
Solution Equilibrium Studies on Salicylidene Aminoguanidine Schiff Base Metal Complexes: Impact of the Hybridization with L-Proline on Stability, Redox Activity and Cytotoxicity

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
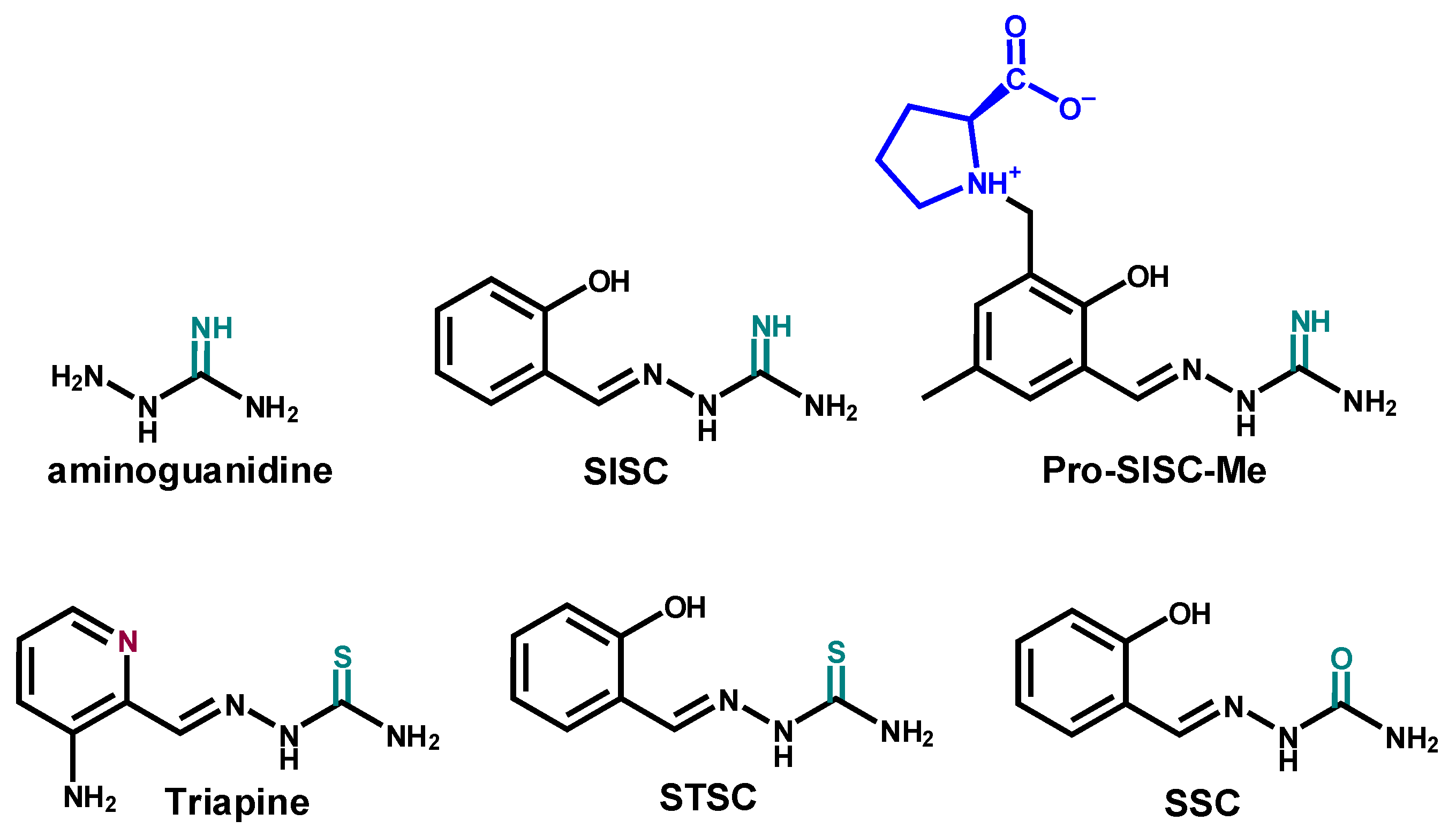
2.1. Synthesis and Characetriazion of Pro-SISC-Me
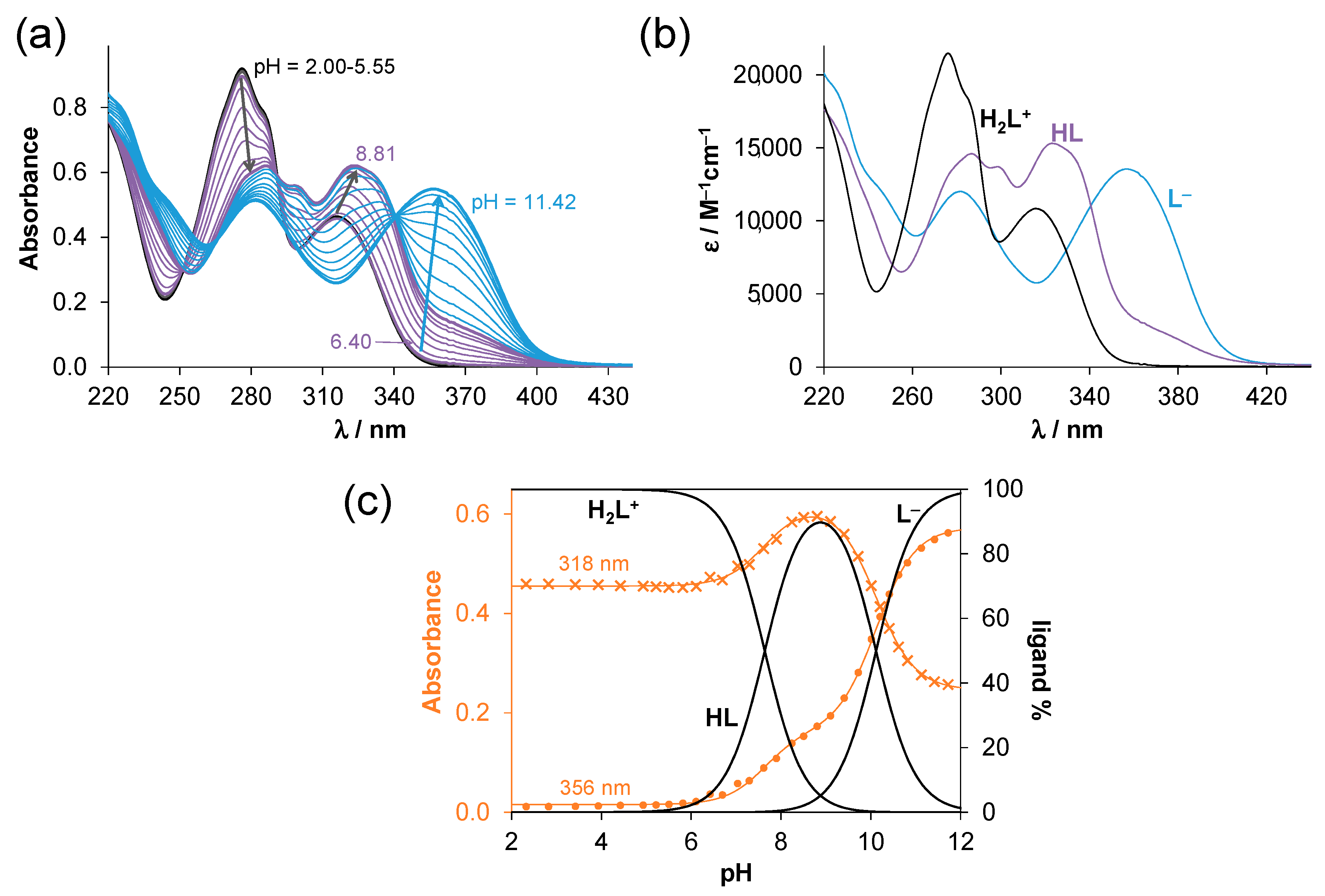
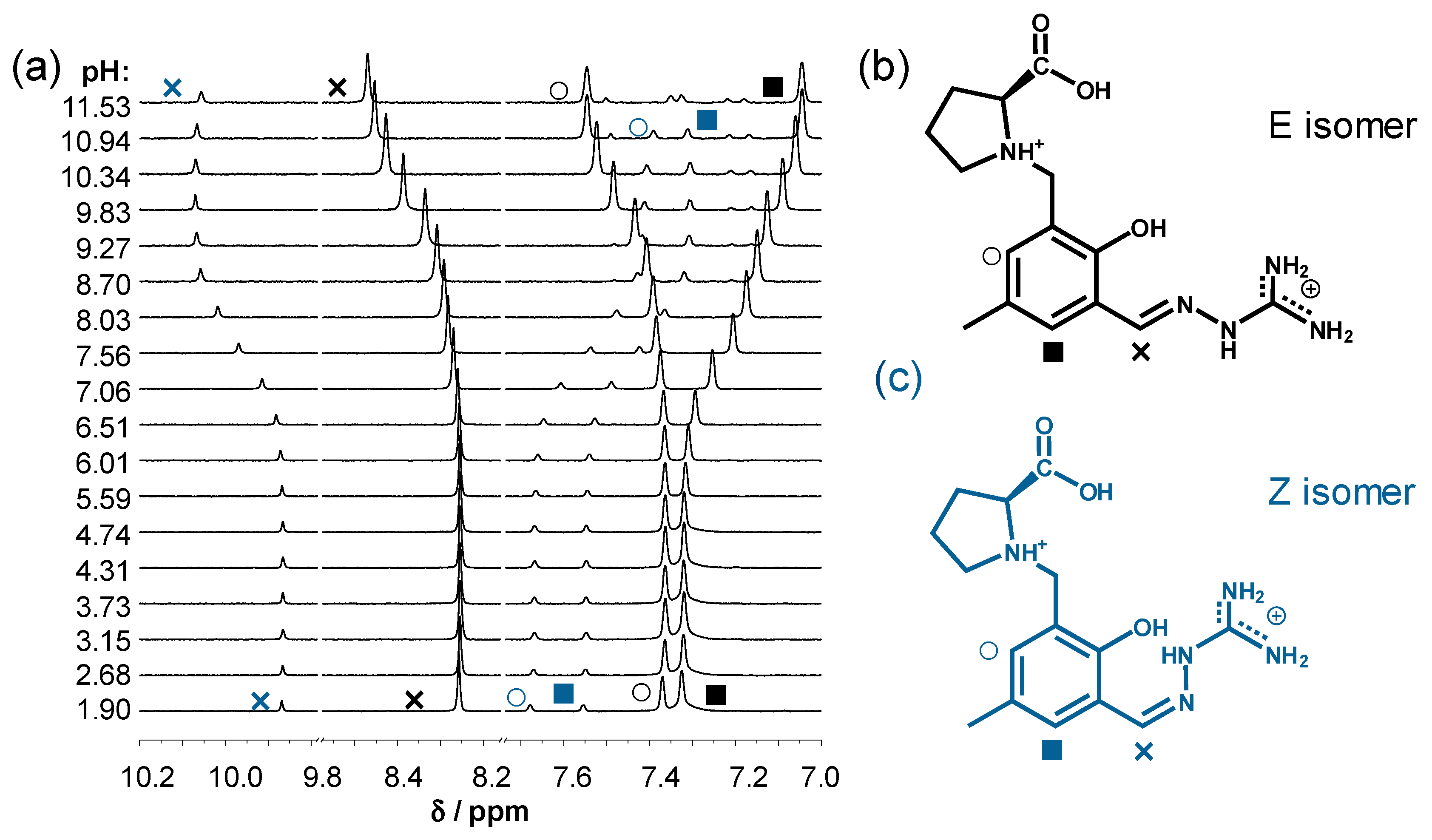
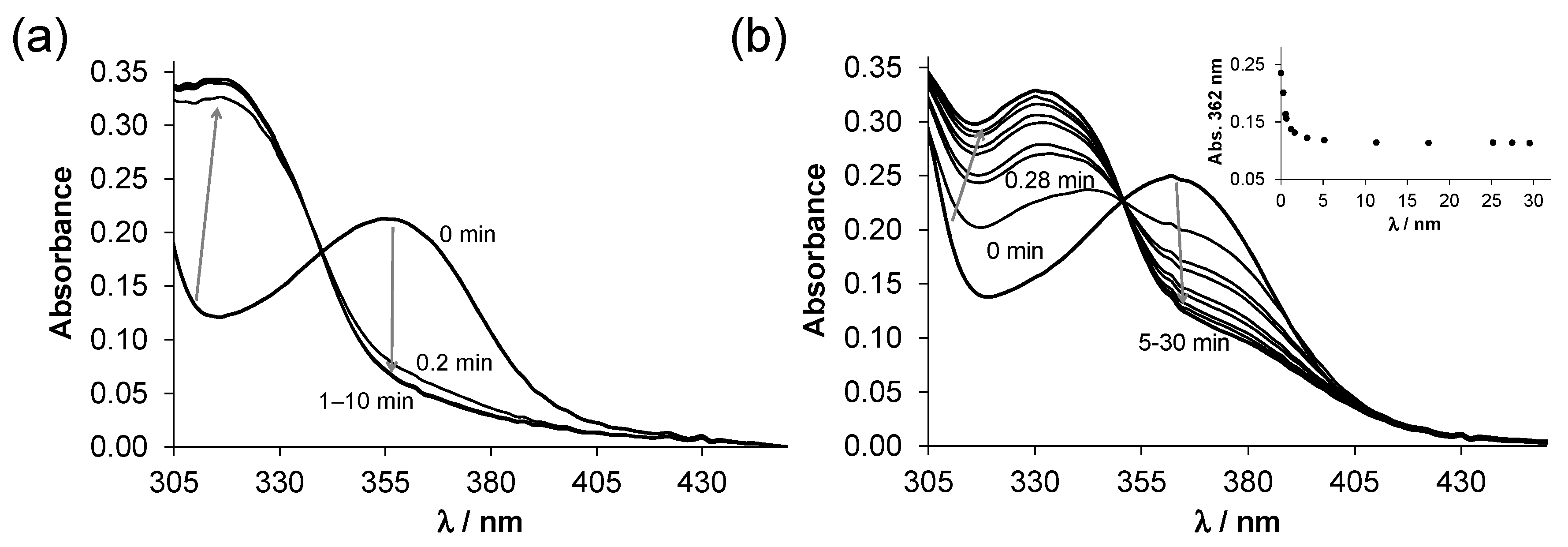
2.2. Solution Chemistry of SISC and Pro-SISC-Me
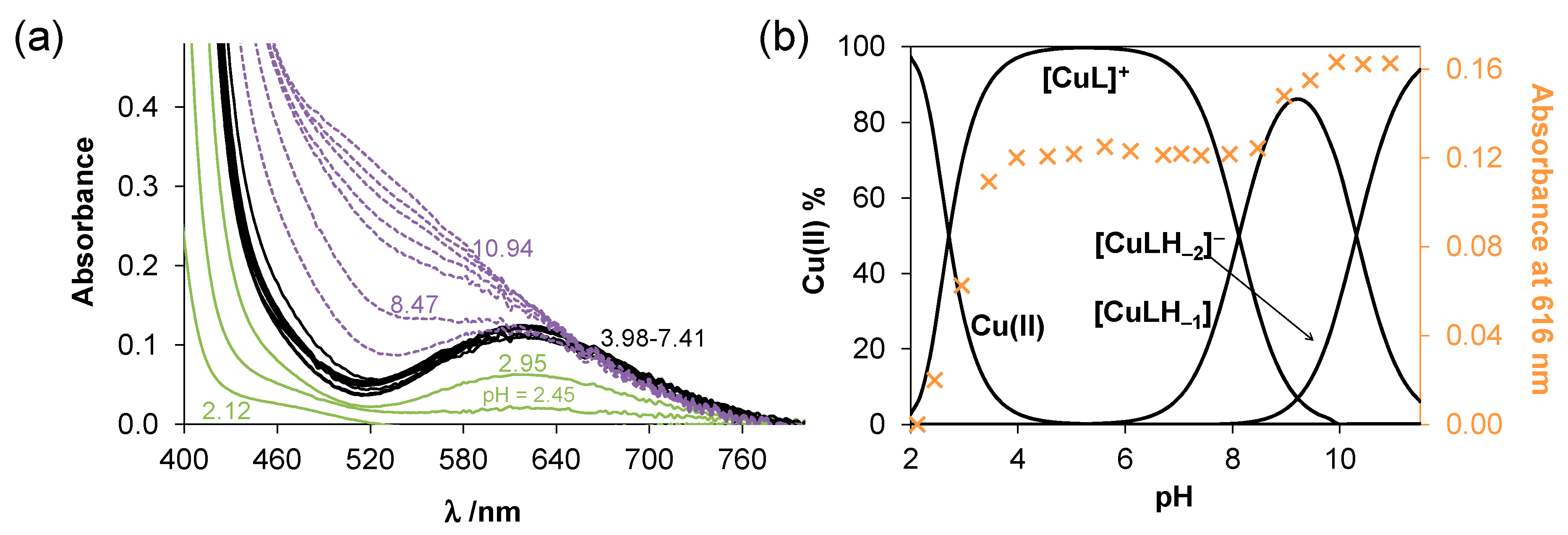
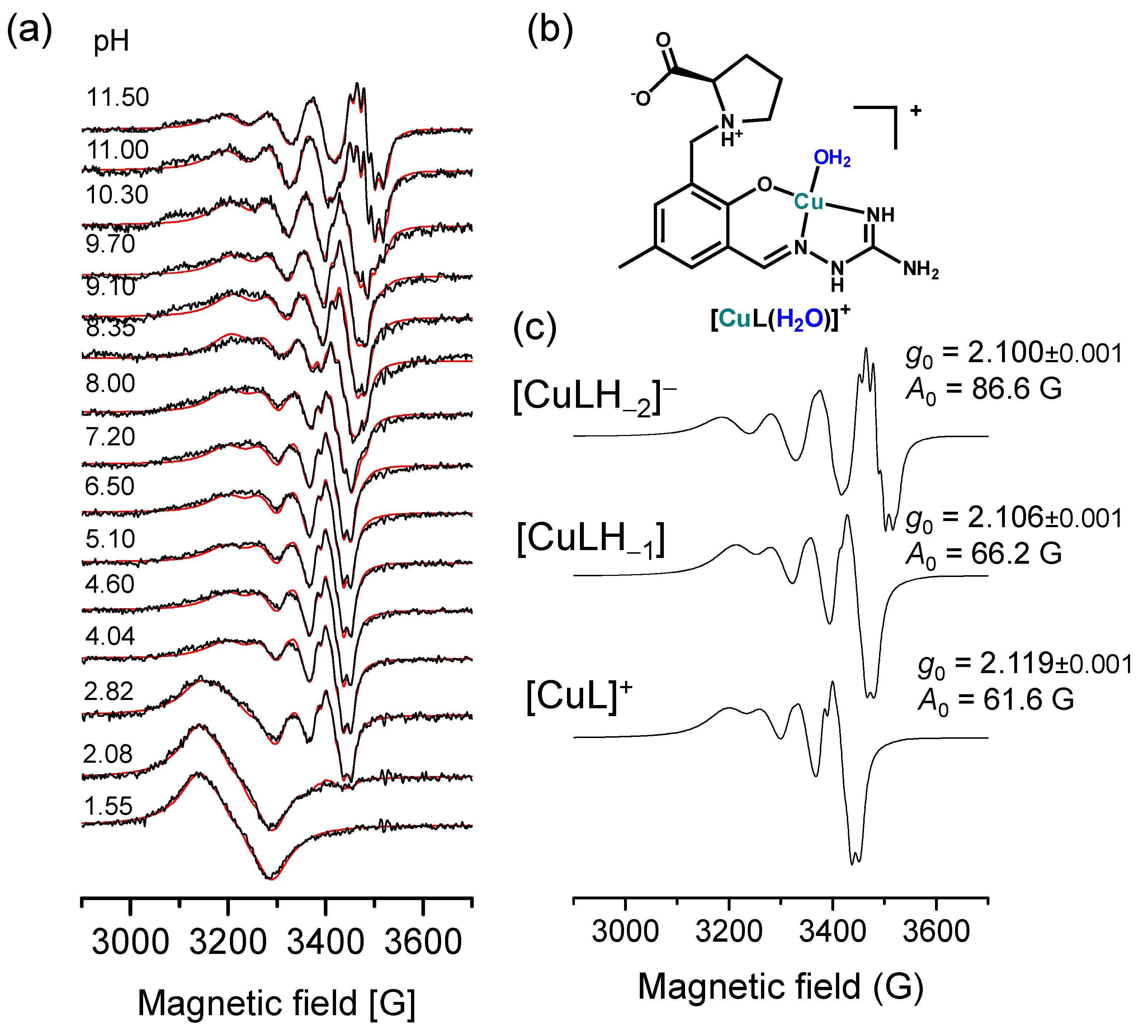
2.3. Complex Formation of SISC and Pro-SISC-Me with Cu(II) Ions
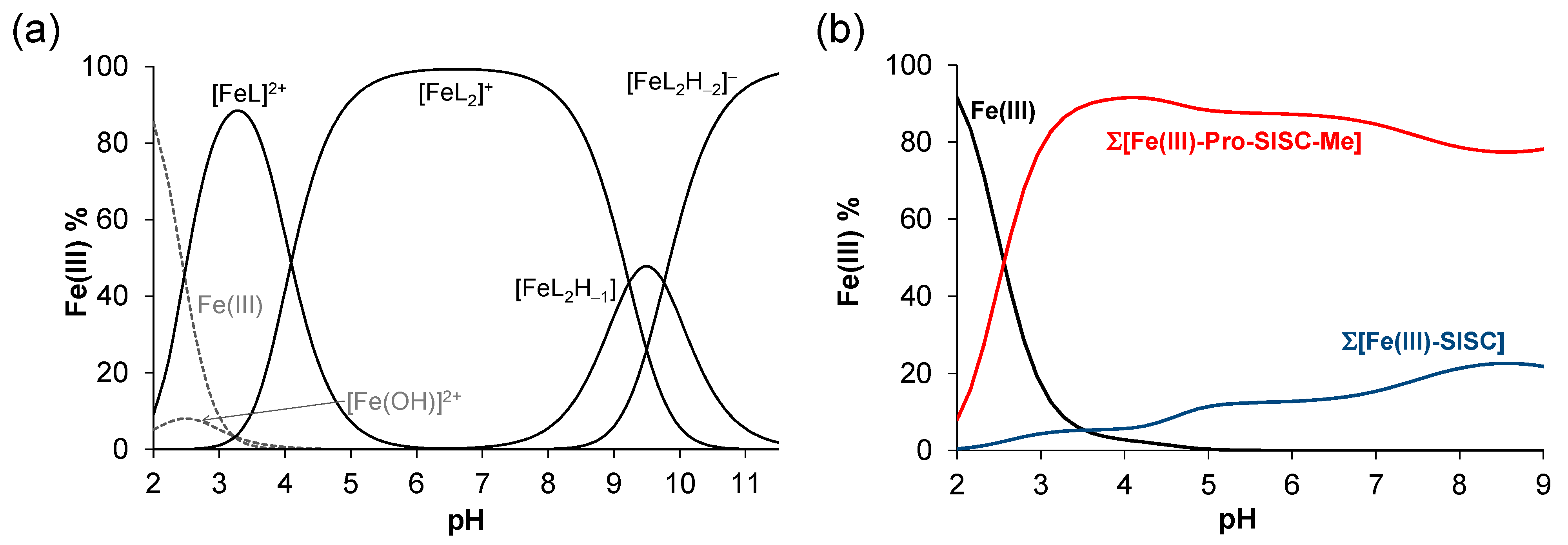
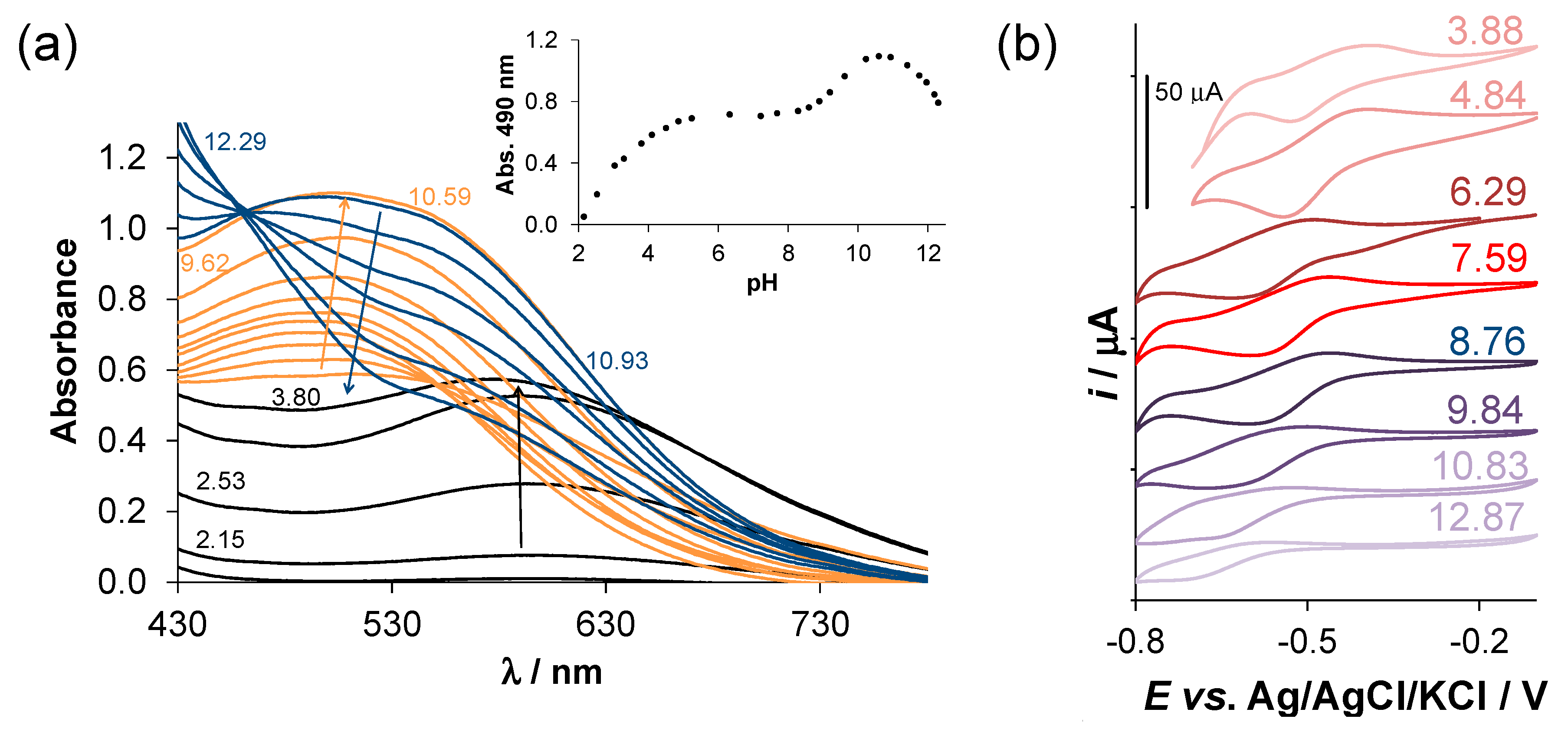
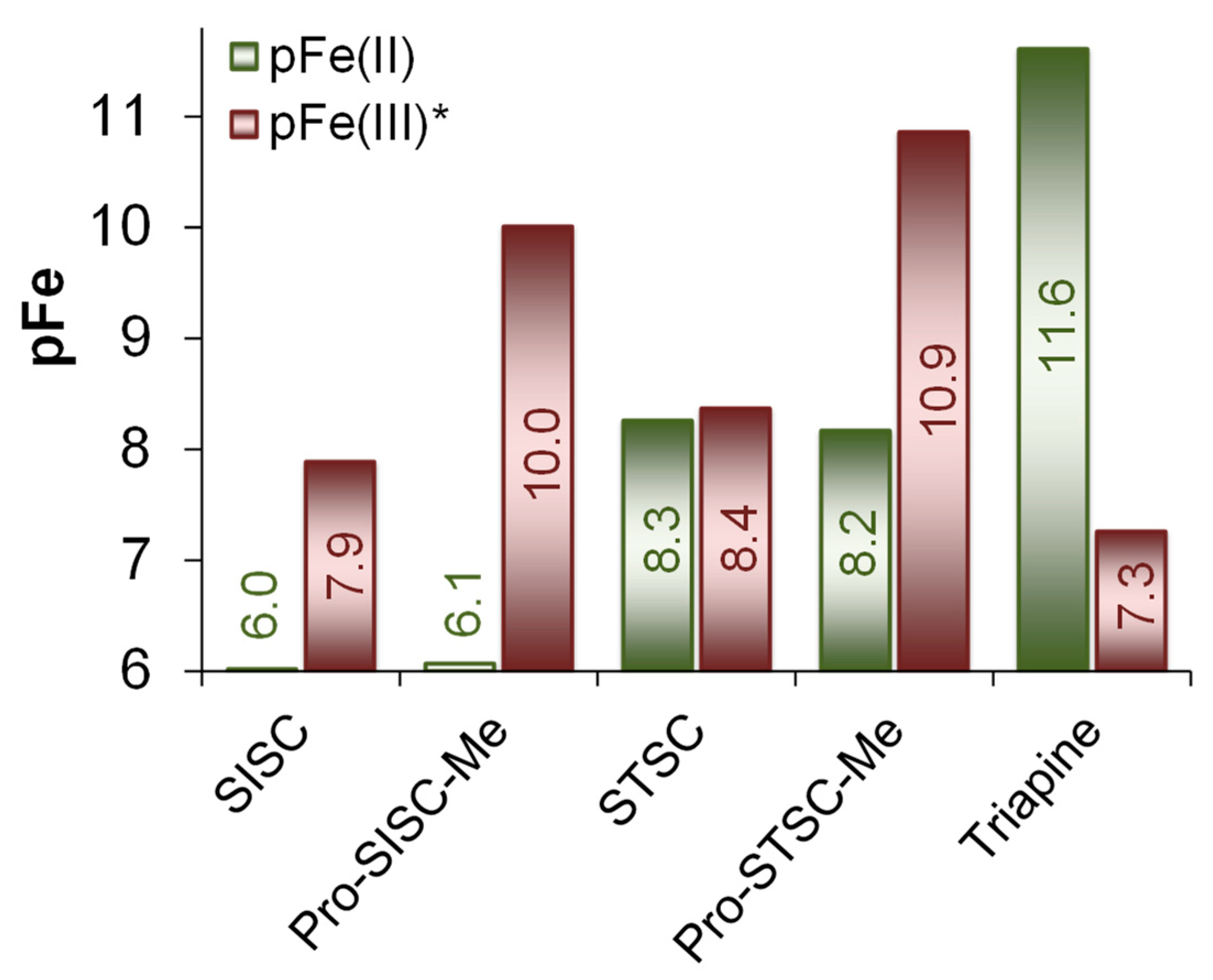
2.4. Complex Formation of SISC and Pro-SISC-Me with Fe(III) and Fe(II) Ions
2.5. In Vitro Cytotoxicity of the Compounds
3. Materials and Methods
3.1. Chemicals
3.2. Synthesis of SISC and Pro-SISC-Me
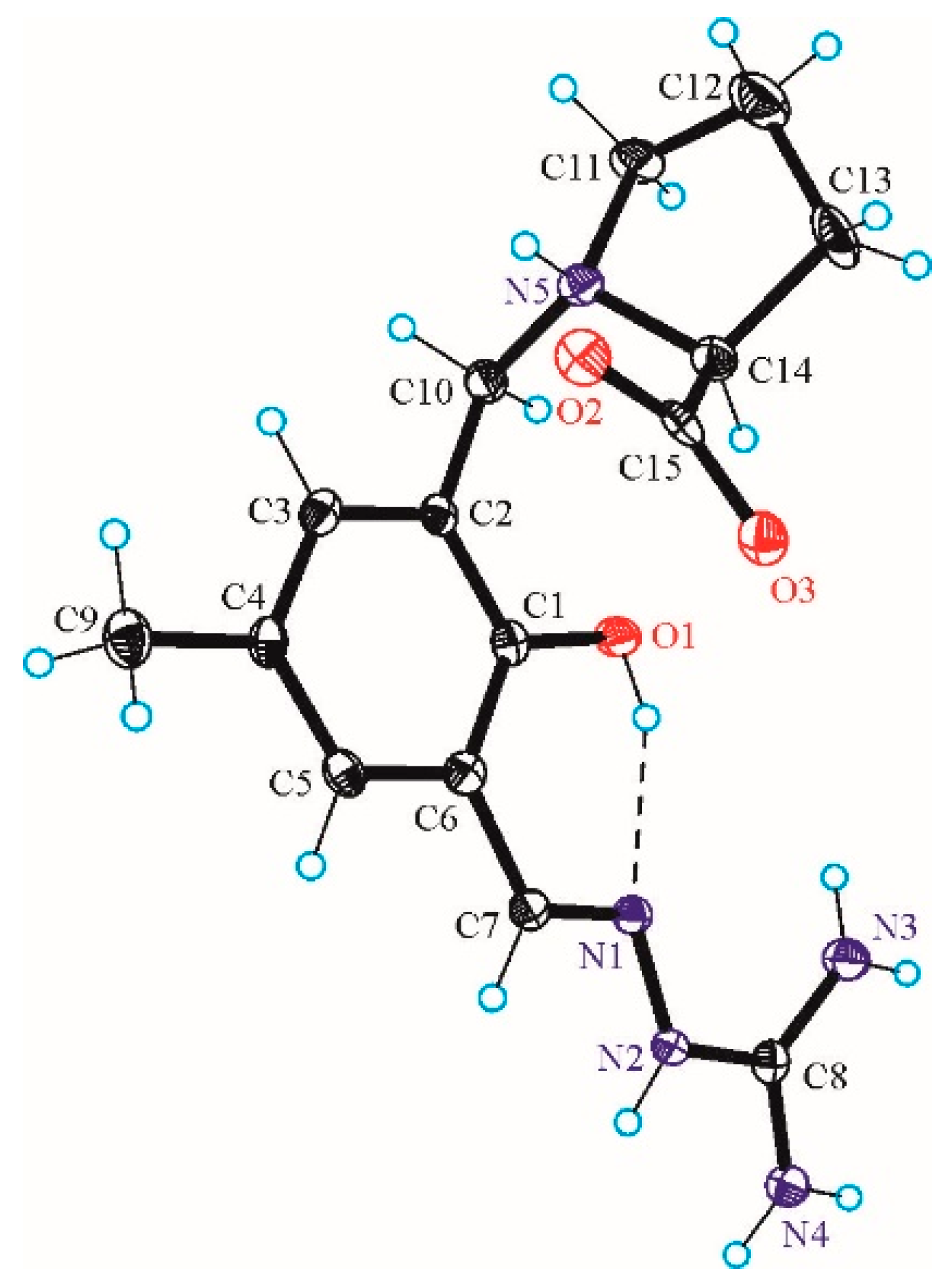
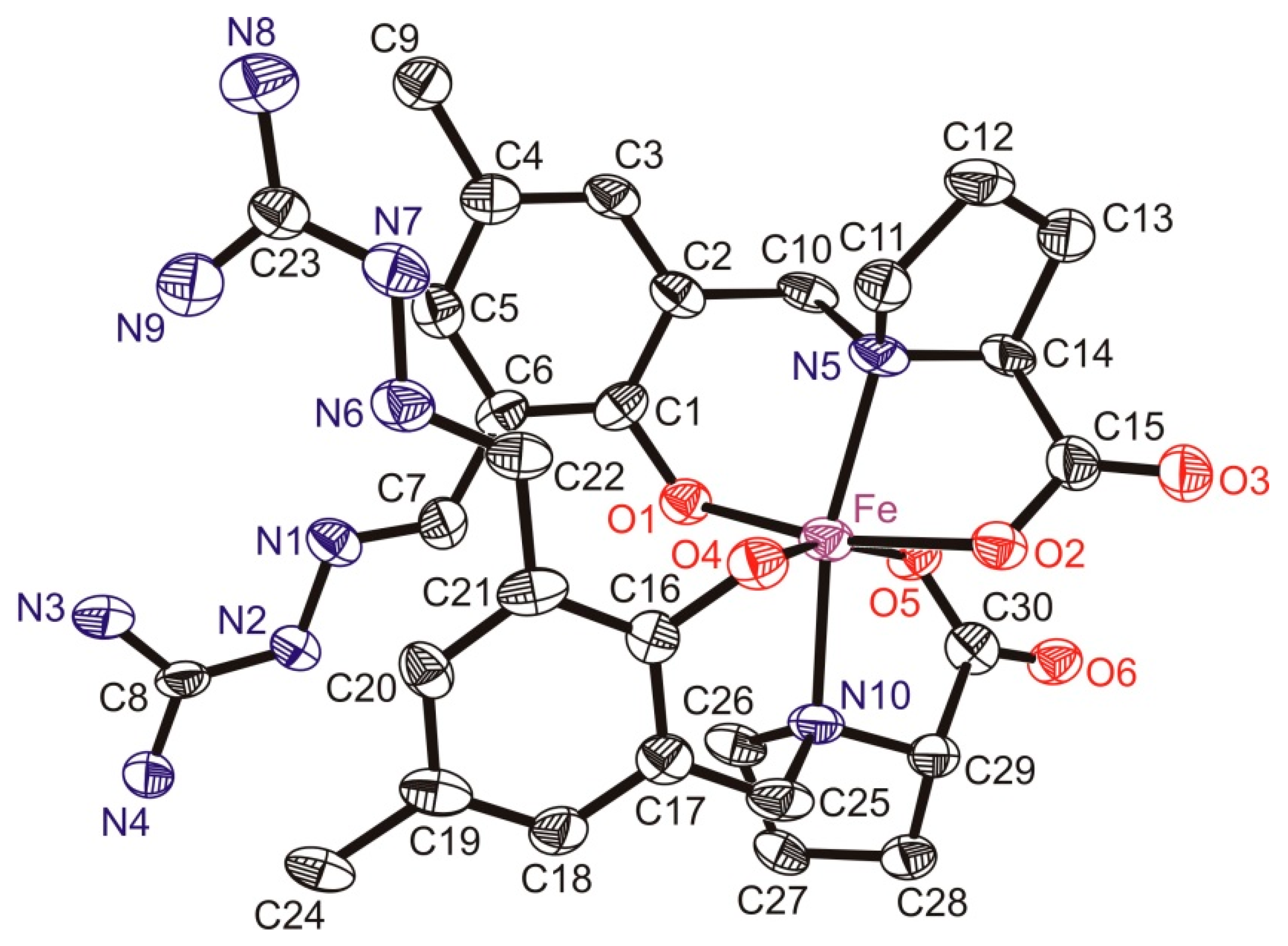
3.3. X-ray Data Collection, Structure Solution and Refinement for Pro-SISC-Me·HBr·H2O (1) and the Complex Na[Fe(III)(Pro-SISC-MeH−2)2](H2O)4 (2)
3.4. pH-Potentiometry
3.5. UV-Vis Spectrophotometry and Fluorometry
3.6. 1H NMR Spectroscopic Titrations
3.7. EPR Spectroscopy
3.8. Cyclic Voltammetry
3.9. In Vitro Cell Studies: Cell Lines and Culture Conditions and MTT Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Thornalley, P.J. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch. Biochem. Biophys. 2003, 419, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Sugiura, M.; Hamada, Y.; Miwa, I. In vivo formation of a Schiff base of aminoguanidine with pyridoxal phosphate. Biochem. Pharmacol. 1998, 55, 1667–1671. [Google Scholar] [CrossRef]
- Mondal, A.; Das, C.; Corbella, M.; Bauzá, A.; Frontera, A.; Saha, M.; Mondal, S.; Das Saha, K.; Chattopadhyay, S.K. Biological promiscuity of a binuclear Cu (ii) complex of aminoguanidine Schiff base: DNA binding, anticancer activity and histidine sensing ability of the complex. New J. Chem. 2020, 44, 7319–7328. [Google Scholar] [CrossRef]
- Chakraborty, M.; Mohanty, M.; Dinda, R.; Sengupta, S.; Chattopadhyay, S.K. Cu(II) complexes of a bio-compatible aminoguanidine Schiff base: Histidine sensing and DNA-binding studies. Polyhedron 2022, 211, 115554. [Google Scholar] [CrossRef]
- Chumakov, Y.M.; TsapKov, V.I.; Bocelli, G.; Antosyak, B.Y.; Shova, S.G.; Gulea, A.P. Crystal structures of salicylideneguanylhydrazinium chloride and its copper(II) and cobalt(III) chloride complexes. Crystallogr. Rep. 2006, 51, 60–67. [Google Scholar] [CrossRef]
- Vojinović-Ješić, L.S.; Radanović, M.M.; Rodić, M.V.; Živković-Radovanović, V.; Jovanović, L.S.; Leovac, V.M. Syntheses and characterization of 2-acetylpyridine-aminoguanidine and its copper(II) complexes: Crystallographic and antimicrobial study. Polyhedron 2016, 117, 526–534. [Google Scholar] [CrossRef]
- Jakuš, V.; Hrnčiarová, M.; Čársky, J.; Krahulec, B.; Rietbrock, N. Inhibition of nonenzymatic protein glycation and lipid peroxidation by drugs with antioxidant activity. Life Sci. 1999, 65, 1991–1993. [Google Scholar] [CrossRef]
- Matela, G. Schiff Bases and Complexes: A Review on Anti-Cancer Activity. Anticancer Agents Med. Chem. (Formerly Curr. Med. Chem. Anticancer. Agents) 2020, 20, 1908–1917. [Google Scholar] [CrossRef]
- Iacopetta, D.; Ceramella, J.; Catalano, A.; Saturnino, C.; Bonomo, M.G.; Franchini, C.; Sinicropi, M.S. Schiff Bases: Interesting Scaffolds with Promising Antitumoral Properties. Appl. Sci. 2021, 11, 1877. [Google Scholar] [CrossRef]
- Vojinović-Ješić, L.S.; Radanović, M.M.; Rodić, M.V.; Jovanović, L.S.; Češljević, V.I.; Joksović, M.D. Syntheses, structures and spectral characterization of copper(II) and dioxidovanadium(V) complexes with salicylidene-aminoguanidine. Polyhedron 2014, 80, 90–96. [Google Scholar] [CrossRef]
- Lalović, M.M.; Vojinović-Ješić, L.S.; Jovanović, L.S.; Leovac, V.M.; Češljević, V.I.; Divjaković, V. Synthesis, characterization and crystal structure of square-pyramidal copper(II) complexes with pyridoxylidene aminoguanidine. Inorg. Chim. Acta 2012, 388, 157–162. [Google Scholar] [CrossRef]
- Buvaylo, E.A.; Kasyanova, K.A.; Vassilyeva, O.Y.; Skelton, B.W. Crystal structure of bis {4-bromo-2-[(carbamimidamidoimino) methyl] phenolato-κ3N, N′, O} cobalt(III) nitrate dimethylformamide monosolvate. Acta Crystallogr. E Crystallogr. Commun. 2016, 72, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Vassilyeva, O.Y.; Buvaylo, E.A.; Kokozay, V.N.; Studzinsky, S.L.; Skelton, B.W.; Vasyliev, G.S. NiII molecular complex with a tetradentate aminoguanidine-derived Schiff base ligand: Structural, spectroscopic and electrochemical studies and photoelectric response. Acta Crystallogr. E Crystallogr. Commun. 2022, E78, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, J.R.; Hueting, R. Metal complexes of thiosemicarbazones for imaging and therapy. Inorg. Chim. Acta 2012, 389, 3–15. [Google Scholar] [CrossRef]
- Heffeter, P.; Pape, V.F.S.; Enyedy, É.A.; Keppler, B.K.; Szakács, G.; Kowol, C.R. Anticancer thiosemicarbazones: Chemical properties, interaction with iron metabolism, and resistance development. Antioxid. Redox Signal. 2019, 30, 1062–1082. [Google Scholar] [CrossRef]
- Lobana, T.S.; Sharma, R.; Bawa, G.; Khanna, S. Bonding and structure trends of thiosemicarbazone derivatives of–An overview. Coord. Chem. Rev. 2009, 253, 977–1055. [Google Scholar] [CrossRef]
- Kalinowski, D.S.; Quach, P.; Richardson, D.R. Thiosemicarbazones: The new wave in cancer treatment. Future Med. Chem. 2009, 1, 1143–1151. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT02466971 (accessed on 27 February 2022).
- Shao, J.; Zhou, B.; Di Bilio, A.J.; Zhu, L.; Wang, T.; Shih, C.Q.J.; Yen, Y. A Ferrous-Triapine complex mediates formation of reactive oxygen species that inactivate human ribonucleotide reductase. Mol. Cancer Ther. 2006, 5, 586–592. [Google Scholar] [CrossRef]
- Enyedy, E.A.; May, N.V.; Pape, V.F.S.; Heffeter, P.; Szakács, G.; Keppler, B.K.; Kowol, C.R. Complex formation and cytotoxicity of Triapine derivatives: A comparative solution study on the effect of the chalcogen atom and NH-methylation. Dalton Trans. 2020, 49, 16887. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Nagy, N.V.; Zsigó, É.; Kowol, C.R.; Arion, V.B.; Roller, A.; Keppler, B.K.; Kiss, T. Comparative solution equilibrium study of the interactions of copper(II), iron(II) and zinc(II) with Triapine (3-aminopyridine-2-carbaldehyde thiosemicarbazone) and related ligands. Eur. J. Inorg. Chem. 2010, 2010, 1717–1728. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Zsigó, É.; Nagy, N.V.; Kowol, C.R.; Roller, A.; Keppler, B.K.; Kiss, T. Complex-formation ability of salicylaldehyde thiosemicarbazone towards ZnII, CuII, FeII, FeIII and GaIII Ions. Eur. J. Inorg. Chem. 2012, 2012, 4036–4047. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Bognár, G.M.; Nagy, N.V.; Jakusch, T.; Kiss, T.; Gambino, D. Solution speciation of potential anticancer metal complexes of salicylaldehyde semicarbazone and its bromo derivative. Polyhedron 2014, 67, 242–252. [Google Scholar] [CrossRef]
- Petrasheuskaya, T.V.; Kiss, M.A.; Dömötör, O.; Holczbauer, T.; May, N.V.; Spengler, G.; Kincses, A.; Čipak Gašparović, A.; Frank, É.; Enyedy, É.A. Salicylaldehyde thiosemicarbazone copper complexes: Impact of hybridization with estrone on cytotoxicity, solution stability and redox activity. New J. Chem. 2020, 44, 12154–12168. [Google Scholar] [CrossRef]
- Lee, W.Y.; Lee, P.P.F.; Yan, Y.K.; Lau, M. Cytotoxic copper (II) salicylaldehyde semicarbazone complexes: Mode of action and proteomic analysis. Metallomics 2010, 2, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Pósa, V.; Hajdu, B.; Tóth, G.; Dömötör, O.; Kowol, C.R.; Keppler, B.K.; Spengler, G.; Gyurcsik, B.; Enyedy, É.A. Variation in the coordination modes of copper(II) complexes of (thio)semicarbazones affecting the solution chemical properties and mechanism of the biological activity. J. Inorg. Biochem. 2022, 231, 1111786. [Google Scholar] [CrossRef] [PubMed]
- Milunovic, M.N.; Enyedy, É.A.; Nagy, N.V.; Kiss, T.; Trondl, R.; Jakupec, M.A.; Keppler, B.K.; Krachler, R.; Novitchi, G.; Arion, V.B. L-and D-proline thiosemicarbazone conjugates: Coordination behavior in solution and the effect of copper(II) coordination on their antiproliferative activity. Inorg. Chem. 2012, 51, 9309–9321. [Google Scholar] [CrossRef]
- Dobrova, A.; Platzer, S.; Bacher, F.; Milunovic, M.N.; Dobrov, A.; Spengler, G.; Enyedy, É.A.; Novitchi, G.; Arion, V.B. Structure–antiproliferative activity studies on L-proline-and homoproline-4-N-pyrrolidine-3-thiosemicarbazone hybrids and their nickel(II), palladium(II) and copper(II) complexes. Dalton Trans. 2016, 45, 13427–13439. [Google Scholar] [CrossRef]
- Milunović, M.N.; Dobrova, A.; Ghenadie, N.; Gligorijević, N.; Radulović, S.; Kožišek, J.; Rapta, P.; Enyedy, É.A.; Arion, V.B. Effects of terminal substitution and iron coordination on antiproliferative activity of L-proline-salicylaldehyde-thiosemicarbazone hybrids. Eur. J. Inorg. Chem. 2017, 2017, 4773–4783. [Google Scholar] [CrossRef]
- Bacher, F.; Enyedy, E.A.; Nagy, N.V.; Rockenbauer, A.; Bognár, G.M.; Trondl, R.; Novak, M.S.; Klapproth, E.; Kiss, T.; Arion, V.B. Copper (II) complexes with highly water-soluble L-and D-Proline–thiosemicarbazone conjugates as potential inhibitors of topoisomerase IIα. Inorg. Chem. 2013, 52, 8895–8908. [Google Scholar] [CrossRef]
- Bacher, F.; Dömötör, O.; Kaltenbrunner, M.; Mojovic, M.; Popovic-Bijelic, A.; Graslund, A.; Ozarowski, A.; Filipovic, L.; Radulović, S.; Enyedy, É.A.; et al. Effects of terminal dimethylation and metal coordination of proline-2-formylpyridine thiosemicarbazone hybrids on lipophilicity, antiproliferative activity, and hR2 RNR inhibition. Inorg. Chem. 2014, 53, 12595–12609. [Google Scholar] [CrossRef]
- Bacher, F.; Dömötör, O.; Enyedy, É.A.; Filipović, L.; Radulović, S.; Smith, G.S.; Arion, V.B. Complex formation reactions of gallium (III) and iron (III/II) with L-proline-thiosemicarbazone hybrids: A comparative study. Inorg. Chim. Acta 2017, 455, 505–513. [Google Scholar] [CrossRef][Green Version]
- Mészáros, J.P.; Poljarevic, J.M.; Szatmári, I.; Csuvik, O.; Fülöp, F.; Szoboszlai, N.; Spengler, G.; Enyedy, É.A. An 8-hydroxyquinoline–proline hybrid with multidrug resistance reversal activity and the solution chemistry of its half-sandwich organometallic Ru and Rh complexes. Dalton. Trans. 2020, 49, 7977–7992. [Google Scholar] [CrossRef] [PubMed]
- Enyedy, É.A.; Primik, M.F.; Kowol, C.R.; Arion, V.B.; Kiss, T.; Keppler, B.K. Interaction of Triapine and related thiosemicarbazones with iron(III)/(II) and gallium(III): A comparative solution equilibrium study. Dalton Trans. 2011, 40, 5895–5905. [Google Scholar] [CrossRef] [PubMed]
- Lalović, M.M.; Jovanović, L.S.; Vojinović-Ješić, L.S.; Leovac, V.M.; Češljević, V.I.; Rodić, M.V.; Divjaković, V. Syntheses, crystal structures, and electrochemical characterizations of two octahedral iron(III) complexes with Schiff base of pyridoxal and aminoguanidine. J. Coord. Chem. 2012, 65, 4217–4229. [Google Scholar] [CrossRef]
- Bruker. Saint-Plus; Version 7.06a, and APEX2; Bruker Nonius AXS Inc.: Madison, WI, USA, 2004. [Google Scholar]
- Rigaku Corporation. CrystalClear SM 1.4.0; Rigaku Corporation: Tokyo, Japan, 2008. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Burnett, M.N.; Johnson, G.K. ORTEPIII, Report ORNL-5138; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1996.
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement viaSIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A study of some problems in determining the stoicheiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Zékány, L.; Nagypál, I. PSEQUAD. In Computational Methods for the Determination of Stability Constants; Leggett, L., Ed.; Plenum Press: New York, NY, USA, 1985; p. 291. [Google Scholar]
- Rockenbauer, A.; Korecz, L. Automatic computer simulations of ESR spectra. Appl. Magn. Reson. 1996, 10, 29–43. [Google Scholar] [CrossRef]
- Rockenbauer, A.; Szabó-Plánka, T.; Árkosi, Z.; Korecz, L. A two-dimensional (magnetic field and concentration) electron paramagnetic resonance method for analysis of multispecies complex equilibrium systems. Information content of EPR spectra. J. Am. Chem. Soc. 2001, 123, 7646–7654. [Google Scholar] [CrossRef] [PubMed]
- GraphPad Prism Version 7.00 for Windows; Graph Pad Software: La Jolla, CA, USA, 2018; Available online: http://www.graphpad.com (accessed on 27 February 2022).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SISC | Pro-SISC-Me | |||||
|---|---|---|---|---|---|---|
| Method | Medium | pK1 (H2L+) | pK2 (HL) | pK1 (H3L2+) | pK2 (H2L+) | pK3 (HL) |
| pH-pot. a | H2O | 7.64 ± 0.09 | 9.96 ± 0.09 | 2.52 ± 0.08 | 7.25 ± 0.06 | 9.61 ± 0.05 |
| UV-vis | 7.64 ± 0.03 | 10.12 ± 0.03 | n.d. b | 7.30 ± 0.03 | 9.67 ± 0.03 | |
| 1H NMR | - | - | ≤2.5 c | 7.25 ± 0.03 c | 9.75 ± 0.03 c | |
| pH-pot. a | 30% DMSO/H2O | 7.62 ± 0.03 | 10.63 ± 0.02 | 1.80 ± 0.08 | 7.08 ± 0.02 | 9.91 ± 0.02 |
| UV-vis | 7.56 ± 0.03 | 10.67 ± 0.03 | n.d. b | 7.07 ± 0.03 | 9.84 ± 0.03 | |
| 1H NMR | 7.68 ± 0.03 | 10.72 ± 0.03 | - | - | - | |
| logD7.4 | n-octanol/H2O | +0.83 ± 0.03 | −0.88 ± 0.05 | |||
| λEX(max) | H2O | 285 nm, 324 nm | 286 nm, 378 nm | |||
| λEM(max) | H2O | 480 nm | 495 nm | |||
| SISC | Pro-SISC-Me | Pro-SISC-Me | |
|---|---|---|---|
| 30% DMSO | 30% DMSO | H2O | |
| logβ [CuL]+ | 14.74 ± 0.02 | 15.08 ± 0.02 | 14.99 ± 0.02 |
| logβ [CuLH−1] | 6.28 ± 0.07 | 6.57 ± 0.05 | 6.96 ± 0.03 |
| logβ [CuLH−2]− | - | −4.42 ± 0.06 | −3.22 ± 0.04 |
| pCu(II) a | 12.08 | 13.39 | 13.59 |
| SISC | Pro-SISC-Me | |
|---|---|---|
| logβ [Fe(II)L]+ | 7.20 ± 0.06 | 6.83 ± 0.04 |
| logβ [Fe(II)LH−1] | −0.73 ± 0.07 | −1.08 ± 0.05 |
| logβ [Fe(II)LH−2]− | −8.94 ± 0.08 | <−11 |
| logβ [Fe(III)L]2+ | 14.96 ± 0.05 | 15.25 ± 0.04 |
| logβ [Fe(III)L2]+ | 27.46 ± 0.05 | 27.63 ± 0.05 |
| logβ [Fe(III)L2H−1] | - | 18.40 ± 0.07 |
| logβ [Fe(III)L2H−2]− | - | 8.64 ± 0.08 |
| IC50/μM | Colo-205 | Colo-320 | MRC-5 |
|---|---|---|---|
| SISC | >80 | >80 | >80 |
| Cu(II)—SISC | 18.7 ± 1.2 | 34.0 ± 7.6 | 58.9 ± 2.1 |
| Pro-SISC-Me | >80 | >80 | >80 |
| Cu(II)—Pro-SISC-Me | >80 | >80 | >80 |
| STSC a | 69.7 ± 1.9 | 52.2 ± 6.8 | 88 ± 19 |
| Cu(II)—STSC a | 0.89 ± 0.09 | 0.64 ± 0.01 | 0.65 ± 0.10 |
| Triapine a | 3.28 ± 0.63 | 2.17 ± 0.15 | 31.4 ± 9.2 |
| Cu(II)—Triapine a | 11.4 ± 1.4 | 26.00 ± 0.73 | 35.61 ± 0.69 |
| CuCl2 | 44.1 ± 0.4 | 48.7 ± 0.4 | 43.9 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dömötör, O.; May, N.V.; Gál, G.T.; Spengler, G.; Dobrova, A.; Arion, V.B.; Enyedy, É.A. Solution Equilibrium Studies on Salicylidene Aminoguanidine Schiff Base Metal Complexes: Impact of the Hybridization with L-Proline on Stability, Redox Activity and Cytotoxicity. Molecules 2022, 27, 2044. https://doi.org/10.3390/molecules27072044
Dömötör O, May NV, Gál GT, Spengler G, Dobrova A, Arion VB, Enyedy ÉA. Solution Equilibrium Studies on Salicylidene Aminoguanidine Schiff Base Metal Complexes: Impact of the Hybridization with L-Proline on Stability, Redox Activity and Cytotoxicity. Molecules. 2022; 27(7):2044. https://doi.org/10.3390/molecules27072044
Chicago/Turabian StyleDömötör, Orsolya, Nóra V. May, G. Tamás Gál, Gabriella Spengler, Aliona Dobrova, Vladimir B. Arion, and Éva A. Enyedy. 2022. "Solution Equilibrium Studies on Salicylidene Aminoguanidine Schiff Base Metal Complexes: Impact of the Hybridization with L-Proline on Stability, Redox Activity and Cytotoxicity" Molecules 27, no. 7: 2044. https://doi.org/10.3390/molecules27072044
APA StyleDömötör, O., May, N. V., Gál, G. T., Spengler, G., Dobrova, A., Arion, V. B., & Enyedy, É. A. (2022). Solution Equilibrium Studies on Salicylidene Aminoguanidine Schiff Base Metal Complexes: Impact of the Hybridization with L-Proline on Stability, Redox Activity and Cytotoxicity. Molecules, 27(7), 2044. https://doi.org/10.3390/molecules27072044