthesis, experimental, theoretical study and molecular docking of 1-ethylpiperazine-1,4-diium bis(nitrate). Solid State Sci. 2020, 106, 106326. [Google Scholar] [CrossRef]
- Pokhodylo, N.T.; Slyvka, Y.; Pavlyuk, V. Synthesis, crystal structure and Hirshfeld surface analysis of 5-cyclopropyl-N-(2-hydroxyethyl)-1-(4-methylphenyl)-1H-1,2,3-triazole-4-carboxamide. Acta Cryst. 2021, E77, 1043–1047. [Google Scholar] [CrossRef]
- Çelik, F.; Ustabaş, R.; Süleymanoğlu, N.; Direkel, Ş.; Güler, H.İ.; Ünver, Y. 1,2,3-triazole derivative: Synthesis, characterization, DFT, molecular docking study and antibacterial-antileishmanial activities. J. Indian Chem. Soc. 2021, 98, 100105. [Google Scholar] [CrossRef]
- Moreira, O.B.; Freitas, M.C.R.; Souza, K.C.; Jordão, A.K.; Resende, J.A. Crystal structure of 1-anilino-5-methyl-1H-1,2,3-triazole-4-carboxylic acid monohydrate. Acta Cryst. 2019, E75, 738–741. [Google Scholar] [CrossRef]
- Issaoui, N.; Rekik, N.; Oujia, B.; Wójcik, M.J. Theoretical infrared line shapes of H-bonds within the strong Anharmonic coupling theory and Fermi resonances effects. Int. J. Quantum Chem. 2010, 110, 2583–2602. [Google Scholar] [CrossRef]
- Yıldırım, A.Ö.; Yıldırım, M.H.; Kaştaş, Ç.A. Studies on the synthesis, spectroscopic analysis and DFT calculations on (E)-4, 6-dichloro-2-[(2-chlorophenylimino) methyl]-3-methoxyphenol as a novel Schiff’s base. J. Mol. Struct. 2016, 1113, 1–8. [Google Scholar] [CrossRef]
- İnkaya, E.; Dinçer, M.; Ekici, Ö.; Cukurovali, A. N′-(2-methoxy-benzylidene)-N-[4-(3-methyl-3-phenyl-cyclobutyl)-thiazol-2-yl]-chloro-acetic hydrazide: X-ray structure, spectroscopic characterization and DFT studies. J. Mol. Struct. 2012, 1026, 117–126. [Google Scholar] [CrossRef]
- Özdemir, N.; İnkaya, E.; Sarıpınar, E.; Akyüz, L.; İlhan, İ.Ö.; Aydın, S.; Dinçer, M.; Büyükgüngör, O. Synthesis, spectroscopic (FT-IR/NMR) characterization, X-ray structure and DFT studies on (E)-2-(1-phenylethylidene) hydrazinecarboximidamide nitrate hemimethanol. Spectrochim. Acta A 2013, 114, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, M. Parst: A system of fortran routines for calculating molecular structure parameters from results of crystal structure analyses. Comput. Chem. 1983, 7, 95–98. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView; Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Wolff, S.; Grimwood, D.; McKinnon, J.; Turner, M.; Jayatilaka, D.; Spackman, M. Crystal Explorer; Version 3.1; University of Western Australia: Perth, Australia, 2013. [Google Scholar]
- Katritzky, A. The infrared spectra of heteroaromatic compounds. Q. Rev. Chem. Soc. 1959, 13, 353–373. [Google Scholar] [CrossRef]
- Segneanu, A.E.; Gozescu, I.; Dabici, A.; Sfirloaga, P.; Szabadai, Z. Organic compounds FT-IR spectroscopy. In Macro to Nano Spectroscopy; Uddin, J., Ed.; Intech Open: London, UK, 2012; Chapter 9; pp. 145–164. [Google Scholar]
- Young, D. Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems; Wiley: New York, NY, USA, 2004. [Google Scholar]
- Jamroz, M.H. Vibrational Energy Distribution Analysis VEDA 4; Veda: Warsaw, Poland, 2004. [Google Scholar]
- Jamróz, M.H. Vibrational energy distribution analysis (VEDA): Scopes and limitations. Spectrochim. Acta A 2013, 114, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Varsányi, G.; Láng, L. Assignments for Vibrational Spectra of Seven Hundred Benzene Derivatives; Wiley: New York, NY, USA, 1974. [Google Scholar]
- Silverstein, R.; Webster, F. Spectroscopic Identification of Organic Compounds; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Sundaraganesan, N.; Meganathan, C.; Joshua, B.D.; Mani, P.; Jayaprakash, A. Molecular structure and vibrational spectra of 3-chloro-4-fluoro benzonitrile by ab initio HF and density functional method. Spectrochim. Acta A 2008, 71, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Karrouchi, K.; Brandán, S.A.; Sert, Y.; El-marzouqi, H.; Radi, S.; Ferbinteanu, M.; Faouzi, M.E.A.; Garcia, Y.; Ansar, M. Synthesis, X-ray structure, vibrational spectroscopy, DFT, biological evaluation and molecular docking studies of (E)-N’-(4-(dimethylamino) benzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 2020, 1219, 128541. [Google Scholar] [CrossRef]
- Sagaama, A.; Issaoui, N.; Bardak, F.; Al-Dossary, O.; Kazachenko, A.S.; Karrouchi, K.; Atac, A.; Wojcik, M.J. Non covalent interactions analysis and spectroscopic characterization combined with molecular docking study of N’-(4-Methoxybenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. J. King Saud. Univ. Sci. 2021, 34, 101778. [Google Scholar] [CrossRef]
- Zimmermann, F.; Lippert, T.; Beyer, C.; Stebani, J.; Nuyken, O.; Wokaun, A. N=N vibrational frequencies and fragmentation patterns of substituted 1-aryl-3,3-dialkyl-triazenes: Comparison with other high-nitrogen compounds. Appl. Spectrosc. 1993, 47, 986–993. [Google Scholar] [CrossRef]
- Yusuf, S. Vibrational, geometrical and HOMO/LUMO/MEP analyses by using DFT/B3LYP and DFT/M06-2X methods: 3-Amino-1,2,4-triazole. Pamukkale Üniv. Mühendis. Bilimleri Derg. 2018, 24, 1272–1277. [Google Scholar] [CrossRef] [Green Version]
- Gökce, H.; Öztürk, N.; Ceylan, Ü.; Alpaslan, Y.B.; Alpaslan, G. Thiol-thione tautomeric analysis, spectroscopic (FT-IR, Laser-Raman, NMR and UV-vis) properties and DFT computations of 5-(3-pyridyl)-4H-1,2,4-triazole-3-thiol molecule. Spectrochim. Acta A 2016, 163, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.J.; Bendell, D.J.; Groundwater, P.W. Organic Spectroscopic Analysis; Royal Society of Chemistry: London, UK, 2004. [Google Scholar] [CrossRef]
- Lambert, J.B.; Shurvell, H.F.; Lightner, D.A.; Cooks, R.G. Introduction to Organic Spectroscopy, 1st ed.; Macmillan Publishing Company: New York, NY, USA, 1987. [Google Scholar]
- Pavia, D.; Lampman, G.; Kriz, G.S.; Vyvyan, J.R. Introduction to Spectroscopy, 4th ed.; Brooks/Cole Publishing: Belmont, CA, USA, 2009; pp. 70–71. [Google Scholar]
- Cordeiro, N.M.; Freitas, R.H.C.N.; Fraga, C.A.M.; Fernandes, P.D. Discovery of novel orally active tetrahydronaphthyl-N-acylhydrazones with in vivo anti-TNF-α effect and remarkable anti-inflammatory properties. PLoS ONE 2016, 11, e0156271. [Google Scholar] [CrossRef] [PubMed]
- O’boyle, N.M.; Tenderholt, A.L.; Langner, K.M. Cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Fukui, K. Role of frontier orbitals in chemical reactions. Science 1982, 218, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Zhan, C.-G.; Nichols, J.A.; Dixon, D.A. Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: Molecular properties from density functional theory orbital energies. J. Phys. Chem. A 2003, 107, 4184–4195. [Google Scholar] [CrossRef] [Green Version]
- Geerlings, P.; de Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness: Applications to organic chemistry. J. Org. Chem. 1989, 54, 1423–1430. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Öztürk, N.; Özdemir, T.; Alpaslan, Y.B.; Gökce, H.; Alpaslan, G. Experimental (FT-IR, Raman and NMR) and theoretical (B3LYP, B3PW91, M06–2X and CAM-B3LYP) analyses of p-tert-butylphenyl salicylate. Bilge Int. J. Sci. Technol. Res. 2018, 2, 56–73. [Google Scholar] [CrossRef]
- Verbiest, T.; Houbrechts, S.; Kauranen, M.; Clays, K.; Persoons, A. Second-order nonlinear optical materials: Recent advances in chromophore design. J. Mater. Chem. 1997, 7, 2175–2189. [Google Scholar] [CrossRef]
- Wolff, J.J.; Wortmann, R. Organic materials for second-order non-linear optics. Adv. Phys. Org. Chem. 1999, 32, 121–217. [Google Scholar] [CrossRef]
- Cassidy, C.; Halbout, J.M.; Donaldson, W.; Tang, C.L. Nonlinear optical properties of urea. Opt. Commun. 1979, 29, 243–246. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- SwissADME. Swiss Institute of Bioinformatics. Available online: http://www.swissadme.ch/ (accessed on 1 December 2021).
- Molsoft: Molecules in Silico. Available online: https://molsoft.com/index.html (accessed on 2 December 2021).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–26. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Comp. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Way2Drug, PASS Online. Available online: http://www.way2drug.com/passonline/ (accessed on 2 December 2021).
- Shafqat, N.; Turnbull, A.; Zschocke, J.; Oppermann, U.; Yue, W.W. Crystal structures of human HMG-CoA synthase isoforms provide insights into inherited ketogenesis disorders and inhibitor design. J. Mol. Biol. 2010, 398, 497–506. [Google Scholar] [CrossRef] [PubMed]
- RCSB Protein Data Bank: A Structural View of Biology. Available online: https://www.rcsb.org/ (accessed on 2 December 2021).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BS BIOVIA: Model the Biosphere. Available online: https://www.3dsbiovia.com/ (accessed on 2 December 2021).
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; The University of Western Australia: Crawley, WA, Australia, 2017. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D–H···A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| N2–H2A···N3 | 0.86 | 2.39 | 2.763 (5) | 107 |
| N7–H7···N8 | 0.86 | 2.38 | 2.752 (5) | 107 |
| C17–H17B···O2 | 0.96 | 2.55 | 3.105 (7) | 117 |
| C41–H41B···O5 | 0.96 | 2.59 | 3.133 (7) | 116 |
| C13–H13···O2 i | 0.93 | 2.45 | 3.316 (5) | 155 |
| C41–H41C···N8i | 0.96 | 2.62 | 3.444 (6) | 144 |
| C37–H37···O5 ii | 0.93 | 2.45 | 3.334 (5) | 158 |
| C17–H17C···N3 ii | 0.96 | 2.65 | 3.486 (6) | 147 |
| Structural Parameters | X-ray Diffraction | DFT/B3LYP/6-31++G(d,p) |
|---|---|---|
| Bond Distances (Å) | ||
| C1–O1 | 1.392 (6) | 1.38203 |
| C7–O1 | 1.388 (6) | 1.38124 |
| C9–C13 | 1.441 (6) | 1.46417 |
| C13–N1 | 1.287 (5) | 1.28080 |
| N1–N2 | 1.369 (5) | 1.35305 |
| N2–C14 | 1.363 (5) | 1.38069 |
| C14–O2 | 1.215 (5) | 1.21697 |
| C14–C15 | 1.463 (6) | 1.48180 |
| C15–N3 | 1.363 (5) | 1.36541 |
| N3–N4 | 1.303 (5) | 1.29101 |
| N4–N5 | 1.372 (5) | 1.36720 |
| N5–C18 | 1.427 (6) | 1.42794 |
| C21–O3 | 1.343 (6) | 1.35941 |
| O3–C24 | 1.425 (8) | 1.42349 |
| Bond angles (°) | ||
| C1–O1–C7 | 117.3 (4) | 120.61383 |
| C9–C13–N1 | 120.9 (4) | 121.94730 |
| C13–N1–N2 | 115.0 (4) | 117.12147 |
| N1–N2–C14 | 120.8 (4) | 121.49785 |
| N2–C14–O2 | 124.0 (4) | 124.86186 |
| N2–C14–C15 | 113.4 (4) | 112.05835 |
| C14–C15–C16 | 127.9 (4) | 128.42862 |
| C16–N5–C18 | 130.5 (4) | 129.54940 |
| N4–N5–C18 | 118.6 (4) | 119.44218 |
| C21–O3–C24 | 117.2 (5) | 118.83652 |
| Torsion angles (°) | ||
| C1–O1–C7–C12 | 35.0 (8) | 41.52414 |
| O2–C14–C15–C16 | 14.0 (8) | 2.42954 |
| C16–N5–C18–C19 | 137.2 (5) | 128.19985 |
| O3–C21–C22–C23 | 177.2 (5) | 179.59354 |
| O2–C14–N2–N1 | 1.6 (8) | 1.64013 |
| Atom | δexp. | δcalc. | Atom | δexp. | δcalc. |
|---|---|---|---|---|---|
| C1 | 156.76 | 168.30 | H3 | 7.18 | 7.45 |
| C2 | 119.47 | 127.10 | H5 | 7.44 | 7.49 |
| C4 | 130.71 | 137.00 | H7 | 7.21 | 7.31 |
| C6 | 124.32 | 130.10 | H9 | 7.44 | 7.51 |
| C8 | 130.71 | 136.70 | H11 | 7.18 | 7.11 |
| C10 | 119.47 | 124.50 | H14 | 7.57 | 8.32 |
| C12 | 157.77 | 167.30 | H17 | 7.50 | 7.38 |
| C13 | 115.98 | 122.10 | H19 | 7.33 | 7.51 |
| C15 | 137.44 | 144.80 | H21 | 7.15 | 7.02 |
| C16 | 123.23 | 133.10 | H23 | 8.58 | 8.21 |
| C18 | 128.46 | 137.50 | H28 | 2.11 | 2.00 |
| C20 | 120.75 | 126.30 | H29 | 2.11 | 3.27 |
| C22 | 147.68 | 152.40 | H30 | 2.11 | 2.37 |
| C24 | 157.78 | 164.00 | H33 | 7.60 | 7.53 |
| C25 | 136.92 | 145.10 | H35 | 7.10 | 7.28 |
| C26 | 138.53 | 149.20 | H38 | 7.10 | 7.07 |
| C27 | 9.89 | 12.40 | H40 | 7.60 | 7.40 |
| C31 | 131.10 | 136.20 | H42 | 3.86 | 3.82 |
| C32 | 127.42 | 135.40 | H43 | 3.86 | 4.16 |
| C34 | 115.22 | 125.30 | H44 | 3.86 | 3.84 |
| C36 | 160.68 | 170.00 | H47 | 12.18 | 10.29 |
| C37 | 115.22 | 115.20 | |||
| C39 | 127.42 | 134.80 | |||
| C41 | 55.85 | 58.60 |
| Solvent | λexp. (nm) | Transitions | λcalc. (nm) | f | ΔE (eV) | Computed Major Transitions |
|---|---|---|---|---|---|---|
| CHCl3 | 293 and 301 | π→π* | 327.30 | 0.6290 | 3.7881 | H→L (93%) |
| π→π* | 308.20 | 0.7463 | 4.0228 | H-1→L (90%) | ||
| π→π* | 287.25 | 0.0088 | 4.3163 | H-2→L (94%) | ||
| π→π* | 271.87 | 0.0001 | 4.5604 | H-6→L (44%), H-7→L (35%) | ||
| π→π* | 271.00 | 0.0078 | 4.5750 | H-3→L (72%) | ||
| π→π* | 263.06 | 0.0236 | 4.7132 | H→L + 1 (83%), H-1→L + 1 (12%) | ||
| MeOH | 293 and 299 | π→π* | 327.61 | 0.5721 | 3.7845 | H→L (92%) |
| π→π* | 308.08 | 0.7446 | 4.0244 | H-1→L (69%), H-2→L (24%) | ||
| π→π* | 293.23 | 0.0560 | 4.2283 | H-2→L (71%), H-1→L (27%) | ||
| π→π* | 271.83 | 0.0079 | 4.5612 | H-3→L (71%), H-4→L (11%) | ||
| π→π* | 269.00 | 0.0003 | 4.6091 | H-7→L (73%), H-6→L (13%) | ||
| π→π* | 259.93 | 0.0572 | 4.7699 | H→L + 1 (68%), H-1→L + 1 (22%) | ||
| MeCN | 292 and 298 | π→π* | 327.75 | 0.5797 | 3.7829 | H→L (93%) |
| π→π* | 308.21 | 0.7388 | 4.0227 | H-1→L (68%), H-2→L (25%) | ||
| π→π* | 293.35 | 0.0552 | 4.2264 | H-2→L (71%), H-1→L (28%) | ||
| π→π* | 271.85 | 0.0080 | 4.5607 | H-3→L (71%), H-4→L (11%) | ||
| π→π* | 268.96 | 0.0003 | 4.6098 | H-7→L (73%), H-6→L (13%) | ||
| π→π* | 259.91 | 0.0594 | 4.7703 | H→L + 1 (67%), H-1→L + 1 (23%) | ||
| DMF | 299 | π→π* | 328.38 | 0.6195 | 3.7757 | H→L (94%) |
| π→π* | 308.81 | 0.7133 | 4.0149 | H-1→L (67%), H-2→L (27%) | ||
| π→π* | 293.55 | 0.0459 | 4.2236 | H-2→L (69%), H-1→L (29%) | ||
| π→π* | 271.90 | 0.0083 | 4.5599 | H-3→L (71%), H-4→L (11%) | ||
| π→π* | 268.95 | 0.0003 | 4.6099 | H-7→L (73%), H-6→L (13%) | ||
| π→π* | 259.98 | 0.0656 | 4.7690 | H→L + 1 (66%), H-1→L + 1 (24%) |
| Parameters | CHCl3 (ε = 4.7113) | MeOH (ε = 32.613) | MeCN (ε = 35.688) | DMF (ε = 37.219) |
|---|---|---|---|---|
| ELUMO (eV) | −2.0169 | −2.1089 | −2.1105 | −2.1111 |
| EHOMO (eV) | −6.2902 | −6.3705 | −6.3718 | −6.3724 |
| Energy bandgap |EHOMO−ELUMO| (eV) | 4.2733 | 4.2616 | 4.2613 | 4.2613 |
| Ionization potential (I = −EHOMO) (eV) | 6.2902 | 6.3705 | 6.3718 | 6.3724 |
| Electron affinity (A = −ELUMO) (eV) | 2.0169 | 2.1089 | 2.1105 | 2.1111 |
| Chemical hardness (η = (I − A)/2) (eV) | 2.1366 | 2.1308 | 2.1307 | 2.1307 |
| Chemical softness (ζ = 1/2η) (eV−1) | 0.2340 | 0.2347 | 0.2347 | 0.2347 |
| Electronegativity (χ = (I + A)/2) (eV) | 4.1535 | 4.2397 | 4.2412 | 4.2417 |
| Chemical potential (μ = −(I + A)/2) (eV) | −4.1535 | −4.2397 | −4.2412 | −4.2417 |
| Electrophilicity index (w = μ2/2η) (eV) | 4.0372 | 4.2179 | 4.2211 | 4.2222 |
| Maximum charge transfer index (ΔNmax. = −μ/η) | 1.9440 | 1.9897 | 1.9905 | 1.9908 |
| MeCN | CHCl3 | ||||||
|---|---|---|---|---|---|---|---|
| P | Value (esu) | P | Value (esu) | P | Value (esu) | P | Value (esu) |
| αxx | 1.06054264 × 10−22 | βxxx | 2.7232392281 × 10−29 | αxx | 1.02029387 × 10−22 | βxxx | 2.3952996201 × 10−29 |
| αxy | 3.34406970 × 10−24 | βxyy | −6.7163351322 × 10−30 | αxy | 2.71384369 × 10−24 | βxyy | −5.3041591257 × 10−30 |
| αxz | −1.96406718 × 10−25 | βxzz | 3.9775500915 × 10−31 | αxz | −1.74450518 × 10−26 | βxzz | 3.4264850753 × 10−31 |
| αyy | 6.14781650 × 10−23 | βyyy | 2.5742850132 × 10−30 | αyy | 5.53813340 × 10−23 | βyyy | 1.9066353494 × 10−30 |
| αyz | 5.61089698 × 10−24 | βyxx | −4.2660804480 × 10−30 | αyz | 4.95659385 × 10−24 | βyxx | −3.8201464576 × 10−30 |
| αzz | 4.89191925 × 10−23 | βyzz | 4.1525419899 × 10−31 | αzz | 4.34111794 × 10−23 | βyzz | 2.7472139962 × 10−31 |
| αtotal | 72.1505403 × 10−24 | βzzz | −7.2495596707 × 10−31 | αtotal | 66.9406335 × 10−24 | βzzz | −4.4045838309 × 10−31 |
| Δα | 53.2230869 × 10−24 | βzxx | −6.2103327791 × 10−31 | Δα | 54.5298916 × 10−23 | βzxx | −4.5456621571 × 10−31 |
| μx | −1.9477408 | βzyy | 4.0011609701 × 10−31 | μx | −1.9675829 | βzyy | 2.6810099604 × 10−31 |
| μy | 0.2212355 | β0 | 209.74073818 × 10−31 | μy | 0.2212355 | β0 | 190.72367172 × 10−31 |
| μz | 0.079071 | μz | 0.0277634 | ||||
| μtotal | 1.9618592 | μtotal | 1.9801764 | ||||
| MeOH | DMF | ||||||
| P | Value (esu) | P | Value (esu) | P | Value (esu) | P | Value (esu) |
| αxx | 1.05991420 × 10−22 | βxxx | 2.7186313156 × 10−29 | αxx | 1.06081731 × 10−22 | βxxx | 2.7252481965 × 10−29 |
| αxy | 3.33205805 × 10−24 | βxyy | −6.6900564567 × 10−30 | αxy | 3.34934748 × 10−24 | βxyy | −6.7278538996 × 10−30 |
| αxz | −1.93663892 × 10−25 | βxzz | 3.9658089890 × 10−31 | αxz | −1.97595431 × 10−25 | βxzz | 3.9828977750 × 10−31 |
| αyy | 6.13687028 × 10−23 | βyyy | 2.5625263928 × 10−30 | αyy | 6.15261523 × 10−23 | βyyy | 2.5794420947 × 10−30 |
| αyz | 5.60026327 × 10−24 | βyxx | −4.2586482569 × 10−30 | αyz | 5.61554695 × 10−24 | βyxx | −4.2692974780 × 10−30 |
| αzz | 4.88130109 × 10−23 | βyzz | 4.1222501073 × 10−31 | αzz | 4.89658696 × 10−23 | βyzz | 4.1658927840 × 10−31 |
| αtotal | 72.0577113 × 10−24 | βzzz | −7.1870785944 × 10−31 | αtotal | 72.1912509 × 10−24 | βzzz | −7.2771077289 × 10−31 |
| Δα | 53.2598403 × 10−24 | βzxx | −6.1728143986 × 10−31 | Δα | 53.2068187 × 10−24 | βzxx | −6.2270660838 × 10−31 |
| μx | −1.9482024 | βzyy | 3.9736307175 × 10−31 | μx | −1.9475465 | βzyy | 4.0132371478 × 10−31 |
| μy | 0.2190976 | β0 | 209.53283113 × 10−31 | μy | 0.2221764 | β0 | 209.83099781 × 10−31 |
| μz | 0.0777472 | μz | 0.0796567 | ||||
| μtotal | 1.9620247 | μtotal | 1.9617963 | ||||
| Value | MeCN | CHCl3 | MeOH | DMF |
|---|---|---|---|---|
| αtotal | 14.21 | 13.19 | 14.19 | 14.22 |
| ∆α | 24.92 | 25.53 | 24.94 | 24.91 |
| β0 | 29.04 | 26.41 | 29.01 | 29.05 |
| SMILES | COc1ccc(cc1)n1nnc(c1C)C(=O)N/N=C/c1cccc(c1)Oc1ccccc1 |
|---|---|
| Physicochemical Properties | Values |
| Molecular weight (MW) | 427.16 g/mol |
| Number of heavy atoms | 32 |
| Number of aromatic heavy atoms | 23 |
| Fraction Csp3 | 0.08 |
| Number of rotatable bonds | 8 |
| Number of H-bond acceptors (HBA) | 6 |
| Number of H-bond donors (HBD) | 1 |
| Molar refractivity (MR) | 120.50 |
| Topological polar surface area (TPSA) | 90.63 Å2 |
| Lipophilicity | Values |
| LogPo/w (iLOGP) | 3.82 |
| LogPo/w (XLOGP3) | 4.49 |
| LogPo/w (WLOGP) | 4.14 |
| LogPo/w (MLOGP) | 3.00 |
| LogPo/w (SILICOS-IT) | 3.59 |
| Consensus LogPo/w | 3.81 |
| Water solubility | Values |
| Log S (ESOL) | −5.32 |
| Solubility | 2.03 × 10−3 mg/mL; 4.76 × 10−6 mol/L |
| Class | Moderately soluble |
| LogS (Ali) | −6.11 |
| Solubility | 3.29 × 10−4 mg/mL; 7.70 × 10−7 mol/L |
| Class | Poorly soluble |
| LogS (SILICOS-IT) | −7.83 |
| Solubility | 6.25 × 10−6 mg/mL; 1.46 × 10−8 mol/L |
| Class | Poorly soluble |
| Pharmacokinetics | Values |
| Gastrointestinal (GI) absorption | High |
| Blood–brain barrier (BBB) permeant | No |
| P-glycoprotein (P-gp) substrate | No |
| CYP1A2 inhibitor | No |
| CYP2C19 inhibitor | Yes |
| CYP2C9 inhibitor | Yes |
| CYP2D6 inhibitor | No |
| CYP3A4 inhibitor | Yes |
| LogKp (skin permeation) | −5.72 cm/s |
| Drug-likeness | Values |
| Lipinski | Yes; zero violation |
| Ghose | Yes |
| Veber | Yes |
| Egan | Yes |
| Muegge | Yes |
| Bioavailability score | 0.55 |
| Medical chemistry | Values |
| PAINS | 0 alert |
| Brenk | 1 alert: imine_1 |
| Lead-likeness | No; three violations: MW > 350, rotors > 7; XLOGP3 > 3.5 |
| Synthetic accessibility | 3.52 |
| Mode | Binding Affinity (kcal/mol) | Distance from the Best Mode (Å) | |
|---|---|---|---|
| RMSD/lb | RMSD/ub | ||
| 1 | −10.10 | 0.000 | 0.000 |
| 2 | −9.90 | 2.271 | 9.987 |
| 3 | −9.80 | 1.851 | 10.125 |
| 4 | −9.60 | 2.361 | 10.036 |
| 5 | −9.50 | 3.371 | 9.958 |
| 6 | −9.10 | 1.826 | 10.625 |
| 7 | −9.00 | 14.962 | 17.748 |
| 8 | −8.80 | 2.062 | 2.664 |
| 9 | −8.70 | 2.603 | 4.106 |
| 10 | −8.60 | 15.096 | 18.245 |
| Chemical Formula | C24H21N5O3 |
|---|---|
| Formula weight | 426.46 |
| F(000) | 896 |
| Radiation | CuKα (λ = 1.54184 Å) |
| Crystal system | Triclinic |
| Space group | P |
| a, b, c (Å) | 6.6452 (5), 7.4466 (5), 43.403 (2) |
| α, β, γ (°) | 86.250 (4), 89.193 (5), 80.484 (6) |
| Volume (Å3) | 2113.7 (2) |
| Z | 4 |
| Crystal dimension (mm) | 0.30 × 0.18 × 0.03 |
| Temperature (K) | 293 |
| Dcalc (mg m−3) | 1.343 |
| Measured reflections | 9077 |
| Independent reflections | 9077 |
| Number of reflections with I > 2 σ(I) | 6949 |
| θ range (°) | 6.0–77.1 |
| Range of h, k, l | −8 < h < 8, −9 < k < 9, −54 < l < 52 |
| Refinement on | F2 |
| Calculated weights | W = 1/(σ2(Fo2) + (0.1235P)2 + 1.0892P), where P = (Fo2 + 2Fc2/3) |
| Number of refinement parameters | 582 |
| S | 1.08 |
| R (F2 > 2σ(F2)) | 0.075 |
| wR (F2) | 0.241 |
| (Δρ)max (e Å−3) | 0.30 |
| (Δρ)min (e Å−3) | −0.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gökce, H.; Şen, F.; Sert, Y.; Abdel-Wahab, B.F.; Kariuki, B.M.; El-Hiti, G.A. Quantum Computational Investigation of (E)-1-(4-methoxyphenyl)-5-methyl-N′-(3-phenoxybenzylidene)-1H-1,2,3-triazole-4-carbohydrazide. Molecules 2022, 27, 2193. https://doi.org/10.3390/molecules27072193
Gökce H, Şen F, Sert Y, Abdel-Wahab BF, Kariuki BM, El-Hiti GA. Quantum Computational Investigation of (E)-1-(4-methoxyphenyl)-5-methyl-N′-(3-phenoxybenzylidene)-1H-1,2,3-triazole-4-carbohydrazide. Molecules. 2022; 27(7):2193. https://doi.org/10.3390/molecules27072193
Chicago/Turabian StyleGökce, Halil, Fatih Şen, Yusuf Sert, Bakr F. Abdel-Wahab, Benson M. Kariuki, and Gamal A. El-Hiti. 2022. "Quantum Computational Investigation of (E)-1-(4-methoxyphenyl)-5-methyl-N′-(3-phenoxybenzylidene)-1H-1,2,3-triazole-4-carbohydrazide" Molecules 27, no. 7: 2193. https://doi.org/10.3390/molecules27072193
APA StyleGökce, H., Şen, F., Sert, Y., Abdel-Wahab, B. F., Kariuki, B. M., & El-Hiti, G. A. (2022). Quantum Computational Investigation of (E)-1-(4-methoxyphenyl)-5-methyl-N′-(3-phenoxybenzylidene)-1H-1,2,3-triazole-4-carbohydrazide. Molecules, 27(7), 2193. https://doi.org/10.3390/molecules27072193