
The Use of a Solid Bismuth Microelectrode for Vanadium Quantification by Adsorptive Stripping Voltammetry in Environmental Water Samples

Abstract
:1. Introduction
2. Materials and Methods
2.1. Apparatus
2.2. Reagents
2.3. Standard Procedure of Measurements
3. Results and Discussion
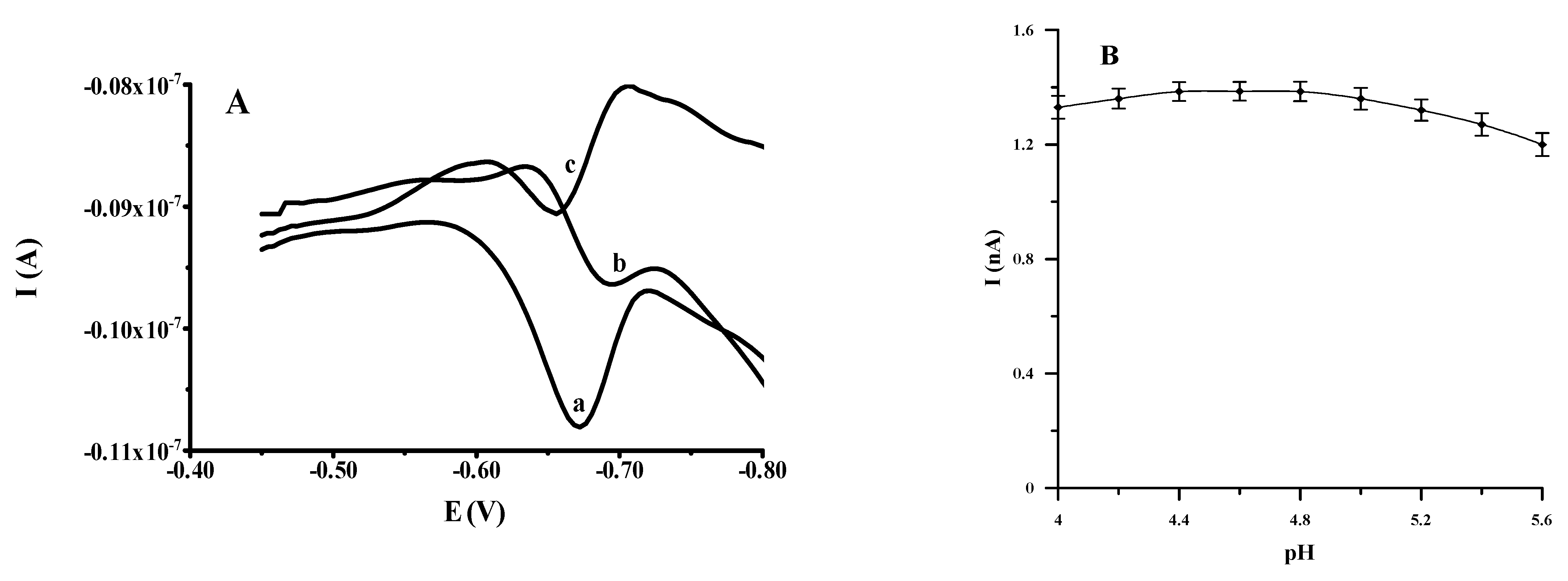
3.1. Optimization of Supporting Electrolyte Composition
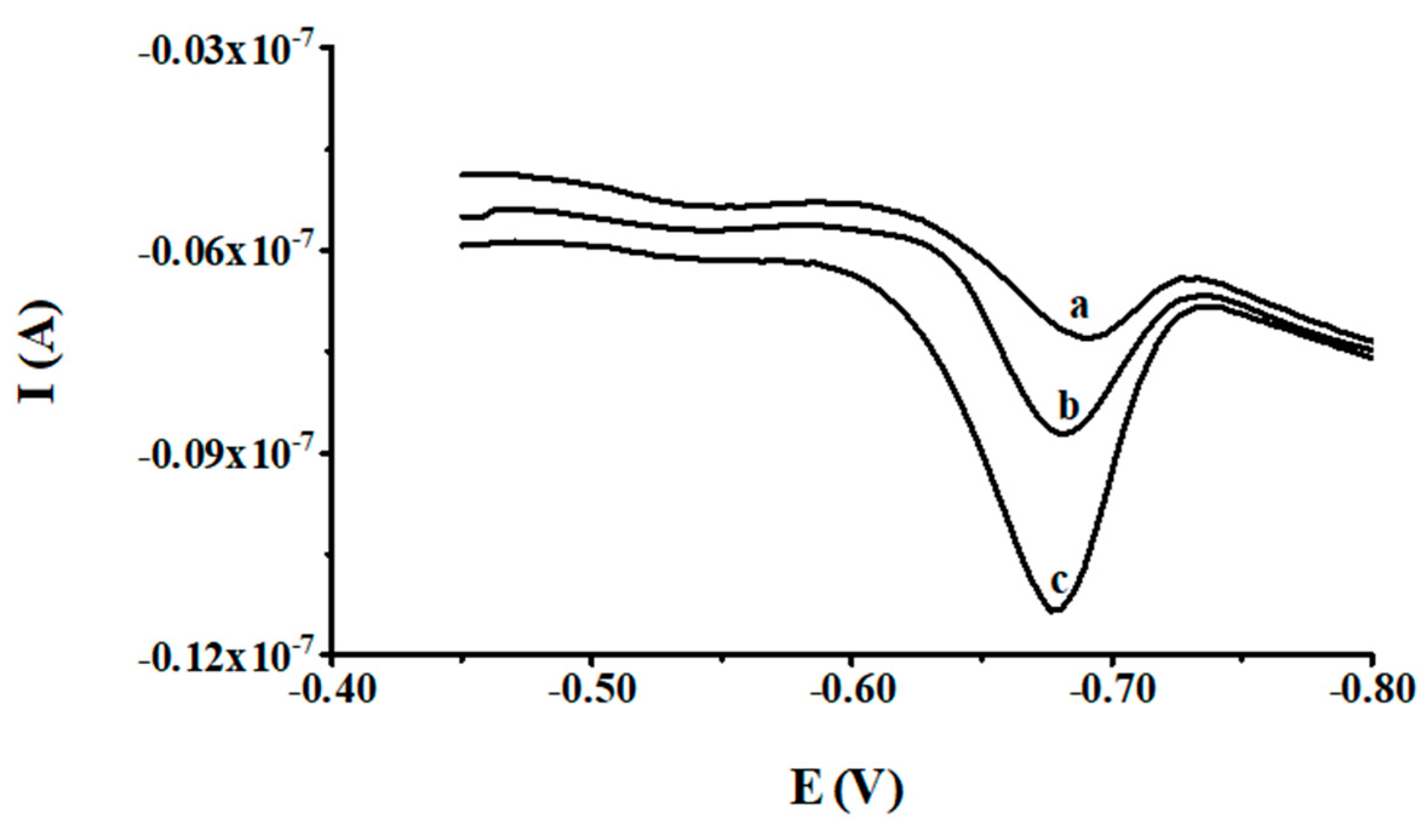
3.2. Optimization of Cupferron Concentration
3.3. Optimization of Activation Potential and Time
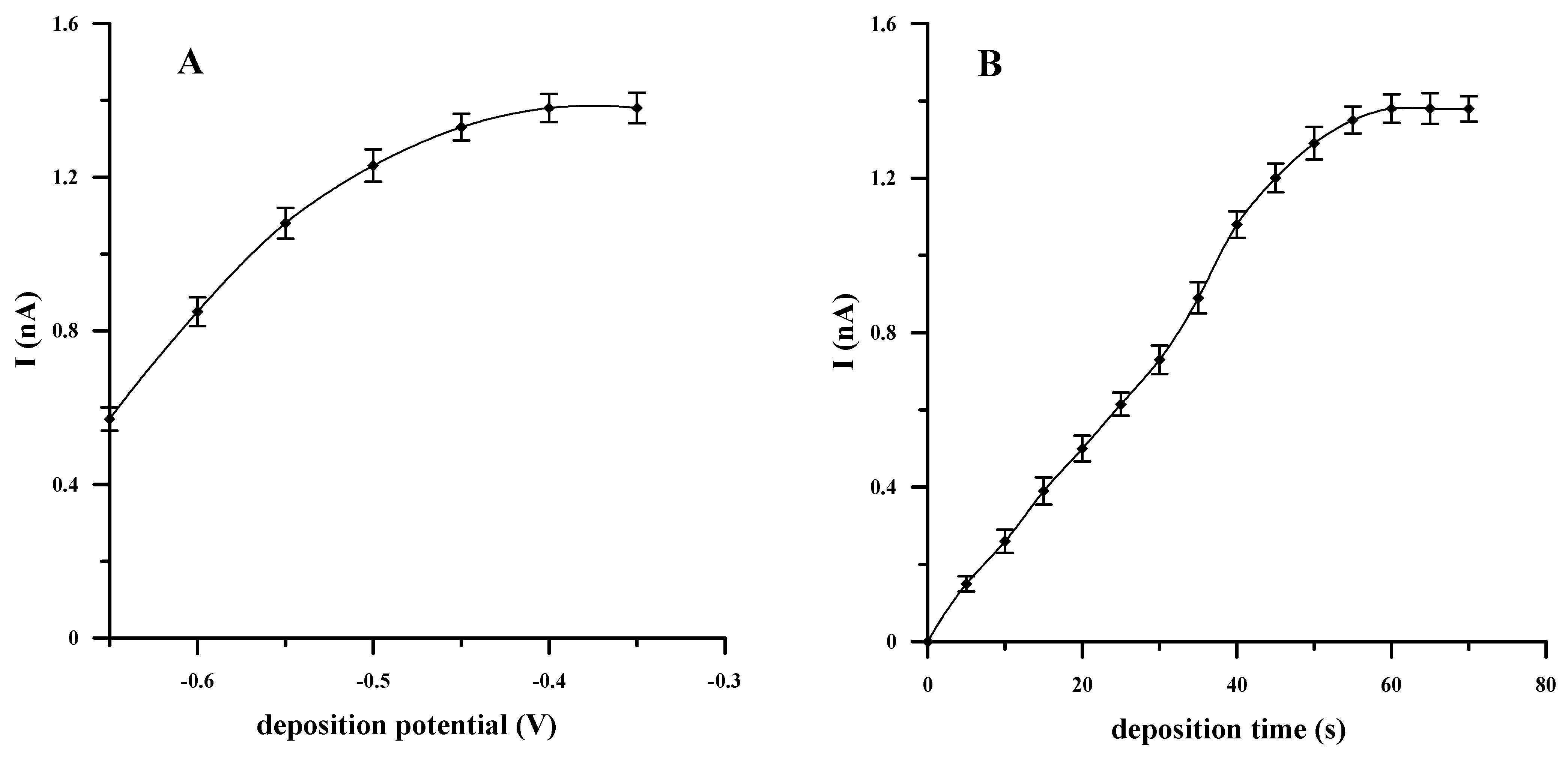
3.4. Optimization of Accumulation Potential and Time
3.5. Characteristics of the Analytical Procedure
3.6. Tolerance to Interfering Species
3.7. Verification of the Procedure Using SPS-WW1 Waste Water CRM and Real Water Sample
4. Conclusions
- The method is simple to implement and makes the analysis eco-friendly, as it allows avoiding the following:
- -
- the use of metallic mercury for electrode formation (thus, the measurements can be carried out without aerating the electrolyte with an inert gas);
- -
- adding bismuth ions to the voltammetric cell when using a film bismuth electrode (therefore, the procedure is considerably simplified and allows for a lesser consumption of reagents).
- The procedure shows a very good analytical performance, such as:
- -
- a low detection limit of 2.5 × 10−10 mol L−1 (60 s);
- -
- a broad linearity range covering more than two orders of magnitude (8 × 10−10 to 1 × 10−7 mol L−1);
- -
- good repeatability and reproducibility with the calculated RSD = 3.4% (n = 8) and 5.0% (n = 5), respectively.
- In the recommended procedure, the height of the V(V) signal is unchanged in the presence of an even 1000-fold excess of many foreign ions in the sample.
- This method makes it possible to carry out vanadium determinations in the presence of 0.5-mg L−1 Triton X-100 and humic acid, while, in other procedures, no signal of V(V) was obtained at the concentration of organic substances.
- During the verification of the procedure using a certified reference material and real water samples, it was shown that the developed procedure is suitable for the analysis of environmental water samples with a complex matrix, even waste water.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Geca, I.; Korolczuk, M. Sensitive Determination of Folic Acid using a Solid Bismuth Microelectrode by Adsorptive Stripping Voltammetry. Electroanalysis 2019, 32, 496–502. [Google Scholar] [CrossRef]
- Gęca, I.; Ochab, M.; Korolczuk, M. Anodic Stripping Voltammetry of Tl(I) Determination with the Use of a Solid Bismuth Microelectrode. J. Electrochem. Soc. 2020, 167, 086506. [Google Scholar] [CrossRef]
- Bond, A. Past, present and future contributions of microelectrodes to analytical studies employing voltammetric detection. A review. Analyst 1994, 119, 1R–21R. [Google Scholar] [CrossRef]
- Wang, J.; Lu, J.; Hocevar, S.B.; Farias, P.A.M.; Ogorevc, B. Bismuth-Coated Carbon Electrodes for Anodic Stripping Voltammetry. Anal. Chem. 2000, 72, 3218–3222. [Google Scholar] [CrossRef] [PubMed]
- Korolczuk, M.; Tyszczuk-Rotko, K.; Grabarczyk, M. Adsorptive stripping voltammetry of nickel and cobalt at in situ plated lead film electrode. Electrochem. Commun. 2005, 7, 1185–1189. [Google Scholar] [CrossRef]
- Hocevar, S.B.; Švancara, I.; Ogorevc, B.; Vytřas, K. Antimony Film Electrode for Electrochemical Stripping Analysis. Anal. Chem. 2007, 79, 8639–8643. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, S.; Khani, H.; Sahraei, R. A Highly Sensitive Adsorptive Stripping Voltammetric Method for Simultaneous Determination of Lead and Vanadium in Foodstuffs. Food Anal. Methods 2011, 5, 272–278. [Google Scholar] [CrossRef]
- Qureshi, M.S.; Yusoff, A.R.B.M.; Shah, A.; Nafady, A. Sirajuddin A new sensitive electrochemical method for the determination of vanadium(IV) and vanadium(V) in Benfield sample. Talanta 2015, 132, 541–547. [Google Scholar] [CrossRef]
- Bobrowski, A.; Nowak, K.; Zarebski, J. Novel voltammetric methods for vanadium determination by exploitation of catalytic adsorptive vanadium–chloranilic acid–bromate system. Anal. Chim. Acta 2005, 543, 150–155. [Google Scholar] [CrossRef]
- Vega, M.; Berg, C.M.V.D. Determination of vanadium in sea water by catalytic adsorptive cathodic stripping voltammetry. Anal. Chim. Acta 1994, 293, 19–28. [Google Scholar] [CrossRef]
- Li, H.; Smart, R.B. Catalytic stripping voltammetry of vanadium in the presence of dihydroxynaphtalene and bromate. Anal. Chim. Acta 1996, 333, 131–138. [Google Scholar] [CrossRef]
- Ensafi, A.A.; Khayamian, T.; Khaloo, S.S. Simultaneous determination of trace amounts of vanadium and molybdenum in water and foodstuff samples using adsorptive cathodic stripping voltammetry. Int. J. Food Sci. Technol. 2008, 43, 416–422. [Google Scholar] [CrossRef]
- Povar, I.; Cazac, T.; Chiriac, L.; Sãndulescu, R. Catalytic-adsorptive determination of vanadium in solutions of 2,3-dihydroxybenzaldehide and bromate-ions. J. Electroanal. Chem. 2011, 661, 275–279. [Google Scholar] [CrossRef]
- Greenway, G.M.; Wolfbauer, G. On-line determination of vanadium by adsorptive stripping voltammetry. Anal. Chim. Acta 1995, 312, 15–25. [Google Scholar] [CrossRef]
- Adeloju, S.B.; Pablo, F. Adsorptive cathodic stripping voltammetric determination of ultra-trace concentrations of vanadium on a glassy carbon mercury film electrode. Anal. Chim. Acta 1994, 288, 157–166. [Google Scholar] [CrossRef]
- Piech, R.; Baś, B.; Paczosa-Bator, B.; Kubiak, W.W. Adsorptive stripping voltammetric determination of vanadium(V) witch chloranilic acid using cyclic renewable mercury film silver based electrode. J. Electroanal. Chem. 2009, 633, 333–338. [Google Scholar] [CrossRef]
- Deng, P.-H.; Fei, J.-J.; Feng, Y.-L. Determination of trace vanadium(V) by adsorptive anodic stripping voltammetry on an acetylene black paste electrode in the presence of alizarin violet. J. Electroanal. Chem. 2010, 648, 85–91. [Google Scholar] [CrossRef]
- Wang, J.; Lu, D.; Thongngamdee, S.; Lin, Y.; Sadik, O.A. Catalytic adsorptive stripping voltammetric measurements of trace vanadium at bismuth film electrodes. Talanta 2006, 69, 914–917. [Google Scholar] [CrossRef] [PubMed]
- Grabarczyk, M.; Adamczyk, M. Application of Electrochemical Sensor Based on Lead Film Electrode in Trace Vanadium (V) Determination by Adsorptive Stripping Voltammetry. IEEE Sens. J. 2019, 19, 5916–5922. [Google Scholar] [CrossRef]
- Rybakowska, I.; Stępińska, M.; Anandc, J.S.; Patocki, M. The beneficial and negative influence of vanadium compounds. Przegląd Lek. 2008, 71, 20–23. [Google Scholar]
- Urban, J.; Antonowicz-Juchniewicz, J.; Andrzejak, R. Wanad-zagrożenia i nadzieje. Med. Pr. 2001, 52, 125–133. [Google Scholar] [PubMed]
- Hosseinzadeh, L.; Abassi, S.; Ahmadi, F. Adsorptive Cathodic Stripping Voltammetry Determination of Ultra Trace of Lead in Different Real Samples. Anal. Lett. 2007, 40, 2693–2707. [Google Scholar] [CrossRef]
- Mocak, J.; Bond, A.M.; Mitchell, S.; Scollary, G. A statistical overview of standard (IUPAC and ACS) and new procedures for determining the limits of detection and quantification: Application to voltammetric and stripping techniques (Technical Report). Pure Appl. Chem. 1997, 69, 297–328. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Working Electrode | Complexing Agent | Detection Limit (mol L−1) | Linear Range (mol L−1) | Accumulation Time (s) | Analytical Application | References |
|---|---|---|---|---|---|---|
| HMDE | chromoxane cyanine R | 3.0 × 10−7–2.4 × 10−5 | 180 | food samples, tap water, rainwater | [12] | |
| MFE | pyrogallol | 180 | certified reference materials | [15] | ||
| BiFE | chloranilic acid | 600 | groundwater | [18] | ||
| ABPE | alizarin violet | 90 | environmental water | [17] | ||
| PbFE | cupferron | 30 | certified reference materials, river water | [19] | ||
| BiFµE | cupferron | 60 | certified reference material, river water, rain water, tap water | this work | ||
| HMDE | cupferron | 50 | food samples, mineral water, environmental water | [7] | ||
| HMDE | dihydroxybenzaldehide | 30 | environmental water | [13] | ||
| MFE | cupferron | 90 | sea water | [14] | ||
| HMDE | cupferron | 9.0 × 10−11 | ND | ND | [8] | |
| HMDE | catechol | 7.0 × 10−11 | ND | 120 | sea water | [10] |
| HMDE | dihydroxynaphthalene | 1.5 × 10−11 | 60 | environmental water | [11] | |
| Hg(Ag)FE | chloranilic acid | 1.0 × 10−11 | 90 | certified reference materials, tap water | [16] | |
| HMDE | chloranilic acid | 9.0 × 10−12 | 100 | certified reference materials | [9] |
| Element | Concentration in ng mL−1 (20 °C) |
|---|---|
| Al | 2000 ± 10 |
| As | 100.0 ± 0.5 |
| Cd | 20.0 ± 0.1 |
| Co | 60.0 ± 0.3 |
| Cr | 200 ± 1 |
| Cu | 400 ± 2 |
| Fe | 1000 ± 5 |
| Mn | 400 ± 2 |
| Ni | 1000 ± 5 |
| P | 1000 ± 5 |
| Pb | 100.0 ± 0.5 |
| V | 100.0 ± 0.5 |
| Zn | 600 ± 6 |
| Sample | Concentration of Added V(V) (nmol L−1) | Concentration of Found V(V) (nmol L−1) | Recovery (%) | RSD (n = 5) (%) |
|---|---|---|---|---|
| Ciemiega river water | 5 10 | 5.2 9.9 | 104 99 | 3.1 3.7 |
| Tap water | 5 10 | 4.9 9.6 | 98 96 | 3.8 4.0 |
| Rain water | 5 10 | 5.3 10.4 | 106 104 | 4.2 3.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabarczyk, M.; Adamczyk, M.; Wlazlowska, E. The Use of a Solid Bismuth Microelectrode for Vanadium Quantification by Adsorptive Stripping Voltammetry in Environmental Water Samples. Molecules 2022, 27, 2168. https://doi.org/10.3390/molecules27072168
Grabarczyk M, Adamczyk M, Wlazlowska E. The Use of a Solid Bismuth Microelectrode for Vanadium Quantification by Adsorptive Stripping Voltammetry in Environmental Water Samples. Molecules. 2022; 27(7):2168. https://doi.org/10.3390/molecules27072168
Chicago/Turabian StyleGrabarczyk, Malgorzata, Marzena Adamczyk, and Edyta Wlazlowska. 2022. "The Use of a Solid Bismuth Microelectrode for Vanadium Quantification by Adsorptive Stripping Voltammetry in Environmental Water Samples" Molecules 27, no. 7: 2168. https://doi.org/10.3390/molecules27072168
APA StyleGrabarczyk, M., Adamczyk, M., & Wlazlowska, E. (2022). The Use of a Solid Bismuth Microelectrode for Vanadium Quantification by Adsorptive Stripping Voltammetry in Environmental Water Samples. Molecules, 27(7), 2168. https://doi.org/10.3390/molecules27072168