
Synthesis of Pyridoxine-Derived Dimethylpyridinols Fused with Aminooxazole, Aminoimidazole, and Aminopyrrole



Abstract
:
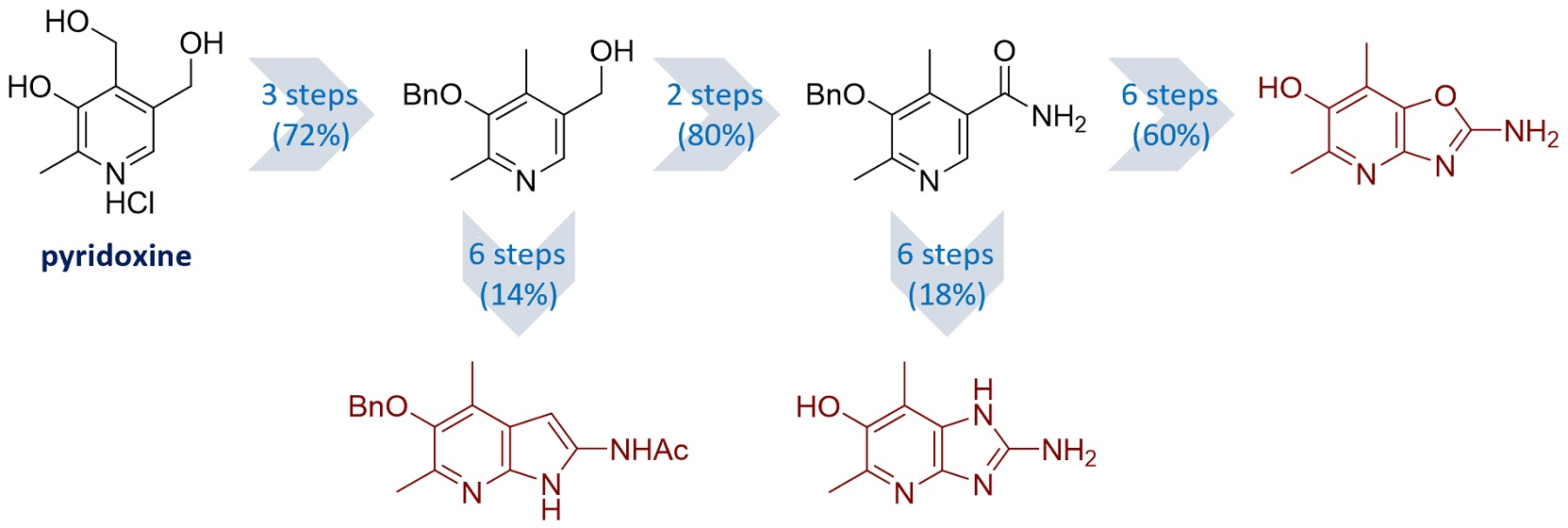{kind=link}
{kind=link}
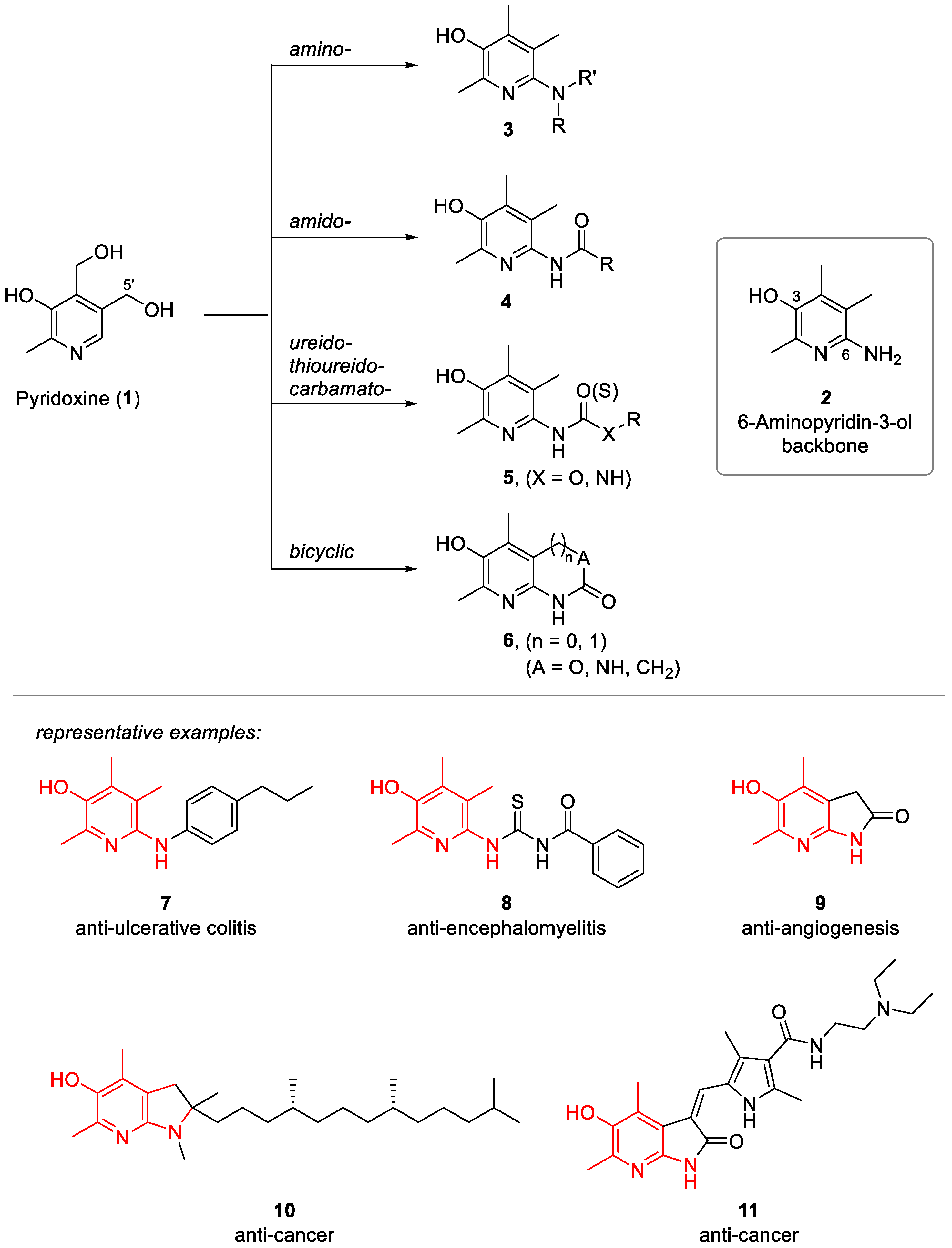
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
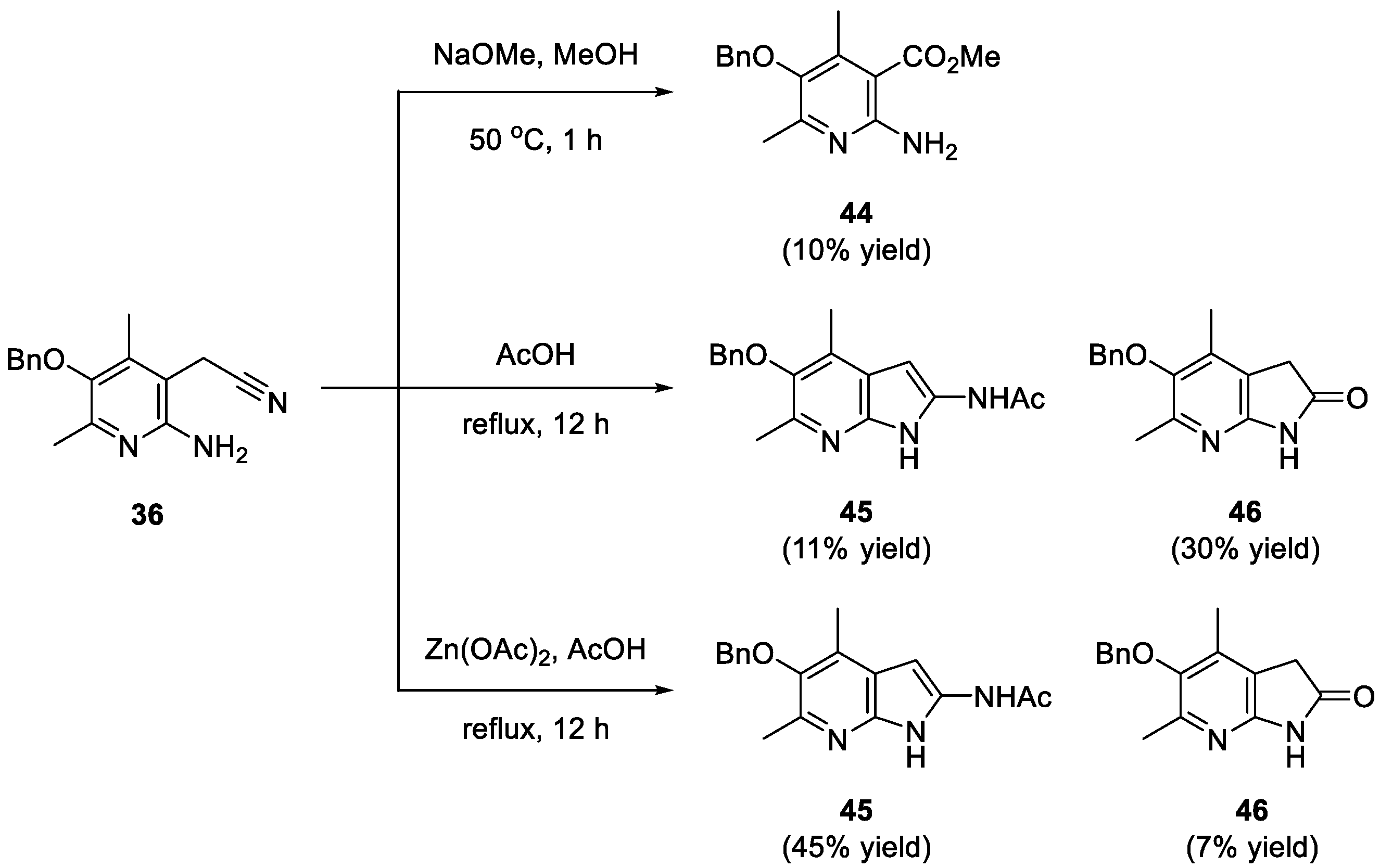{kind=link}
{kind=link}
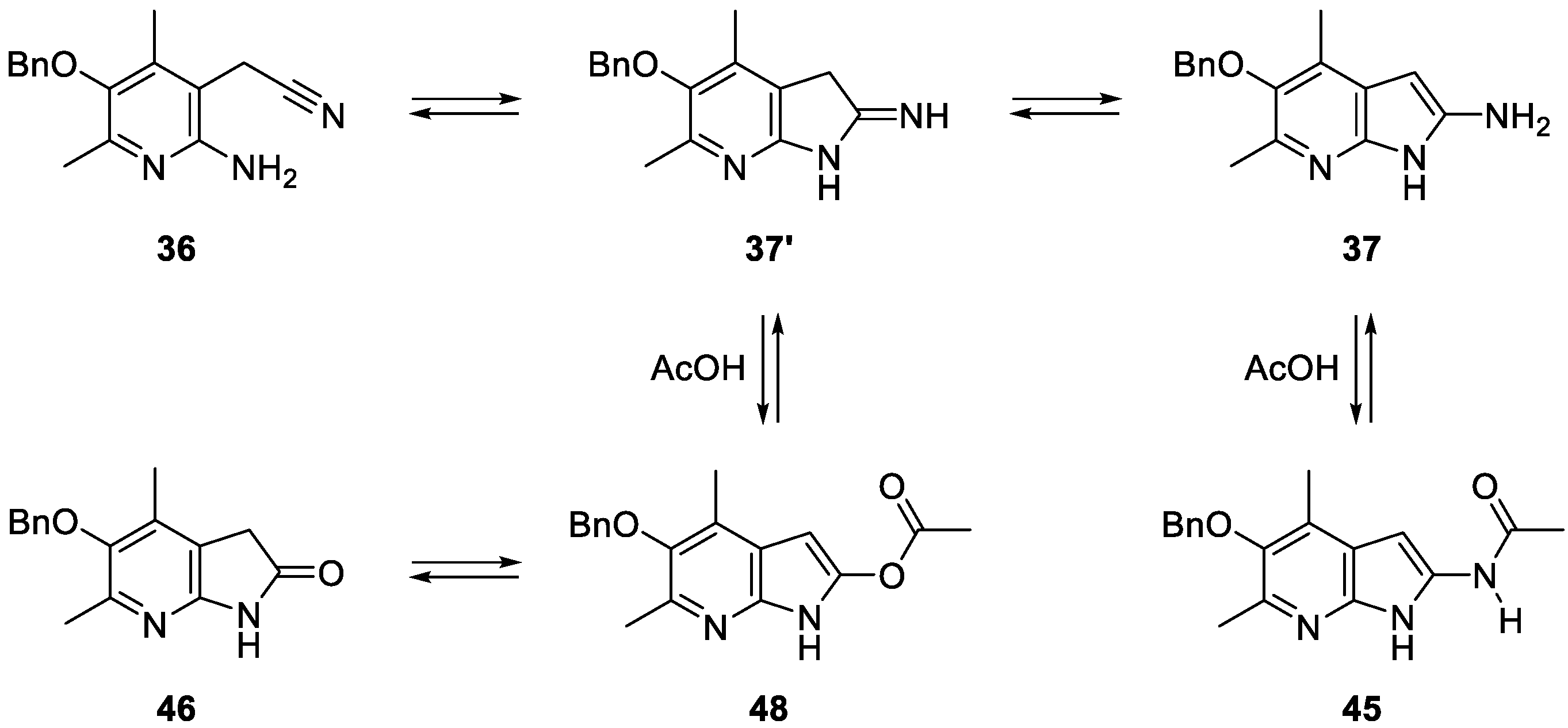{kind=link}
1. Introduction
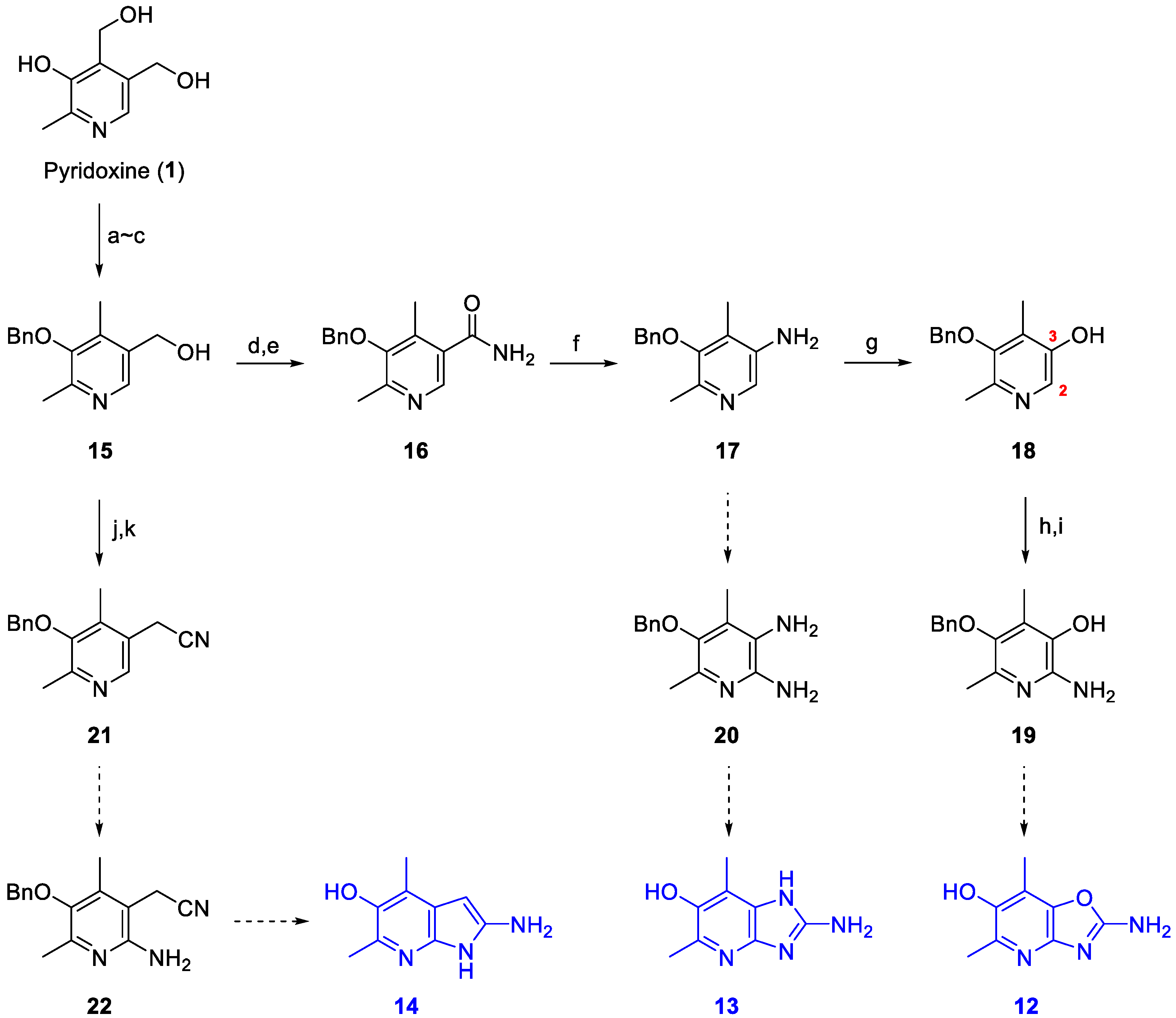
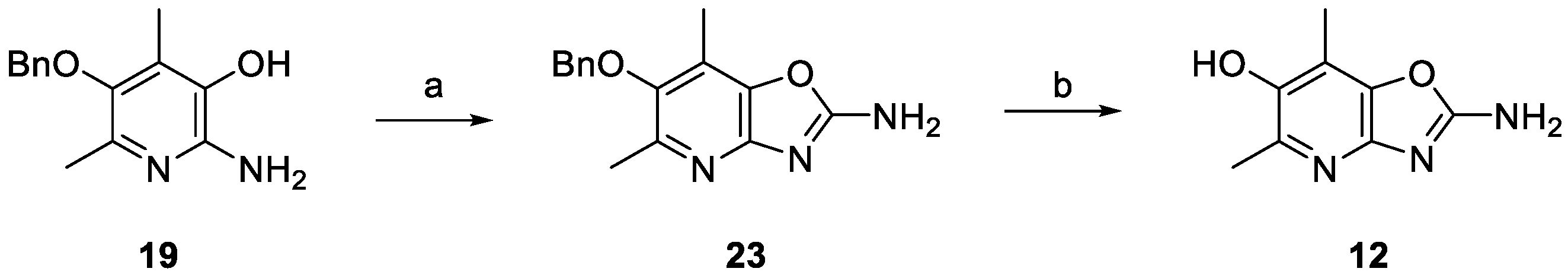
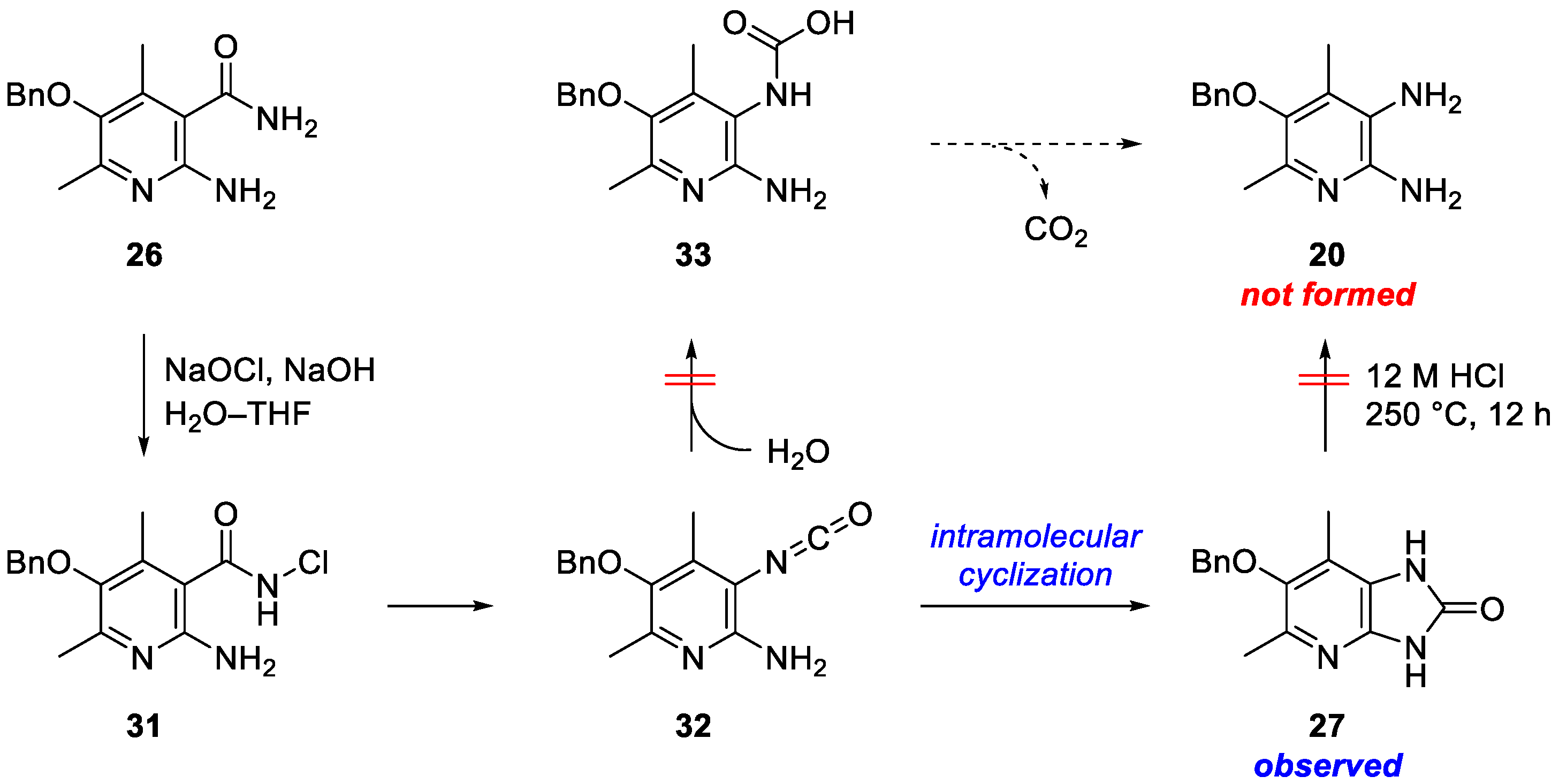
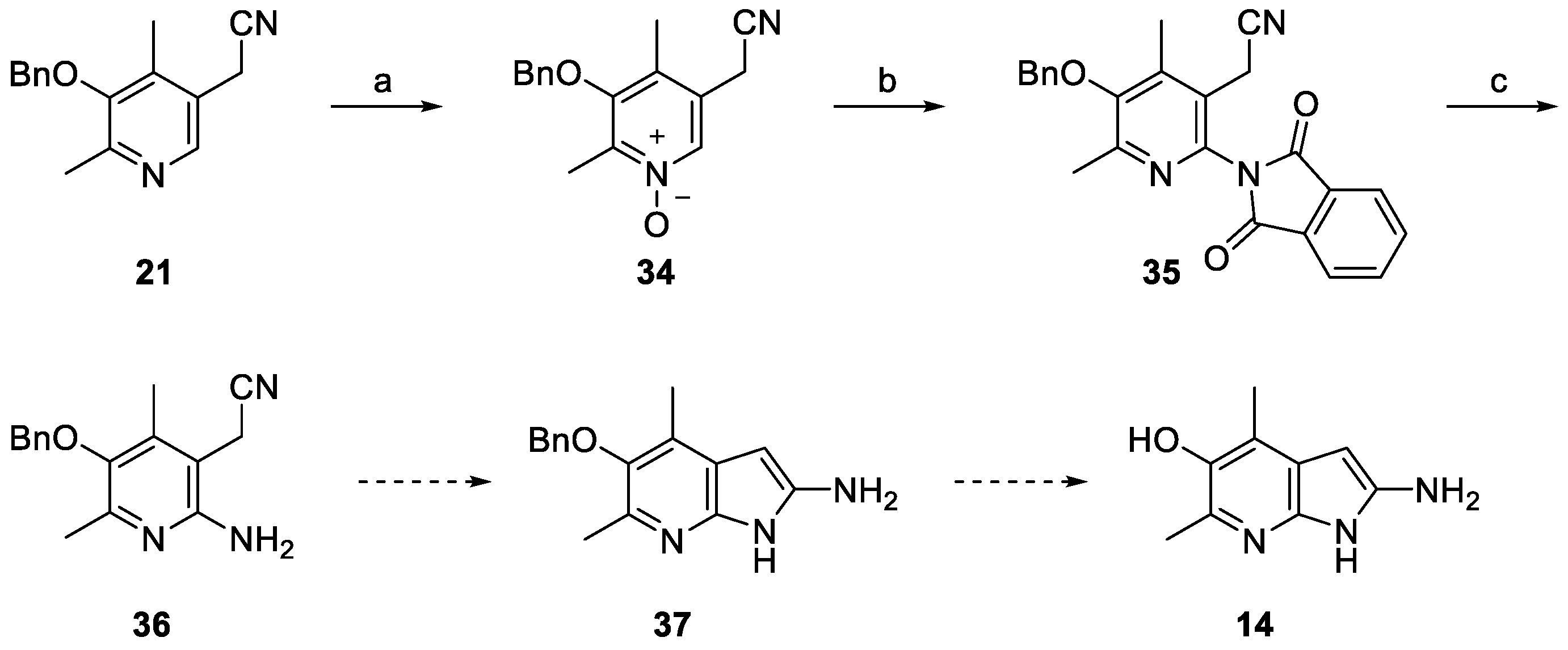
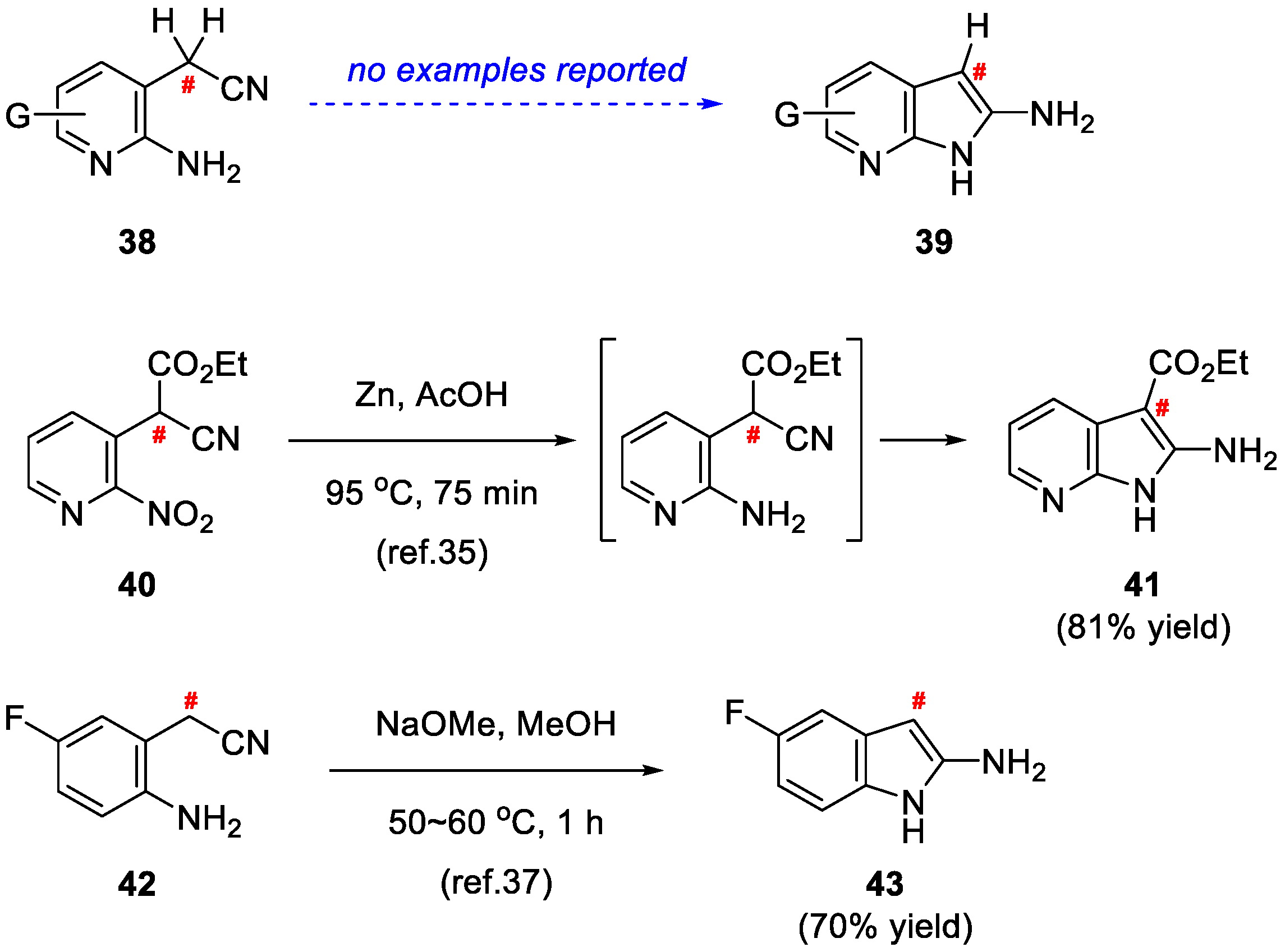
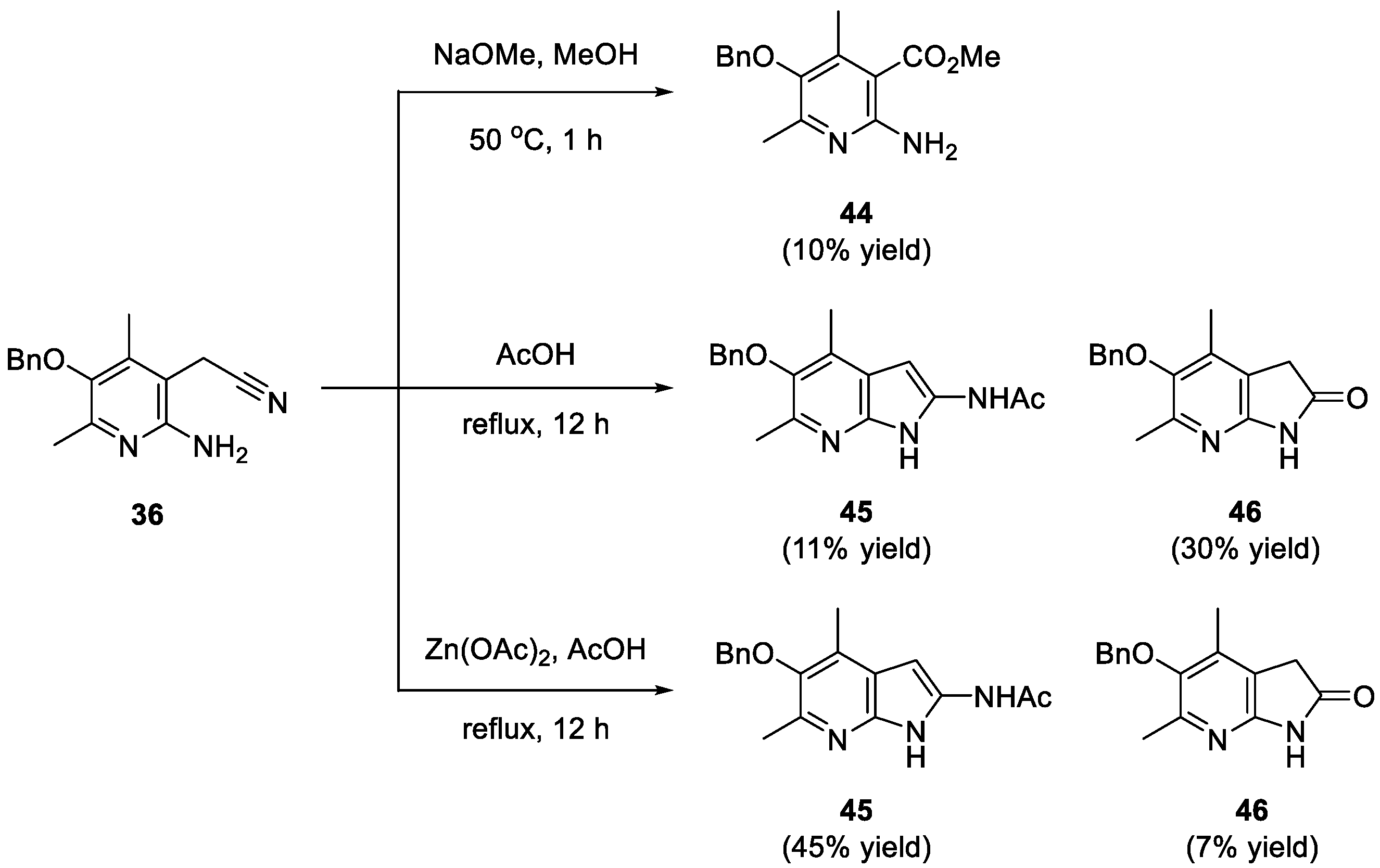
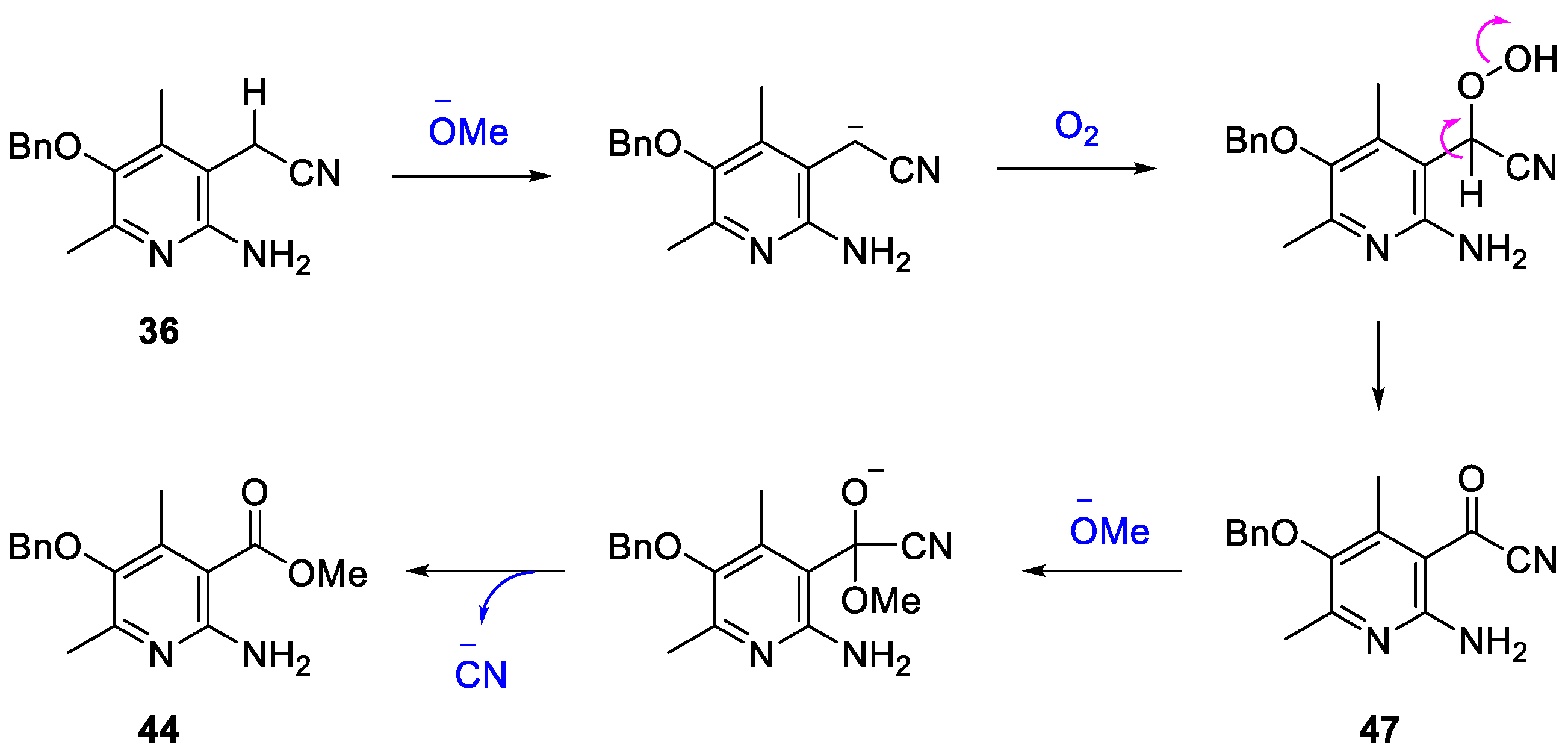
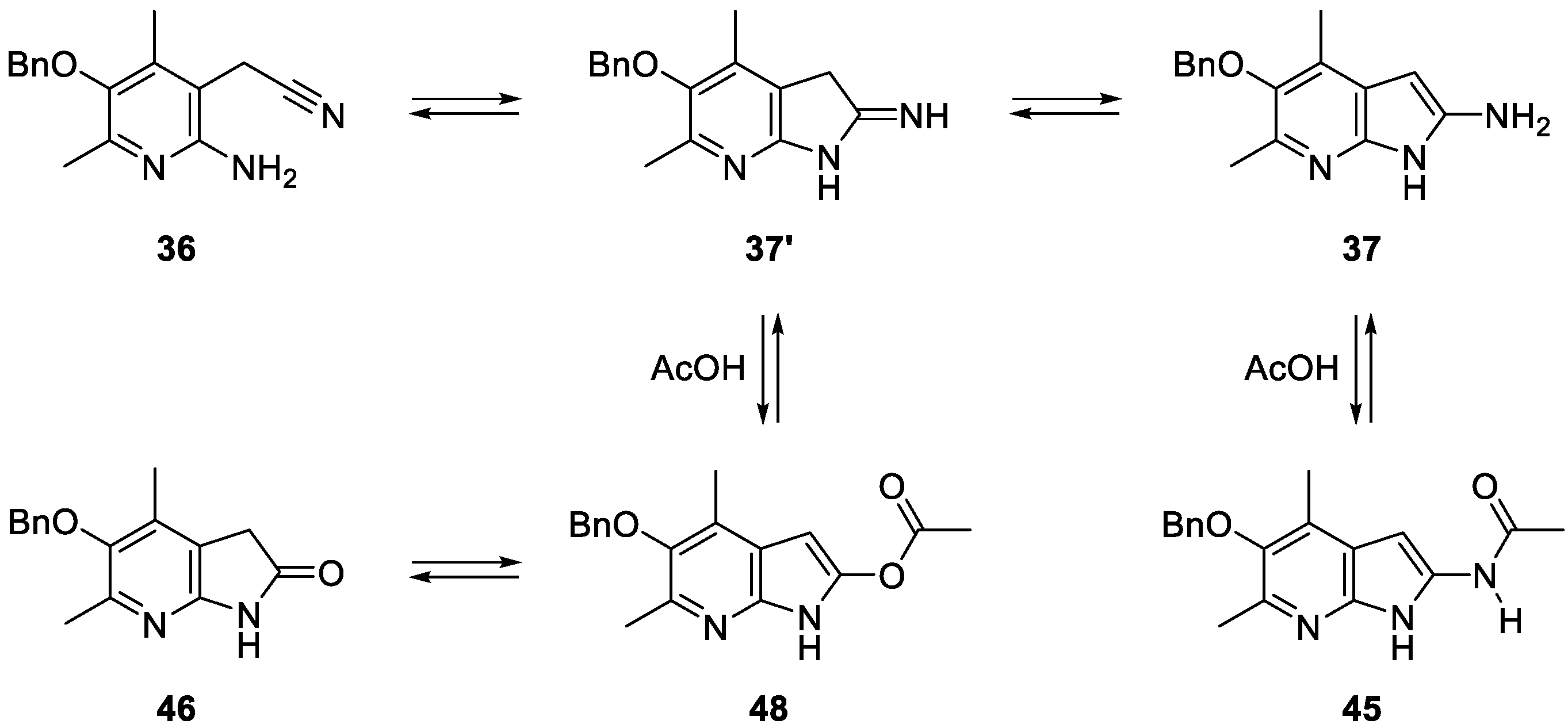
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. 6-(Benzyloxy)-5,7-dimethyloxazolo[4,5-b]pyridin-2-amine (23)
3.3. 2-Amino-5,7-dimethyloxazolo[4,5-b]pyridin-6-ol (12)
3.4. 6-Benzyloxy-5,7-dimethyl-1H-imidazo[4,5-b]pyridin-2(3H)-one (27)
3.5. 5-Amino-3-(benzyloxy)-2,4-dimethylpyridine 1-oxide (28)
3.6. 2-(3-Amino-5-(benzyloxy)-4,6-dimethylpyridin-2-yl)isoindoline-1,3-dione (29)
3.7. 5-(Benzyloxy)-4,6-dimethylpyridine-2,3-diamine (20)
3.8. 6-(Benzyloxy)-5,7-dimethyl-3H-imidazo[4,5-b]pyridin-2-amine (30)
3.9. 2-Amino-5,7-dimethyl-3H-imidazo[4,5-b]pyridin-6-ol hydrochloride (13)
3.10. 3-(Benzyloxy)-5-(cyanomethyl)-2,4-dimethylpyridine 1-oxide (34)
3.11. 2-(5-(Benzyloxy)-2-(1,3-dioxoisoindolin-2-yl)-4,6-dimethylpyridin-3-yl)acetonitrile (35)
3.12. 2-(2-Amino-5-(benzyloxy)-4,6-dimethylpyridin-3-yl)acetonitrile (36)
3.13. Methyl 2-amino-5-(benzyloxy)-4,6-dimethylnicotinate (44)
3.14. N-(5-(benzyloxy)-4,6-dimethyl-1H-pyrrolo[2,3-b]pyridin-2-yl)acetamide (45)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Childers, W.E.; Elokely, K.M.; Abou-Gharbia, M. The Resurrection of Phenotypic Drug Discovery. ACS Med. Chem. Lett. 2020, 11, 1820–1828. [Google Scholar] [CrossRef] [PubMed]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Laraia, L.; Robke, L.; Waldmann, H. Bioactive Compound Collections: From Design to Target Identification. Chem 2018, 4, 705–730. [Google Scholar] [CrossRef]
- Karageorgis, G.; Foley, D.J.; Laraia, L.; Waldmann, H. Principle and design of pseudo-natural products. Nat. Chem. 2020, 12, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Silva, D.; Emery, F.D.S. Strategies towards expansion of chemical space of natural product-based compounds to enable drug discovery. Braz. J. Pharm. Sci. 2018, 54, e01004. [Google Scholar] [CrossRef]
- Mooney, S.; Leuendorf, J.-E.; Hendrickson, C.; Hellmann, H. Vitamin B6: A Long Known Compound of Surprising Complexity. Molecules 2009, 14, 329–351. [Google Scholar] [CrossRef] [Green Version]
- Parra, M.; Stahl, S.; Hellmann, H. Vitamin B6 and Its Role in Cell Metabolism and Physiology. Cells 2018, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Pratt, D.A.; DiLabio, G.A.; Brigati, G.; Pedulli, G.F.; Valgimigli, L. 5-Pyrimidinols: Novel Chain-Breaking Antioxidants More Effective than Phenols. J. Am. Chem. Soc. 2001, 123, 4625–4626. [Google Scholar] [CrossRef]
- Serwa, R.; Nam, T.-G.; Valgimigli, L.; Culbertson, S.; Rector, C.L.; Jeong, B.-S.; Pratt, D.A.; Porter, N.A. Preparation and Investigation of Vitamin B6-Derived Aminopyridinol Antioxidants. Chem. Eur. J. 2010, 16, 14106–14114. [Google Scholar] [CrossRef]
- Omata, Y.; Saito, Y.; Yoshida, Y.; Jeong, B.-S.; Serwa, R.; Nam, T.-G.; Porter, N.A.; Niki, E. Action of 6-amino-3-pyridinols as novel antioxidants against free radicals and oxidative stress in solution, plasma, and cultured cells. Free Radic. Biol. Med. 2010, 48, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-G.; Kang, Y.; Lee, H.; Lee, E.K.; Nam, T.-G.; Kim, J.-A.; Jeong, B.-S. 6-Amino-2,4,5-trimethylpyridin-3-ols: A new general synthetic route and antiangiogenic activity. Eur. J. Med. Chem. 2014, 78, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Banskota, S.; Gautam, J.; Regmi, S.C.; Gurung, P.; Park, M.-H.; Kim, S.J.; Nam, T.-G.; Jeong, B.-S.; Kim, J.-A. BJ-1108, a 6-Amino-2,4,5-Trimethylpyridin-3-ol Analog, Inhibits Serotonin-Induced Angiogenesis and Tumor Growth through PI3K/NOX Pathway. PLoS ONE 2016, 11, e0148133. [Google Scholar] [CrossRef]
- Shah, S.; Lee, C.; Choi, H.; Gautam, J.; Jang, H.; Kim, G.J.; Lee, Y.-J.; Chaudhary, C.L.; Park, S.W.; Nam, T.-G.; et al. 5-Hydroxy-7-azaindolin-2-one, a novel hybrid of pyridinol and sunitinib: Design, synthesis and cytotoxicity against cancer cells. Org. Biomol. Chem. 2016, 14, 4829–4841. [Google Scholar] [CrossRef] [PubMed]
- Banskota, S.; Kang, H.-E.; Kim, D.-G.; Park, S.W.; Jang, H.; Karmacharya, U.; Jeong, B.-S.; Kim, J.-A.; Nam, T.-G. In vitro and in vivo inhibitory activity of 6-amino-2,4,5-trimethylpyridin-3-ols against inflammatory bowel disease. Bioorg. Med. Chem. Lett. 2016, 26, 4587–4591. [Google Scholar] [CrossRef]
- Gautam, J.; Banskota, S.; Regmi, S.C.; Ahn, S.; Jeon, Y.H.; Jeong, H.; Kim, S.J.; Nam, T.-G.; Jeong, B.-S.; Kim, J.-A. Tryptophan hydroxylase 1 and 5-HT7 receptor preferentially expressed in triple-negative breast cancer promote cancer progression through autocrine serotonin signaling. Mol. Cancer 2016, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Timilshina, M.; Nam, T.-G.; Jeong, B.-S.; Chang, J.-H. BJ-1108, a 6-Amino-2,4,5-trimethylpyridin-3-ol analogue, regulates differentiation of Th1 and Th17 cells to ameliorate experimental autoimmune encephalomyelitis. Biol. Res. 2017, 50, 8. [Google Scholar] [CrossRef] [Green Version]
- Acharya, S.; Timilshina, M.; Jiang, L.; Neupane, S.; Choi, D.-Y.; Park, S.W.; Lee, S.Y.; Jeong, B.-S.; Kim, J.-A.; Nam, T.-G.; et al. Amelioration of Experimental autoimmune encephalomyelitis and DSS induced colitis by NTG-A-009 through the inhibition of Th1 and Th17 cells differentiation. Sci. Rep. 2018, 8, 7799. [Google Scholar] [CrossRef]
- Park, S.W.; Banskota, S.; Gurung, P.; Jin, Y.J.; Kang, H.-E.; Chaudhary, C.L.; Lee, S.Y.; Jeong, B.-S.; Kim, J.-A.; Nam, T.-G. Synthesis and evaluation of 6-heteroarylamino-2,4,5-trimethylpyridin-3-ols as inhibitors of TNF-α-induced cell adhesion and inflammatory bowel disease. MedChemComm 2018, 9, 1305–1310. [Google Scholar] [CrossRef]
- Gautam, J.; Banskota, S.; Chaudhary, P.; Dahal, S.; Kim, D.-G.; Kang, H.-E.; Lee, I.-H.; Nam, T.-G.; Jeong, B.-S.; Kim, J.-A. Antitumor activity of BJ-1207, a 6-amino-2,4,5-trimethylpyridin-3-ol derivative, in human lung cancer. Chem. Biol. Interact. 2018, 294, 1–8. [Google Scholar] [CrossRef]
- Lee, H.; Lee, J.S.; Cho, H.J.; Lee, Y.-J.; Kim, E.S.; Kim, S.K.; Nam, T.-G.; Jeong, B.-S.; Kim, J.-A. Antioxidant Analogue 6-Amino-2,4,5-Trimethylpyridin-3-ol Ameliorates Experimental Colitis in Mice. Dig. Dis. Sci. 2021, 66, 1022–1033. [Google Scholar] [CrossRef]
- Karmacharya, U.; Chaudhary, P.; Lim, D.; Dahal, S.; Awasthi, B.P.; Park, H.D.; Kim, J.-A.; Jeong, B.-S. Synthesis and anticancer evaluation of 6-azacyclonol-2,4,6-trimethylpyridin-3-ol derivatives: M3 muscarinic acetylcholine receptor-mediated anticancer activity of a cyclohexyl derivative in androgen-refractory prostate cancer. Bioorg. Chem. 2021, 110, 104805. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Banskota, S.; Kim, D.-G.; Been, J.-H.; Jin, Y.-J.; Gautam, J.; Jang, H.; Nam, T.-G.; Kim, J.-A.; Jeong, B.-S. Synthesis and antiangiogenic activity of 6-amido-2,4,5-trimethylpyridin-3-ols. Bioorg. Med. Chem. Lett. 2014, 24, 3131–3136. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Timilshina, M.; Jeong, B.-S.; Chang, J.-H. BJ-2266 ameliorates experimental autoimmune encephalomyelitis through down-regulation of the JAK/STAT signaling pathway. Eur. J. Immunol. 2017, 47, 1488–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, D.; Gautam, J.; Jang, H.; Banskota, S.; Lee, S.Y.; Jeong, M.-J.; Kim, A.-S.; Kim, H.C.; Lee, I.-H.; Nam, T.-G.; et al. Protective effects of 6-ureido/thioureido-2,4,5-trimethylpyridin-3-ols against 4-hydroxynonenal-induced cell death in adult retinal pigment epithelial-19 cells. Bioorg. Med. Chem. Lett. 2018, 28, 107–112. [Google Scholar] [CrossRef]
- Chaudhary, C.L.; Gurung, P.; Jang, S.; Banskota, S.; Nam, T.-G.; Kim, J.-A.; Jeong, B.-S. Synthesis, activity and mechanism of alkoxy-, carbamato-, sulfonamido-, thioureido-, and ureido-derivatives of 2,4,5-trimethylpyridin-3-ol against inflammatory bowel disease. J. Enzym. Inhib. Med. Chem. 2020, 35, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Karmacharya, U.; Regmi, S.C.; Awasthi, B.P.; Chaudhary, P.; Kim, Y.E.; Lee, I.-H.; Nam, T.-G.; Kim, J.-A.; Jeong, B.-S. Synthesis and activity of N-(5-hydroxy-3,4,6-trimethylpyridin-2-yl)acetamide analogues as anticolitis agents via dual inhibition of TNF-α- and IL-6-induced cell adhesions. Bioorg. Med. Chem. Lett. 2021, 43, 128059. [Google Scholar] [CrossRef]
- Nam, T.-G.; Ku, J.-M.; Park, H.-G.; Porter, N.A.; Jeong, B.-S. New synthetic route to N-tocopherol derivatives: Synthesis of pyrrolopyridinol analogue of α-tocopherol from pyridoxine. Org. Biomol. Chem. 2011, 9, 1749–1755. [Google Scholar] [CrossRef]
- Nam, T.-G.; Ku, J.-M.; Rector, C.L.; Choi, H.; Porter, N.A.; Jeong, B.-S. Pyridoxine-derived bicyclic aminopyridinol antioxidants: Synthesis and their antioxidant activities. Org. Biomol. Chem. 2011, 9, 8475–8482. [Google Scholar] [CrossRef]
- Shchepin, R.V.; Liu, W.; Yin, H.; Zagol-Ikapitte, I.; Amin, T.; Jeong, B.-S.; Roberts, I.L.J.; Oates, J.A.; Porter, N.A.; Boutaud, O. Rational Design of Novel Pyridinol-Fused Ring Acetaminophen Analogues. ACS Med. Chem. Lett. 2013, 4, 710–714. [Google Scholar] [CrossRef]
- Lee, H.; Kim, D.-G.; Banskota, S.; Lee, Y.K.; Nam, T.-G.; Kim, J.-A.; Jeong, B.-S. Pyridoxine-derived bicyclic amido-, ureido-, and carbamato-pyridinols: Synthesis and antiangiogenic activities. Org. Biomol. Chem. 2014, 12, 8702–8710. [Google Scholar] [CrossRef] [PubMed]
- Gautam, J.; Ku, J.-M.; Regmi, S.C.; Jeong, H.; Wang, Y.; Banskota, S.; Park, M.-H.; Nam, T.-G.; Jeong, B.-S.; Kim, J.-A. Dual Inhibition of NOX2 and Receptor Tyrosine Kinase by BJ-1301 Enhances Anticancer Therapy Efficacy via Suppression of Autocrine-Stimulatory Factors in Lung Cancer. Mol. Cancer Ther. 2017, 16, 2144–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, J.; Banskota, S.; Lee, H.; Lee, Y.-J.; Jeon, Y.H.; Kim, J.-A.; Jeong, B.-S. Down-regulation of cathepsin S and matrix metalloproteinase-9 via Src, a non-receptor tyrosine kinase, suppresses triple-negative breast cancer growth and metastasis. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosurkar, U.B.; Dadmal, T.; Appalanaidu, K.; Rao, Y.K.; Nanubolu, J.B.; Kumbhare, R.M. Microwave assisted synthesis of 2-aminooxazolo [4,5-b] pyridine derivatives via intramolecular C–O bond formation in aqueous medium. Tetrahedron Lett. 2014, 55, 1296–1298. [Google Scholar] [CrossRef]
- Veselovská, L.; Kudlová, N.; Gurská, S.; Lišková, B.; Medvedíková, M.; Hodek, O.; Tloušťová, E.; Milisavljevic, N.; Tichý, M.; Perlíková, P.; et al. Synthesis and Cytotoxic and Antiviral Activity Profiling of All-Four Isomeric Series of Pyrido-Fused 7-Deazapurine Ribonucleosides. Chem. Eur. J. 2020, 26, 13002–13015. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Y.; Zhou, B.; Aguilar, A.; Liu, L.; Bai, L.; McEachern, D.; Sun, D.; Wen, B.; Luo, R.; et al. Preparation of 9H-Pyrimido[4,5-b]indoles and Related Analogs as BET Bromodomain Inhibitors. WO2015131005, 3 September 2015. [Google Scholar]
- Shigeyoshi, N.; Kenji, H.; Shuji, Y.; Takeshi, T.; Hiroyuki, O. Preparation of 2-Amino-5-halogenoindoles. JP2003267952, 25 September 2003. [Google Scholar]
- Gilchrist, T.L.; Rees, C.W.; Thomas, C. Reactive intermediates. Part XXVI. Flash vacuum pyrolysis of phenyl-substituted 1,2,4-triazoles; a new synthesis of isoindoles. J. Chem. Soc. Perkin Trans. 1 1975, 1, 12–18. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awasthi, B.P.; Lee, H.; Jeong, B.-S. Synthesis of Pyridoxine-Derived Dimethylpyridinols Fused with Aminooxazole, Aminoimidazole, and Aminopyrrole. Molecules 2022, 27, 2075. https://doi.org/10.3390/molecules27072075
Awasthi BP, Lee H, Jeong B-S. Synthesis of Pyridoxine-Derived Dimethylpyridinols Fused with Aminooxazole, Aminoimidazole, and Aminopyrrole. Molecules. 2022; 27(7):2075. https://doi.org/10.3390/molecules27072075
Chicago/Turabian StyleAwasthi, Bhuwan Prasad, Hyunji Lee, and Byeong-Seon Jeong. 2022. "Synthesis of Pyridoxine-Derived Dimethylpyridinols Fused with Aminooxazole, Aminoimidazole, and Aminopyrrole" Molecules 27, no. 7: 2075. https://doi.org/10.3390/molecules27072075
APA StyleAwasthi, B. P., Lee, H., & Jeong, B.-S. (2022). Synthesis of Pyridoxine-Derived Dimethylpyridinols Fused with Aminooxazole, Aminoimidazole, and Aminopyrrole. Molecules, 27(7), 2075. https://doi.org/10.3390/molecules27072075