
Completion of the Total Synthesis of Several Bioactive Sarpagine/Macroline Alkaloids including the Important NF-κB Inhibitor N4-Methyltalpinine

 ,
,  ,
,  and
and 
Abstract
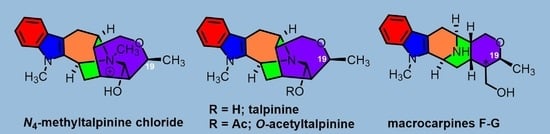
:
1. Introduction
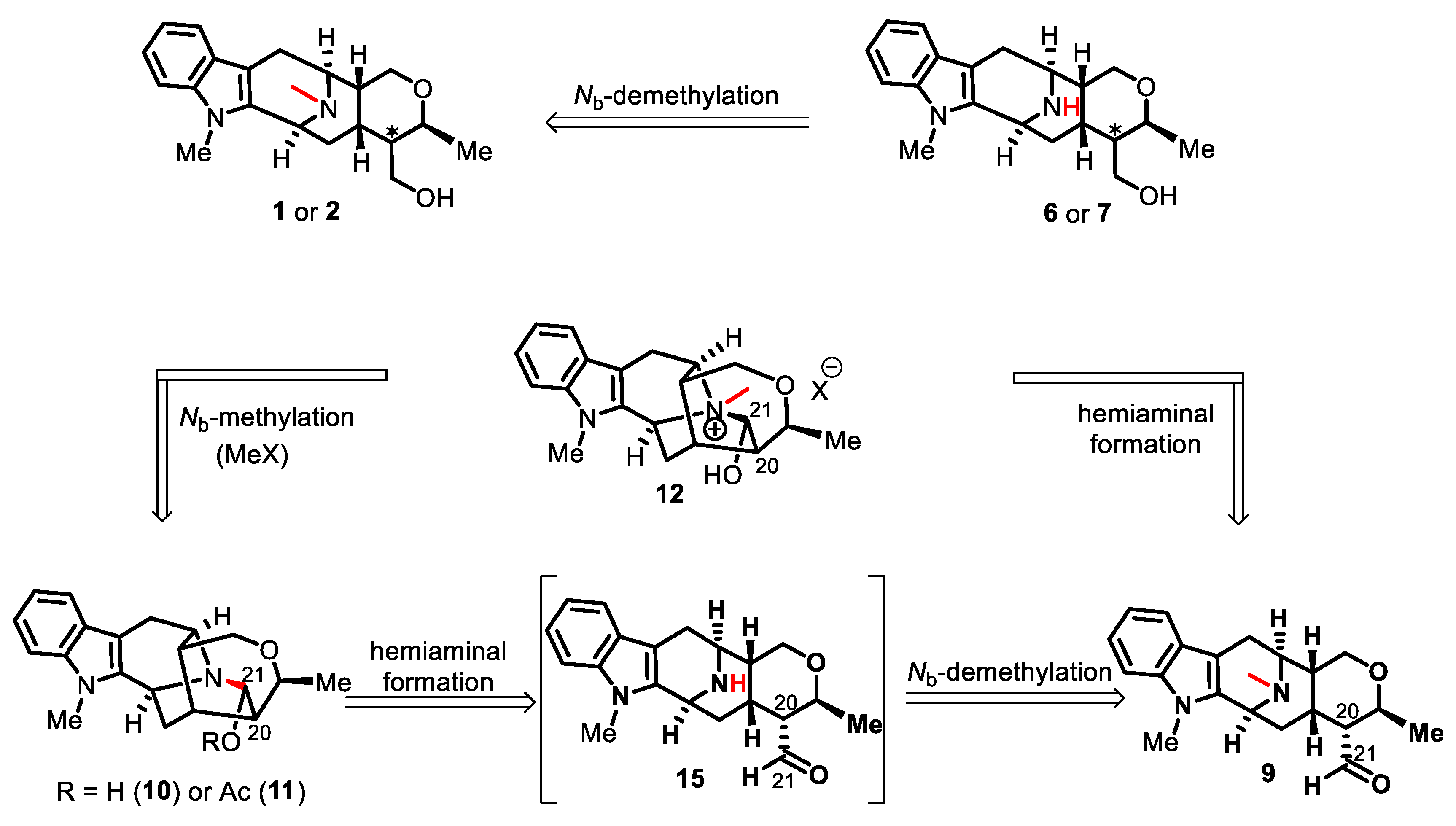
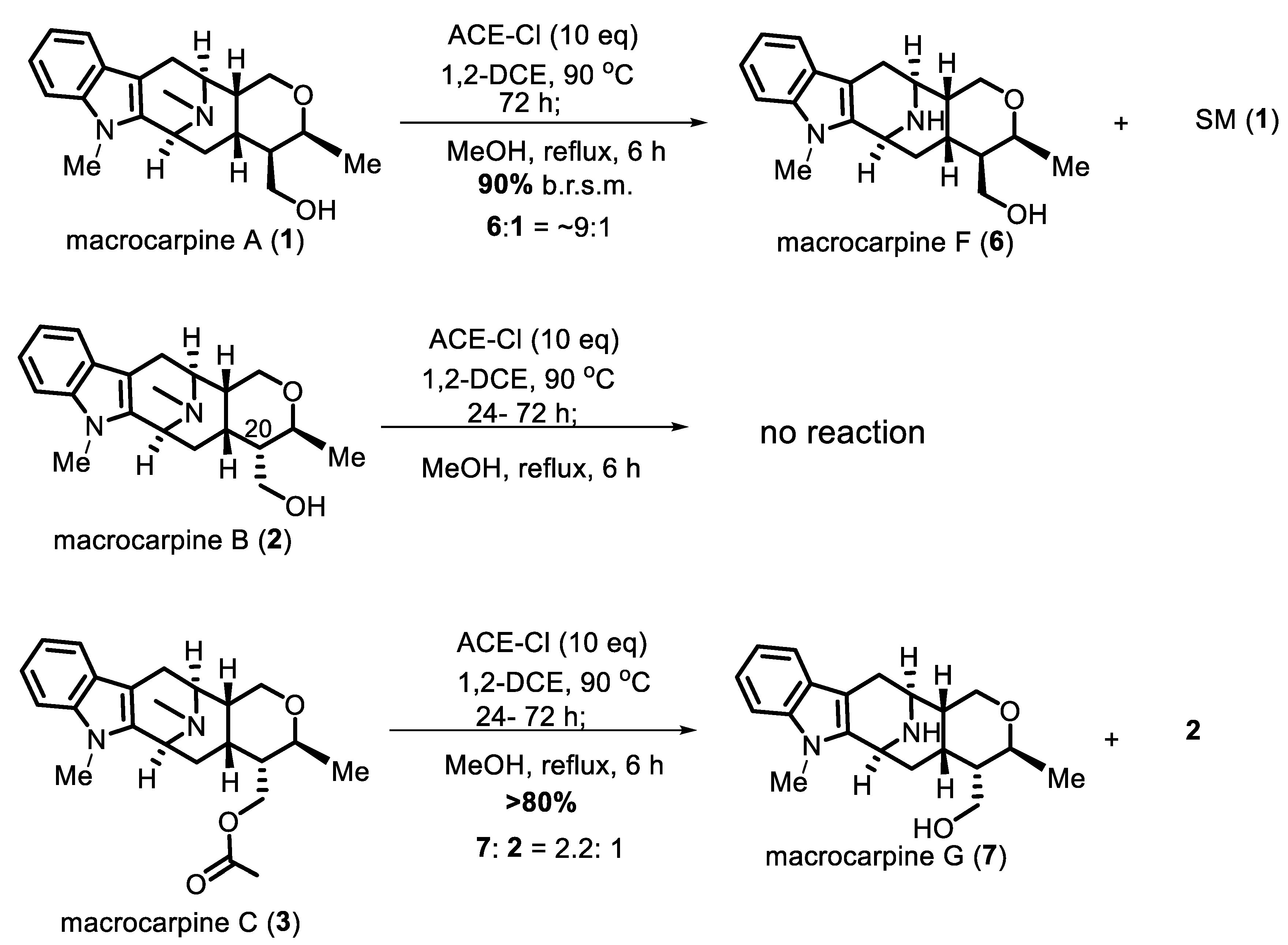
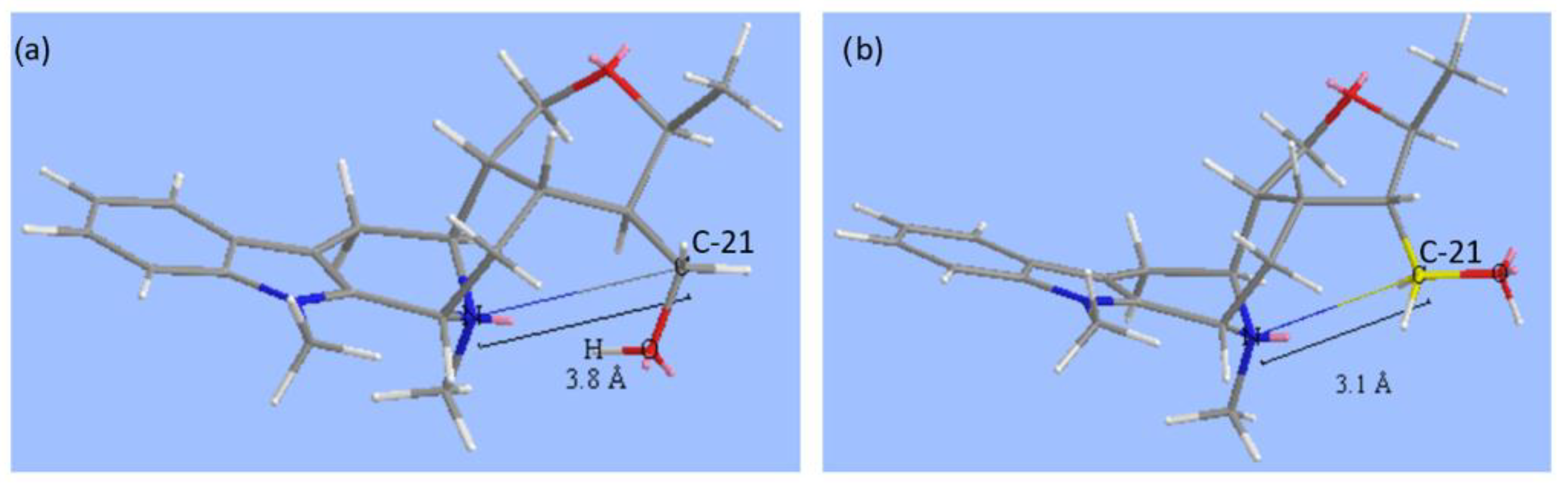
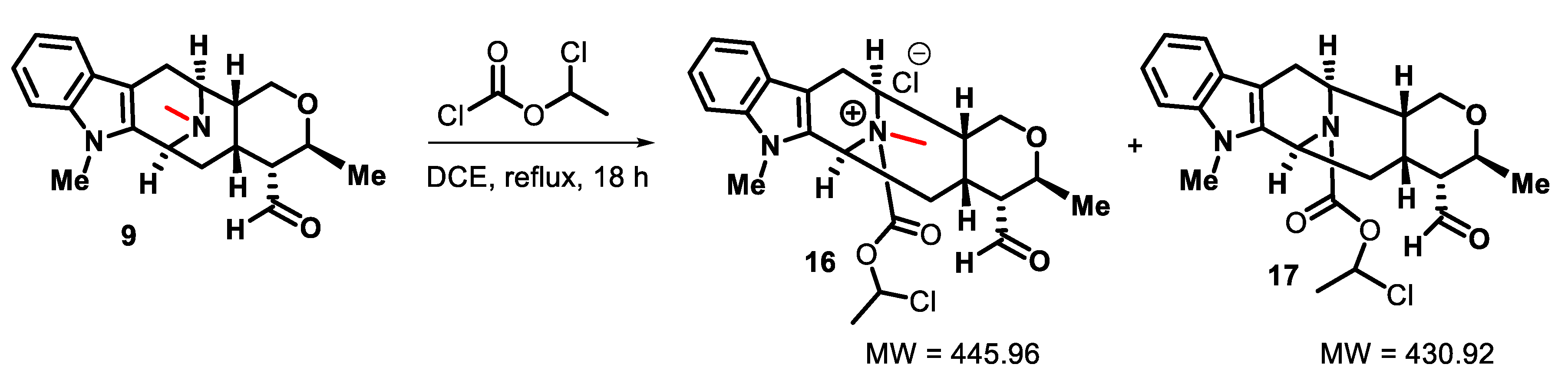
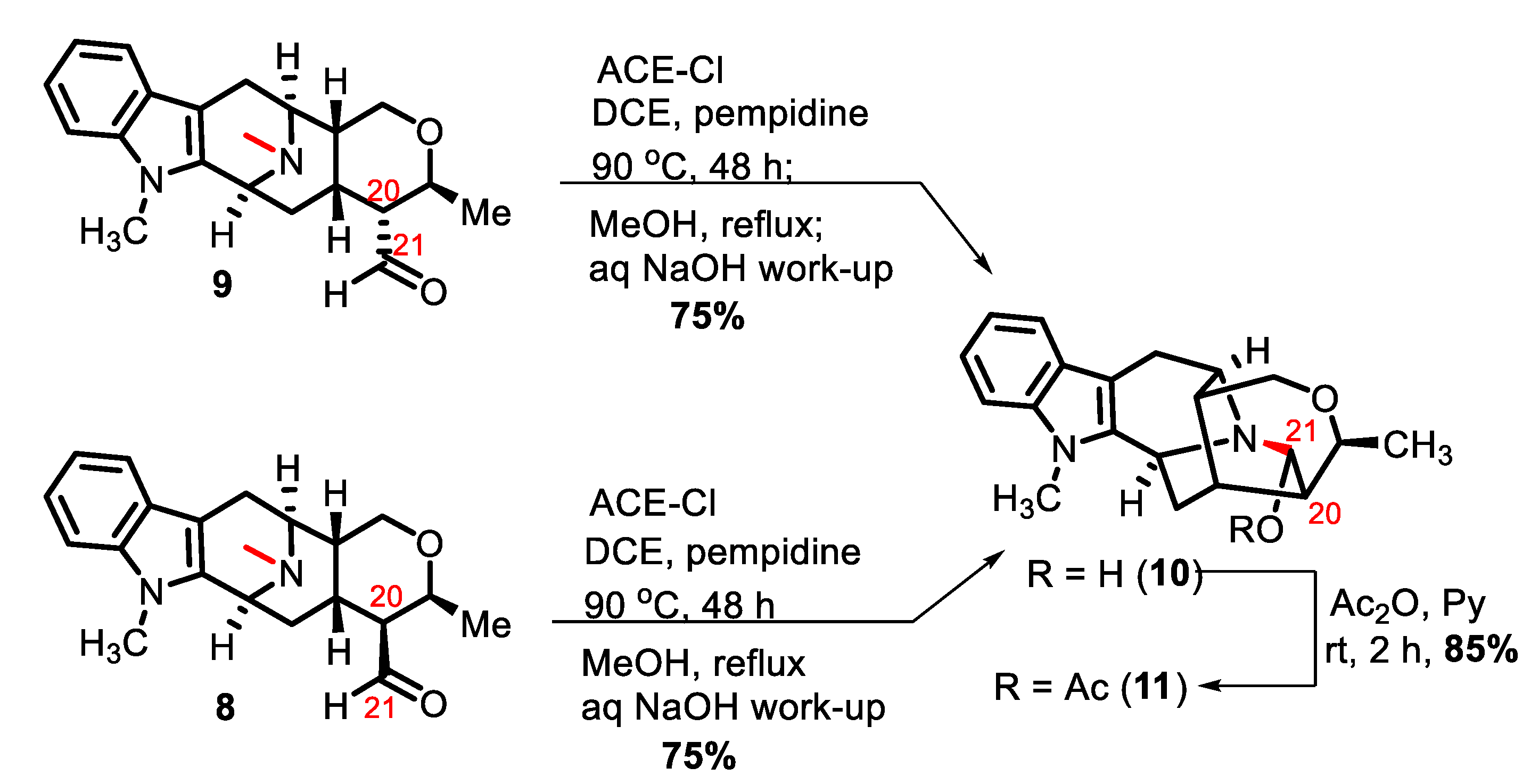
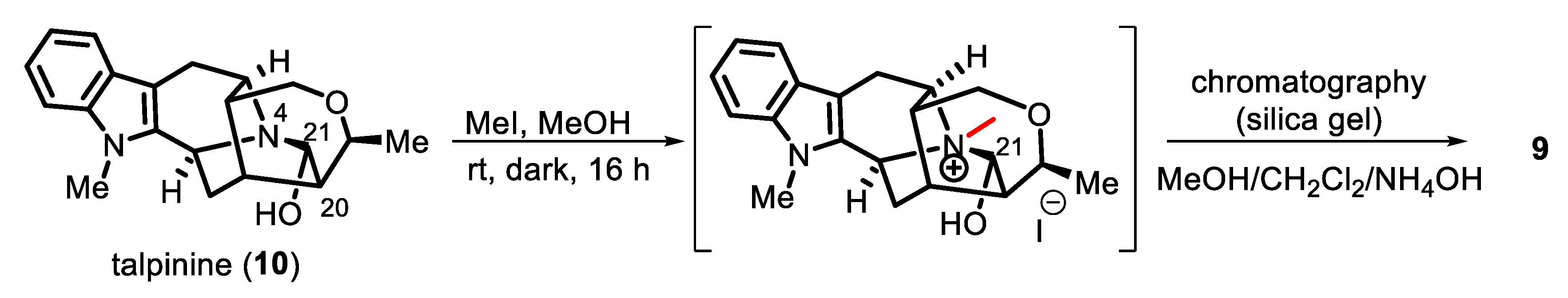
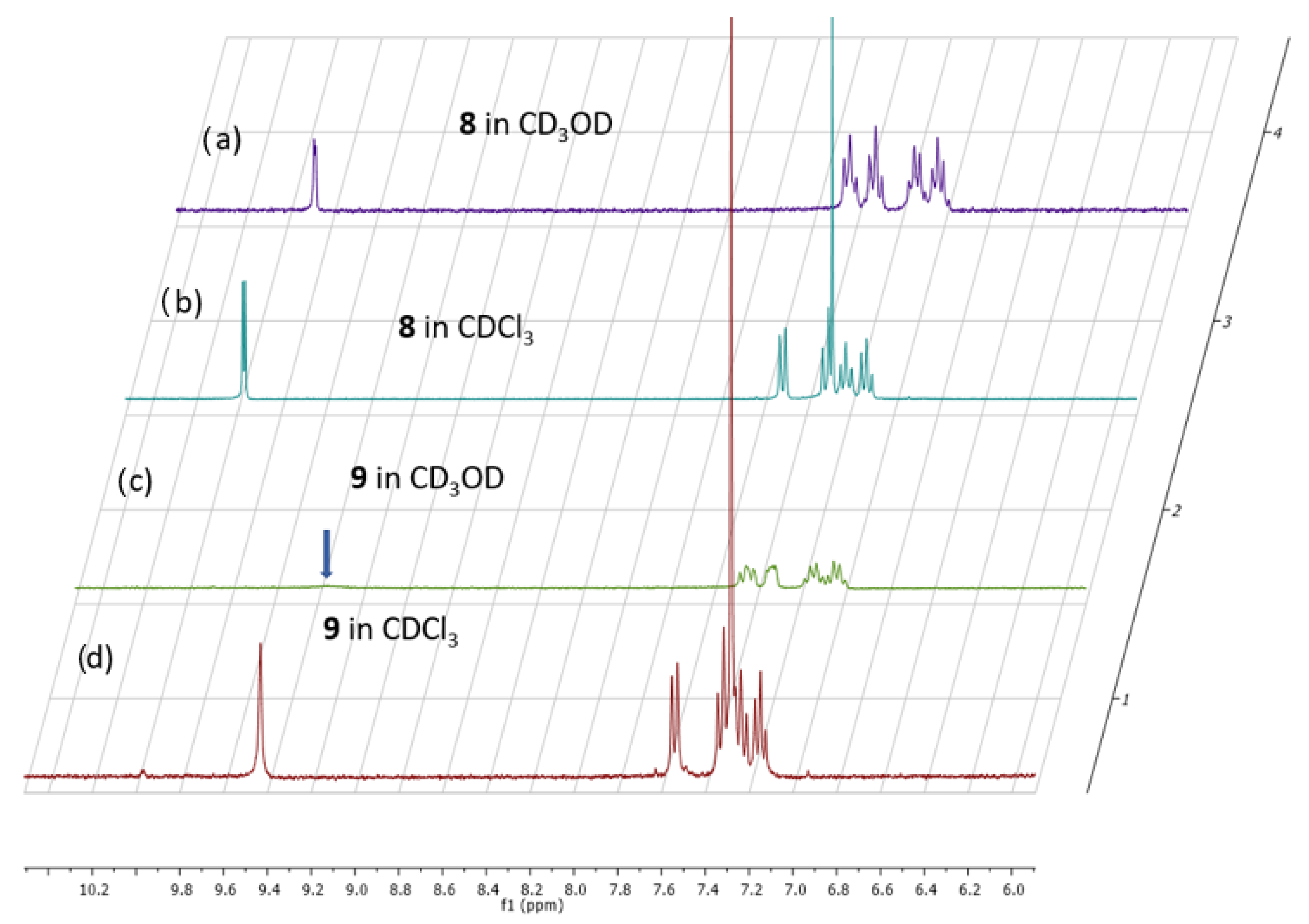
2. Results and Discussion
3. Experimental
3.1. General Experimental Considerations
3.2. Macrocarpine F (6)
3.3. Macrocarpine G (7)
3.4. Talpinine 10
3.5. O-Acetyltalpinine (11)
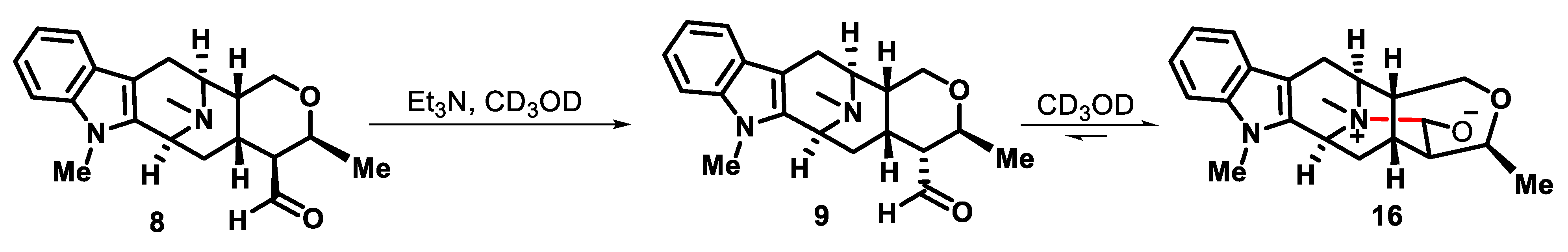
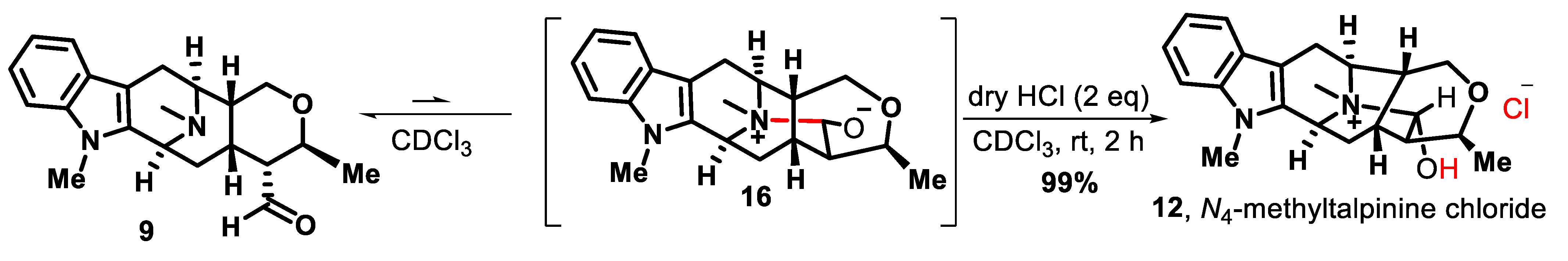
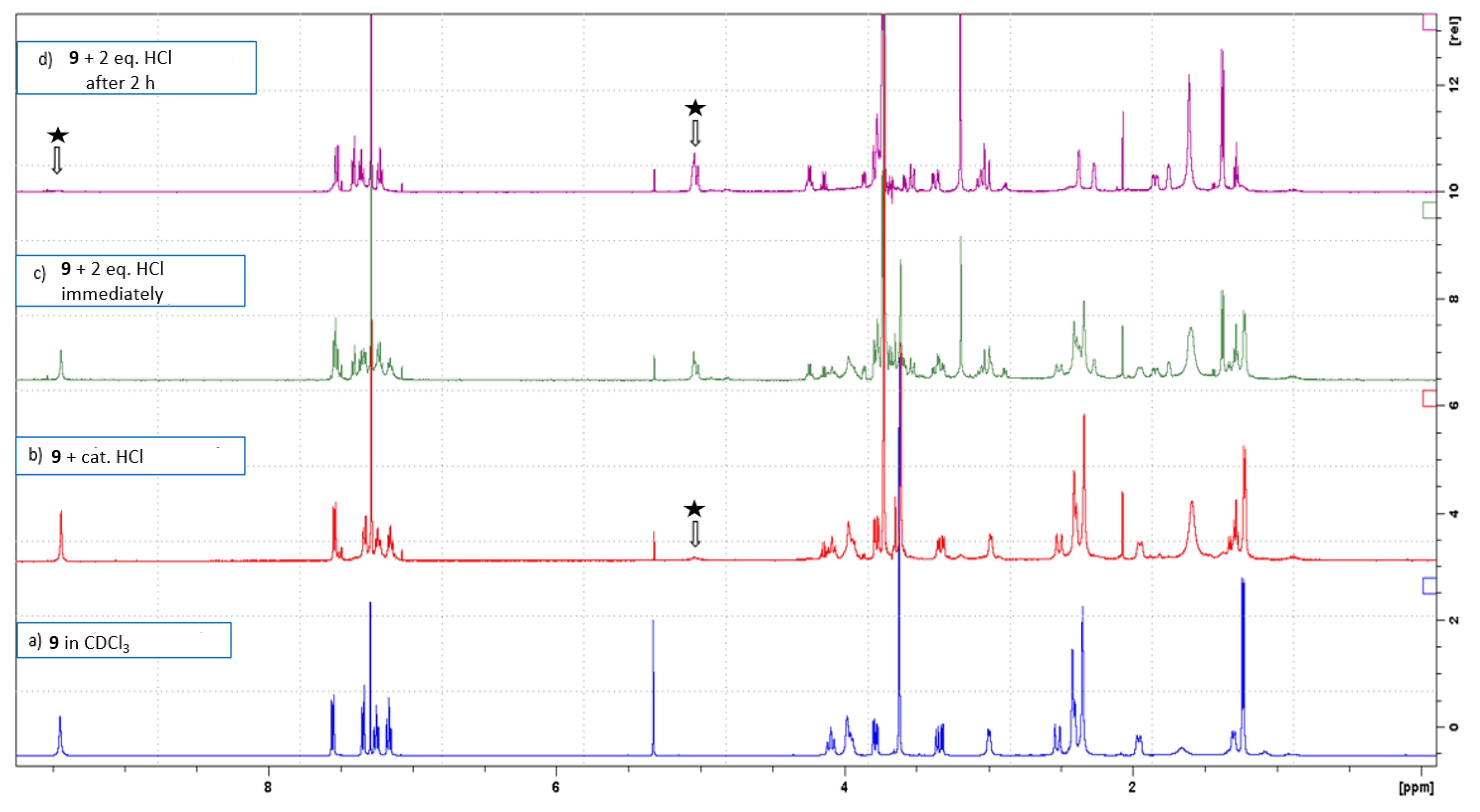
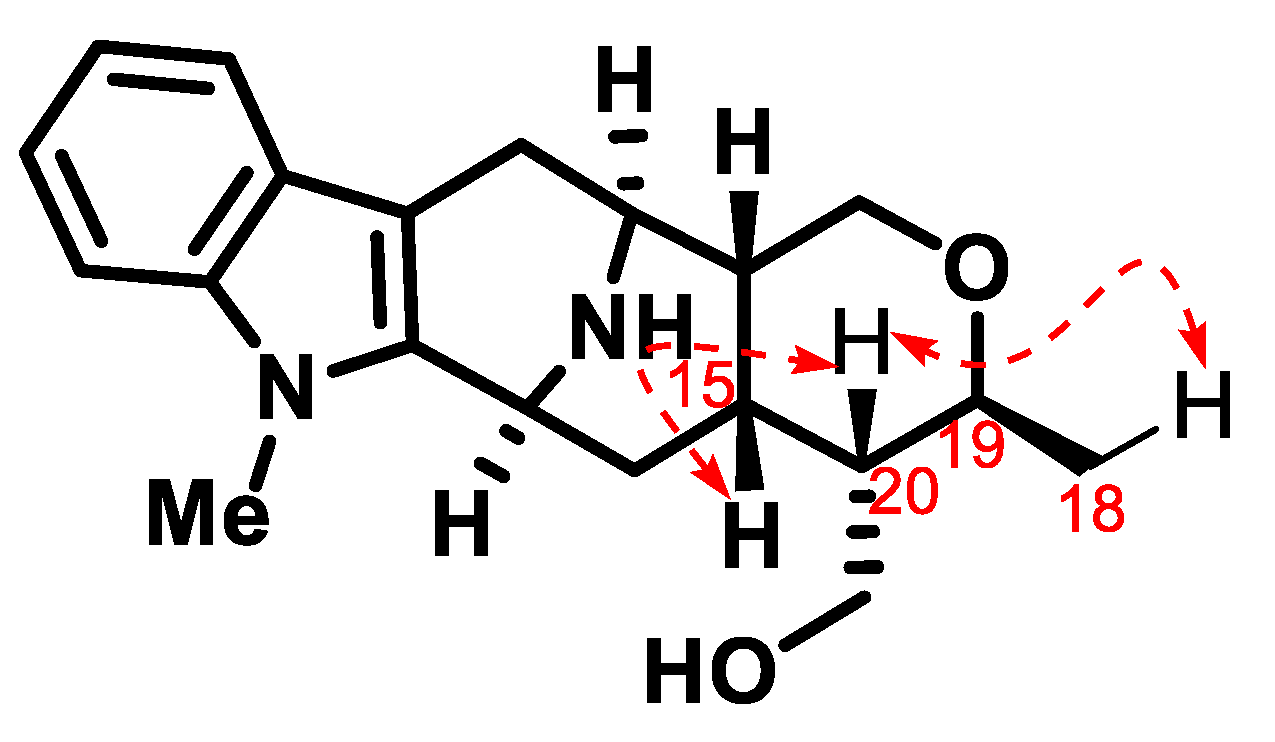
3.6. N4-Methyltalpinine (12) as the Chloride Salt
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pfitzner, A.; Stöckigt, J. Biogenetic Link Between Sarpagine and Ajmaline Type Alkaloids. Tetrahedron Lett. 1983, 24, 5197–5200. [Google Scholar] [CrossRef]
- Lounasmaa, M.; Hanhinen, P.; Westersund, M.; Halonen, N. The Sarpagine Group of Indole Alkaloids. In The Alkaloids: Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: San Diego, CA, USA, 1999; Volume 52, pp. 103–195. [Google Scholar]
- Wu, F.; Kerčmar, P.; Zhang, C.; Stöckigt, J. Sarpagan-Ajmalan-Type Indoles: Biosynthesis, structural biology, and chemo-enzymatic significance. In The Alkaloids: Chemistry and Biology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 76, pp. 1–61. [Google Scholar]
- Namjoshi, O.A.; Cook, J.M. Chapter Two-Sarpagine and Related Alkaloids. Alkaloids Chem. Biol. 2016, 76, 63–169. [Google Scholar] [PubMed] [Green Version]
- Kam, T.-S.; Choo, Y.-M. Bisindole alkaloids. In The Alkaloids: Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: San Diego, CA, USA, 2006; Volume 63, pp. 181–337. [Google Scholar]
- Pandey, K.P.; Rahman, M.T.; Cook, J.M. Bisindole Alkaloids from the Alstonia Species: Recent Isolation, Bioactivity, Biosynthesis, and Synthesis. Molecules 2021, 26, 3459. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.T.; Tiruveedhula, V.V.; Cook, J.M. Synthesis of Bisindole Alkaloids from the Apocynaceae Which Contain a Macroline or Sarpagine Unit: A Review. Molecules 2016, 21, 1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.T.; Cook, J.M. The C-19 Methyl Substituted Sarpagine-Macroline-Ajmaline Alkaloids: Diversity, Occurrence, Bioactivity, and Synthesis. In Studies in Natural Products Chemistry; Atta-ur-Rahman, M.T., Ed.; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Garnick, R.L.; Le Quesne, P.W. Biomimetic Transformations Among Monomeric Macroline-Related Indole Alkaloids. J. Am. Chem. Soc. 1978, 100, 4213–4219. [Google Scholar] [CrossRef]
- Elderfield, R.C.; Gilman, R.E. Alkaloids of Alstonia muelleriana. Phytochemistry 1972, 11, 339–343. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Cao, J.-X.; Yao, Y.-C.; Xu, S.-P. Progress of Pharmacological Studies on Alkaloids from Apocynaceae. J. Asian Nat. Prod. Res. 2013, 15, 166–184. [Google Scholar] [CrossRef]
- Rahman, M.T.; Namjoshi, O.A.; Cook, J.M. The Stereospecific and Enantiospecific Synthesis of Indole Alkaloids which Culminated in the Ambidextrous Pictet–Spengler Reaction for the C-19 Methyl–Substituted Sarpagine Family. In Progress in Heterocyclic Chemistry; Elsevier: Amsterdam, The Netherlands, 2021; Volume 32, pp. 1–26. [Google Scholar]
- Edwankar, R.V.; Edwankar, C.R.; Deschamps, J.R.; Cook, J.M. General Strategy for Synthesis of C-19 Methyl-Substituted Sarpagine/Macroline/Ajmaline Indole Alkaloids Including Total Synthesis of 19 (S), 20 (R)-Dihydroperaksine, 19 (S), 20 (R)-Dihydroperaksine-17-al, and Peraksine. J. Org. Chem. 2014, 79, 10030–10048. [Google Scholar] [CrossRef]
- Edwankar, R.V.; Edwankar, C.R.; Deschamps, J.; Cook, J.M. Regiospecific, Enantiospecific Total Synthesis of C-19 Methyl Substituted Sarpagine Alkaloids Dihydroperaksine-17-al and Dihydroperaksine. Org. Lett. 2011, 13, 5216–5219. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.T.; Deschamps, J.R.; Imler, G.H.; Cook, J.M. Total Synthesis of Sarpagine-Related Bioactive Indole Alkaloids. Chem. Eur. J. 2018, 24, 2354–2359. [Google Scholar] [CrossRef]
- Rahman, M.T.; Cook, J.M. The Ambidextrous Pictet–Spengler Reaction: Access to the (+)-or (–)-Enantiomers of the Bioactive C-19 Methyl-Substituted Sarpagine/Macroline/Ajmaline Alkaloids from Either D-or L-Tryptophan. Synthesis 2019, 51, 1980–1988. [Google Scholar] [CrossRef]
- Rahman, M.T. Shorter and Improved Access to the Key Tetracyclic Core of Sarpagine-Macroline-Ajmaline Indole Alkaloids: The Total Synthesis of Alkaloids Macrocarpines Ag, Talcarpine, N (4)-methyl-n (4), 21-secotalpinine, Deoxyperaksine, Dihydroperaksine, Talpinine, O-acetyltalpinine, and N (4)-methyltalpinine. Ph.D. Thesis, University of Wisconsin-Milwaukee, Milwaukee, WI, USA, 2018. [Google Scholar]
- Rahman, M.T.; Cook, J.M. Unprecedented Stereocontrol in the Synthesis of 1, 2, 3-Trisubstituted Tetrahydro-β-Carbolines via a New Asymmetric Pictet—Spengler Reaction Towards Sarpagine-Type Indole Alkaloids. Eur. J. Org. Chem. 2018, 2018, 3224–3229. [Google Scholar] [CrossRef]
- Rahman, M.T.; Deschamps, J.R.; Imler, G.H.; Schwabacher, A.W.; Cook, J.M. Total Synthesis of Macrocarpines D and E via an Enolate-Driven Copper-Mediated Cross-Coupling Process: Replacement of Catalytic Palladium with Copper Iodide. Org. Lett. 2016, 18, 4174–4177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.-J.; Lim, J.-L.; Low, Y.-Y.; Sim, K.-S.; Lim, S.-H.; Kam, T.-S. Oxidized Derivatives of Macroline, Sarpagine, and Pleiocarpamine Alkaloids from Alstonia angustifolia. J. Nat. Prod. 2014, 77, 2068–2080. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.-S.; Choo, Y.-M.; Komiyama, K. Unusual Spirocyclic Macroline Alkaloids, Nitrogenous Derivatives, and a Cytotoxic Bisindole from Alstonia. Tetrahedron 2004, 60, 3957–3966. [Google Scholar] [CrossRef]
- Naranjo, J.; Pinar, M.; Hesse, M.; Schmid, H. Über die Indolalkaloide von Pleiocarpa talbotii Wernham. 145. Mitteilung über Alkaloide. Helv. Chim. Acta 1972, 55, 752–771. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Terrazas, C.; Acuña, U.M.; Ninh, T.N.; Chai, H.; De Blanco, E.J.C.; Soejarto, D.D.; Satoskar, A.R.; Kinghorn, A.D. Bioactive Indole Alkaloids Isolated from Alstonia angustifolia. Phytochem. Lett. 2014, 10, LIV–LIX. [Google Scholar] [CrossRef] [Green Version]
- Kinghorn, A.D.; Ohio State University, Columbus, OH, USA. Personal communication, 2015.
- Keawpradub, N.; Kirby, G.; Steele, J.; Houghton, P. Antiplasmodial Activity of Extracts and Alkaloids of Three Alstonia Species from Thailand. Planta Med. 1999, 65, 690–694. [Google Scholar] [CrossRef]
- Iwu, M. Stem Bark Alkaloids of Rauwolfia vomitoria. Planta Med. 1982, 45, 105–111. [Google Scholar] [CrossRef]
- Larock, R.C. Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2nd ed.; John Wiley & Sons: New York, NY, USA, 1999. [Google Scholar]
- Wuts, P.G.; Greene, T.W. Greene’s Protective Groups in Organic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Braun, J. Die Einwirkung von Bromcyan auf Tertiäre Amine. Ber. Dtsch. Chem. Ges. 1900, 33, 1438–1452. [Google Scholar] [CrossRef]
- Cooley, J.; Evain, E. Amine Dealkylations with Acyl Chlorides. Synthesis 1989, 1989, 1–7. [Google Scholar] [CrossRef]
- Stephen, M.R.; Rahman, M.T.; Tiruveedhula, V.P.B.; Fonseca, G.O.; Deschamps, J.R.; Cook, J.M. Concise Total Synthesis of (−)-Affinisine Oxindole,(+)-Isoalstonisine,(+)-Alstofoline,(−)-Macrogentine,(+)−Na-Demethylalstonisine,(−)-Alstonoxine A, and (+)-Alstonisine. Chem. Eur. J. 2017, 23, 15805–15819. [Google Scholar] [CrossRef]
- Olofson, R.; Martz, J.T.; Senet, J.P.; Piteau, M.; Malfroot, T. A New Reagent for the Selective, High-Yield N-Dealkylation of Tertiary Amines: Improved Syntheses of Naltrexone and Nalbuphine. J. Org. Chem. 1984, 49, 2081–2082. [Google Scholar] [CrossRef]
- Hu, P.; Chi, H.M.; DeBacker, K.C.; Gong, X.; Keim, J.H.; Hsu, I.T.; Snyder, S.A. Quaternary-Centre-Guided Synthesis of Complex Polycyclic Terpenes. Nature 2019, 569, 703–707. [Google Scholar] [CrossRef]
- Yu, P.; Wang, T.; Li, J.; Cook, J.M. Enantiospecific Total Synthesis of the Sarpagine Related Indole Alkaloids Talpinine and Talcarpine as Well as the Improved Total Synthesis of Alstonerine and Anhydromacrosalhine-methine via the Asymmetric Pictet—Spengler Reaction. J. Org. Chem. 2000, 65, 3173–3191. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H# | 1H NMR Natural (400 MHz) [20] | 1H NMR Synthetic (300 MHz) | C# | 13C NMR Natural (100 MHz) [20] | 13C NMR Synthetic (75 MHz) |
|---|---|---|---|---|---|
| 3 | 4.27 (m) | 4.37 (br s) | 2 | 136.4 | 136.3 |
| 5 | 3.21 (m) | 3.23–3.15 (m) | 3 | 46.5 | 46.5 |
| 6β 6α | 2.63 (d, J = 15 Hz) 3.17 (m) | 2.68 (br d, J = 14.7 Hz) 3.30–3.23 (m) | 5 | 48.2 | 48.2 |
| 9 | 7.46 (br d, J = 8 Hz) | 7.49 (d, J = 7.6 Hz) | 6 | 28.5 | 28.4 |
| 10 | 7.07 (br t, J = 8 Hz) | 7.14–7.06 (m) | 7 | 107.7 | 107.9 |
| 11 | 7.18 (br t, J = 8 Hz) | 7.23–7.16 (m) | 8 | 126.6 | 126.2 |
| 12 | 7.75 (br d, J = 8 Hz) * | 7.32–7.23 (d, J = 8.1 Hz) # | 9 | 118.1 | 118.1 |
| 14β 14α | 1.35 (m) 2.45 (td, J = 12, 4 Hz) | 1.31–1.25 (m) $ 1.46–1.35 (m) | 10 | 119.1 | 119.0 |
| 15 | 2.10 (m) | 2.19–2.11 (m) $ | 11 | 121.2 | 121.1 |
| 16 | 2.08 (m) | 2.28–2.12 (m) $ | 12 | 108.9 | 108.8 |
| 17β 17α | 3.78 (dd, J = 11, 5 Hz) 4.07 (t, J = 11 Hz) | 3.88–3.79 (m) 4.19–4.08 (t, J = 11.6 Hz) | 13 | 136.8 | - § |
| 19 | 3.94 (qd, J = 6.8, 2 Hz) | 4.02–3.90 (m) | 14 | 29.8 | 29.7 |
| 20 | 1.04 (m) | 1.13–1.01 (m) | 15 | 29.0 | 29.5 |
| 21 | 3.65 (dd, J = 11, 4 Hz) | 3.75–3.65 (m) | 16 | 38.1 | 38.3 |
| 21′ | 3.72 (dd, J = 11, 6 Hz) | 3.88–3.79 (m) $ | 17 | 68.6 | 68.6 |
| 18-Me | 1.21 (d, J = 6.8 Hz) | 1.29–1.19 (m) $ | 18 | 18.9 | 18.8 |
| N(1)-Me | 3.52 (s) | 3.61 (s) | 19 | 71.3 | 71.4 |
| 20 | 44.1 | 43.9 | |||
| 21 | 62.5 | 63.1 | |||
| N(1)-Me | 29.0 | 29.0 |
| H# | 1H NMR Natural (400 MHz) [20] | 1H NMR Synthetic (500 MHz) |
|---|---|---|
| 3 | 4.27 (br t, J = 3 Hz) | 4.35 (br s) |
| 5 | 3.25 (m) | 3.31 (d, J = 7.3 Hz) |
| 6β 6α | 2.58 (d, J = 16 Hz) 3.18 (dd, J = 16, 7 Hz) | 2.65 (br d, J = 16.2 Hz) 3.25-3.19 (m) |
| 9 | 7.46 (br d, J = 7.5 Hz) | 7.49 (d, J = 7.9 Hz) |
| 10 | 7.08 (td, J = 7.5, 1 Hz) | 7.10 (t, J = 7.5 Hz) |
| 11 | 7.17 (td, J = 7.5, 1 Hz) | 7.19 (t, J = 7.5 Hz) |
| 12 | 7.24 (br d, J = 7.5 Hz) | 7.29 (d, J = 8.1 Hz) |
| 14β 14α | 1.49 (dt, J = 12, 2 Hz) 2.18 (td, J = 12, 4 Hz) | 1.58-1.53 (m) * 2.28 (td, J = 12.6, 3.8 Hz) |
| 15 | 2.02 (m) | 2.15–2.08 (m) |
| 16 | 1.81 (dt, J = 12, 5 Hz) | 1.91–1.86 (m) * |
| 17β 17α | 3.72 (dd, J = 12, 4 Hz) 4.03 (t, J = 12 Hz) | 3.78 (dd, J = 11.4, 4.4 Hz) 4.10 (br t, J = 12.2 Hz) |
| 19 | 3.46 (m) | 3.58–3.54 (m) * |
| 20 | 1.37 (m) | 1.52–1.45 (m) * |
| 21 | 3.22 (m) | 3.39–3.34 (m) |
| 21′ | 3.40 (dd, J = 11, 5 Hz) | 3.51 (dd, J = 11.0, 5.3 Hz) * |
| 18-Me | 1.11 (d, J = 6 Hz) | 1.17 (d, 6.1 Hz) |
| N(1)-Me | 3.52 (s) | 3.63 (s) |
| H# | 1H NMR Synthetic (400 MHz) | 1H NMR Synthetic (300 MHz) Yu et al. [34] | C# | 13C NMR Synthetic (75 MHz) | 13C NMR Synthetic (75 MHz) Yu et al. [34] |
|---|---|---|---|---|---|
| 3 | 4.43 (d, J = 7.9 Hz) | 4.39 (d, J = 12.1 Hz) | 2 | 139.1 | 138.48 |
| 5 | 3.52–3.41 (m) $ | 3.50 (t, J = 6.3 Hz) | 3 | 40.2 | 40.51 |
| 6β 6α | 2.65 (d, J = 15.6 Hz) 3.21 (dd, J = 15.6, 6.0 Hz) | 2.60 (d, J = 15.6 Hz) 3.20 (dd, J = 15.8, 5.8 Hz) | 5 | 49.8 | 50.18 |
| 9 | 7.47 (m) | 7.45 (d, J = 7.7 Hz) | 6 | 26.3 | 26.63 |
| 10 | 7.12–7.05 (m) | 7.07 (t, J = 7.9 Hz) | 7 | 103.3 | 103.55 |
| 11 | 7.19 (t, J = 7.7 Hz) | 7.17 (t, J = 7.8 Hz) | 8 | 127.3 | 127.50 |
| 12 | 7.31–7.27 (m) | 7.27 (d, J = 8.0 Hz) | 9 | 118.3 | 118.69 |
| 14β 14α | 1.38–1.20 (m) * 1.87–1.79 (m) | 1.30 (d, J = 12.1 Hz) 1.81 (t, J = 12.0 Hz) | 10 | 118.9 | 119.28 |
| 15 | 1.93–1.86 (m) | 1.87 (d, J = 3.0 Hz) | 11 | 120.9 | 121.31 |
| 16 | 1.38–1.20 (m) * | 1.20 (m) | 12 | 108.7 | 109.12 |
| 17β 17α | 3.52–3.41 (m) $ 3.72–3.62 (m) | 3.42 (d, J = 11.0 Hz) 3.65 (t, J = 11.4 Hz) | 13 | 137.5 | 137.73 |
| 19 | 4.10–3.99 (m) | 4.03 (q, J = 6.8 Hz) | 14 | 31.9 | 32.31 |
| 20 | 1.38–1.20 (m) * | 1.25 (m) | 15 | 23.3 | 23.63 |
| 21 | 4.71 (m) | 4.69 (d, J = 1.8 Hz) | 16 | 35.2 | 35.58 |
| 18-Me | 1.38–1.20 (m) * | 1.27 (d, J = 6.0 Hz) | 17 | 64.0 | 64.35 |
| N(1)-Me | 3.59 (s) | 3.55 (s) | 18 | 15.7 | 16.10 |
| 19 | 72.6 | 72.98 | |||
| 20 | 43.6 | 44.13 | |||
| 21 | 87.9 | 88.25 | |||
| N(1)-Me | 29.2 | 29.63 |
| H# | 1H NMR Natural (400 MHz) [20] | 1H NMR Synthetic (500 MHz) | C# | 13C NMR Natural (100 MHz) [20] | 13C NMR Synthetic (125 MHz) |
|---|---|---|---|---|---|
| 3 | 4.48 (br dd, J = 10, 2 Hz) | 4.46 (br d, J = 9.3 Hz) | 2 | 138.9 | - * |
| 5 | 3.52 (br t, J = 5.5 Hz) | 3.56 (br t, J = 5.5 Hz) | 3 | 41.7 | 41.7 |
| 6β 6α | 2.66 (d, J = 15.6 Hz) 3.20 (dd, J = 15.6, 5.5 Hz) | 2.62 (br d, J = 15.8 Hz) 3.18 (dd, J = 15.2, 5.9 Hz) | 5 | 50.3 | 50.3 |
| 9 | 7.47 (br d, J = 7.5 Hz) | 7.47 (d, J = 7.7 Hz) | 6 | 26.6 | 26.6 |
| 10 | 7.09 (br td, J = 7.5 Hz) | 7.09 (t, J = 7.4 Hz) | 7 | 103.1 | 103.1 |
| 11 | 7.19 (td, J = 7.5, 1 Hz) | 7.18 (t, J = 7.6 Hz) | 8 | 127.2 | 127.2 |
| 12 | 7.29 (br d, J = 7.5 Hz) | 7.29 (d, J = 8.2 Hz) | 9 | 118.1 | 118.1 |
| 14β 14α | 1.52 (ddd, J = 12, 4, 2.8 Hz) 1.89 (ddd, J = 12, 10, 1.6 Hz) | 1.53–1.47 (m) 1.91–1.83 (m) * | 10 | 120.9 | 120.9 |
| 15 | 2.00 (m) | 2.03–1.97 (m) | 11 | 118.8 | 118.9 |
| 16 | 1.30 (m) | 1.30 (m) * | 12 | 108.7 | 108.7 |
| 17β 17α | 3.47 (dd, J = 11, 2 Hz) 3.71 (dd, J = 11, 1 Hz) | 3.47 (dd, J = 11.4, 1.9 Hz) 3.71 (dd, J = 11.4, 1.1 Hz) | 13 | 137.4 | 137.4 |
| 19 | 4.34 (q, J = 7 Hz) | 4.34 (q, J = 7.0 Hz) | 14 | 32.3 | 32.3 |
| 20 | 1.34 (m) | 1.34–1.31 (m) * | 15 | 23.2 | 23.2 |
| 21 | 5.63 (br d, J = 2 Hz) | 5.61 (m) | 16 | 35.2 | 35.2 |
| 18-Me | 1.30 (d, J = 7 Hz) | 1.30 (d, 6.8 Hz) | 17 | 63.8 | 63.8 |
| N(1)-Me | 3.66 (s) | 3.66 (s) | 18 | 15.6 | 15.7 |
| 19 | 71.6 | 71.6 | |||
| 20 | 43.6 | 43.6 | |||
| 21 | 88.9 | 88.9 | |||
| N(1)-Me | 29.3 | 29.3 | |||
| 21-OAc | 21.2 | 21.3 | |||
| 21-OAc | 169.6 | 169.6 |
| H# | 1H NMR Natural (400 MHz) [23] | 1H NMR Synthetic (500 MHz) | C# | 13C NMR Natural (100 MHz) [23] | 13C NMR Synthetic (125 MHz) |
|---|---|---|---|---|---|
| 3 | 4.99 (1H, d, J = 10.7 Hz) | 4.99 (1H, d, J = 10.4 Hz) | 2 | 134.2 | 134.2 |
| 5 | 3.91 (1H, t, J = 5.4 Hz) | 3.92 (1H, t, J = 5.5 Hz) | 3 | 53.1 | 53.1 |
| 6β 6α | 3.09 (1H, d, J = 16.6 Hz) 3.36 (1H, dd, J = 17.4, 5.3 Hz) | 3.09 (1H, d, J = 17.4 Hz) 3.36 (1H, dd, J = 17.2, 5.4 Hz) | 5 | 61.3 | 61.3 |
| 9 | 7.53 (1H, d, J = 7.9 Hz) | 7.53 (1H, d, J = 7.9 Hz) | 6 | 24.7 | 24.7 |
| 10 | 7.12 (1H, t, J = 7.8 Hz) | 7.12 (1H, t, J = 7.6 Hz) | 7 | 101.2 | 101.6 |
| 11 | 7.25 (1H, t, J = 7.7 Hz) | 7.25 (1H, t, J = 7.7 Hz) | 8 | 127.3 | 127.3 |
| 12 | 7.43 (1H, d, J = 8.2 Hz) | 7.43 (1H, d, J = 8.3 Hz) | 9 | 119.5 | 119.5 |
| 14β 14α | 1.98 (1H, ddd, J = 13.2, 5.0, 1.8 Hz) 2.47 (1H, br t, J = 12.2 Hz) | 1.96 (1H, ddd, J = 13.3, 5.0, 1.8 Hz) 2.47 (1H, m) | 10 | 121.0 | 121.0 |
| 15 | 2.36 (1H, br s) | 2.36 (1H, m) | 11 | 123.7 | 123.7 |
| 16 | 1.77 (1H, br s) | 1.77 (1H, br s) | 12 | 110.6 | 110.6 |
| 17β 17α | 3.80 (1H, d, J = 11.7 Hz) 3.54 (1H, dd, J = 11.9, 2.1 Hz) | 3.80 (1H, d, J = 11.7 Hz) 3.54 (1H, dd, J = 11.9, 2.2 Hz) | 13 | 139.7 | 139.7 |
| 19 | 4.15 (1H, q, J = 6.8 Hz) | 4.15 (1H, q, J = 6.9 Hz) | 14 | 32.4 | 32.3 |
| 20 | 2.04 (1H, br s) | 2.04 (1H, br s) | 15 | 22.7 | 22.6 |
| 21 | 4.95 (1H, d, J = 1.9 Hz) | 4.95 (1H, d, J = 1.7 Hz) | 16 | 38.2 | 38.2 |
| 18-Me | 1.38 (3H, d, J = 6.8 Hz) | 1.38 (3H, d, J = 6.8 Hz) | 17 | 63.1 | 63.1 |
| N(1)-Me | 3.73 (3H, s) | 3.73 (3H, s) | 18 | 15.7 | 15.7 |
| N(4)-Me | 3.07 (3H, s) | 3.07 (3H, s) | 19 | 72.7 | 72.7 |
| 20 | 48.2 | 48.2 | |||
| 21 | 98.4 | 98.4 | |||
| N(1)-Me | 29.9 | 29.9 | |||
| N(4)-Me | 43.5 | 43.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.T.; Tiruveedhula, V.V.N.P.B.; Stephen, M.R.; Rallapalli, S.K.; Pandey, K.P.; Cook, J.M. Completion of the Total Synthesis of Several Bioactive Sarpagine/Macroline Alkaloids including the Important NF-κB Inhibitor N4-Methyltalpinine. Molecules 2022, 27, 1738. https://doi.org/10.3390/molecules27051738
Rahman MT, Tiruveedhula VVNPB, Stephen MR, Rallapalli SK, Pandey KP, Cook JM. Completion of the Total Synthesis of Several Bioactive Sarpagine/Macroline Alkaloids including the Important NF-κB Inhibitor N4-Methyltalpinine. Molecules. 2022; 27(5):1738. https://doi.org/10.3390/molecules27051738
Chicago/Turabian StyleRahman, Md Toufiqur, Veera Venkata Naga Phani Babu Tiruveedhula, Michael Rajesh Stephen, Sundari K. Rallapalli, Kamal P. Pandey, and James M. Cook. 2022. "Completion of the Total Synthesis of Several Bioactive Sarpagine/Macroline Alkaloids including the Important NF-κB Inhibitor N4-Methyltalpinine" Molecules 27, no. 5: 1738. https://doi.org/10.3390/molecules27051738
APA StyleRahman, M. T., Tiruveedhula, V. V. N. P. B., Stephen, M. R., Rallapalli, S. K., Pandey, K. P., & Cook, J. M. (2022). Completion of the Total Synthesis of Several Bioactive Sarpagine/Macroline Alkaloids including the Important NF-κB Inhibitor N4-Methyltalpinine. Molecules, 27(5), 1738. https://doi.org/10.3390/molecules27051738