
Chemistry of Several Sterically Bulky Molecules with P=P, P=C, and C≡P Bond

Abstract
:
1. Introduction
2. Steric Protection and the First “Phosphobenzene”
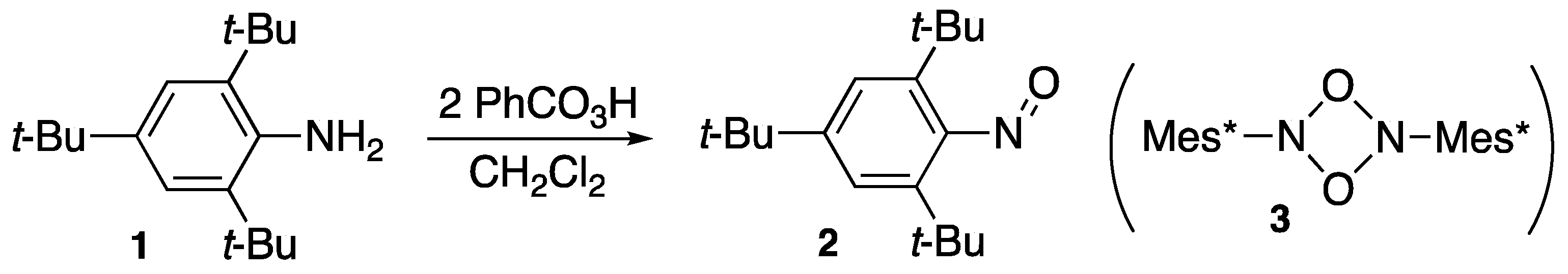
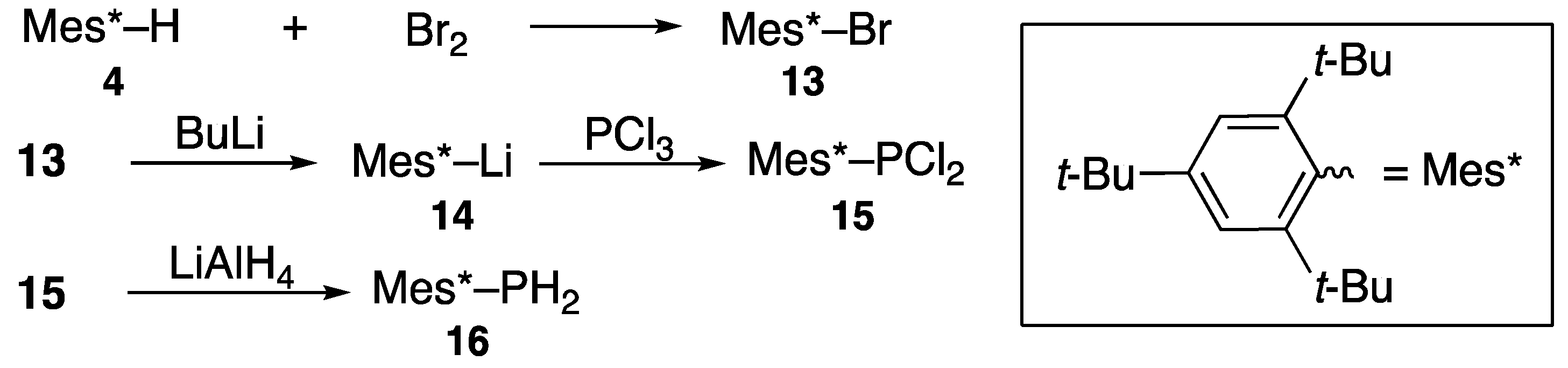
2.1. Steric Protection for Stabilization of Unstable Compounds
2.2. Preparation and Characterization of Diphosphenes
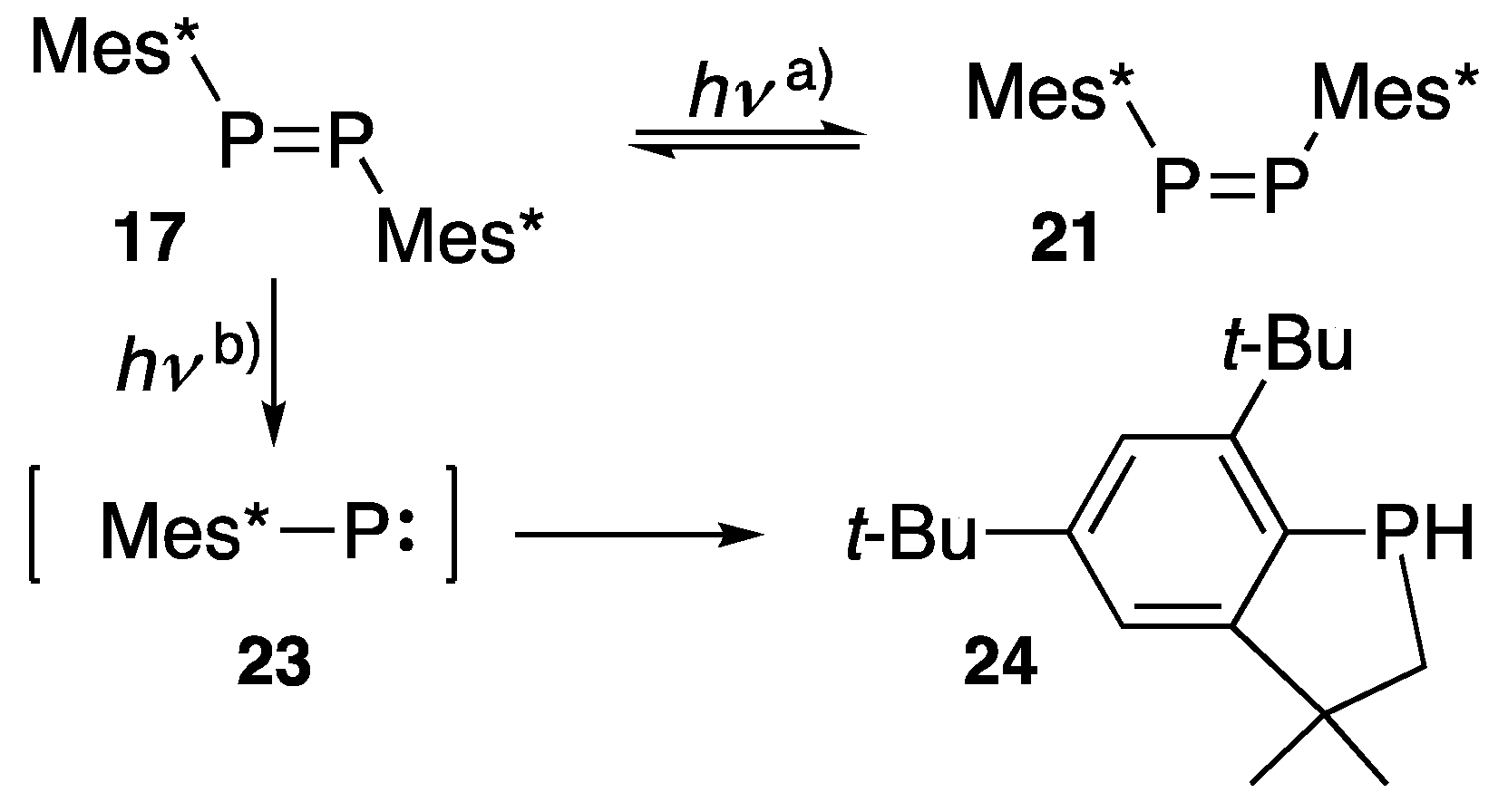
2.3. Chemical Reactivity of Diphosphenes
3. Stable Phosphaalkenes and Phosphaalkynes
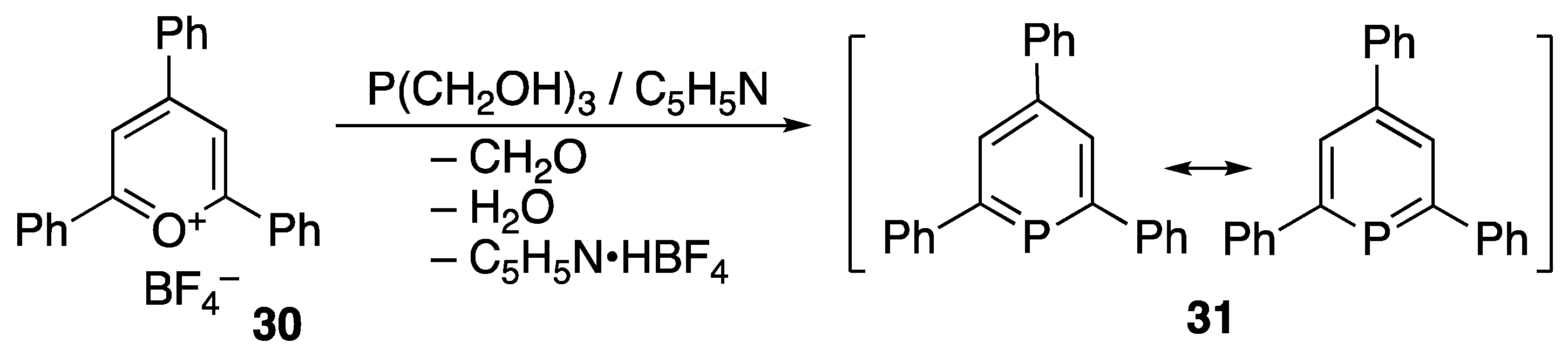
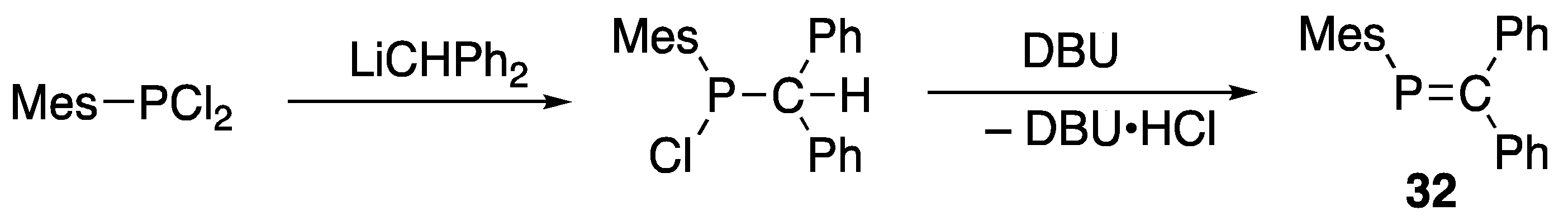
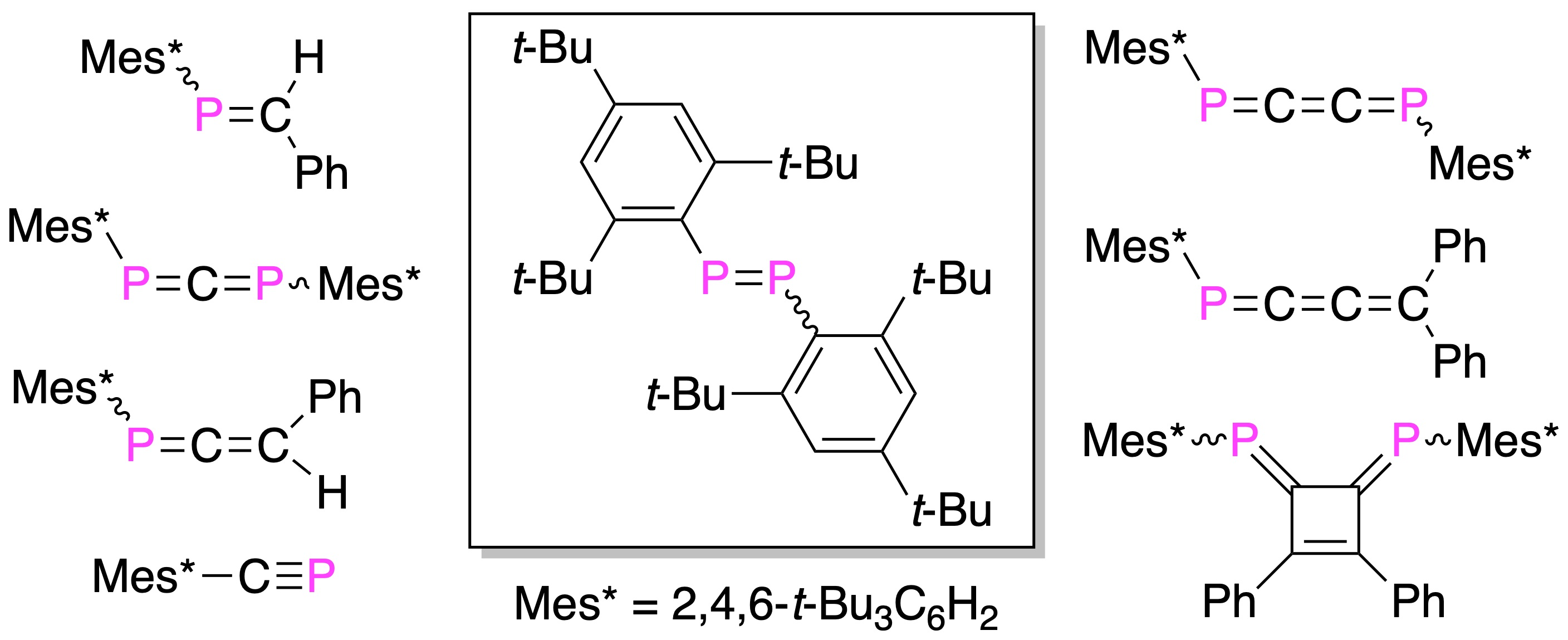
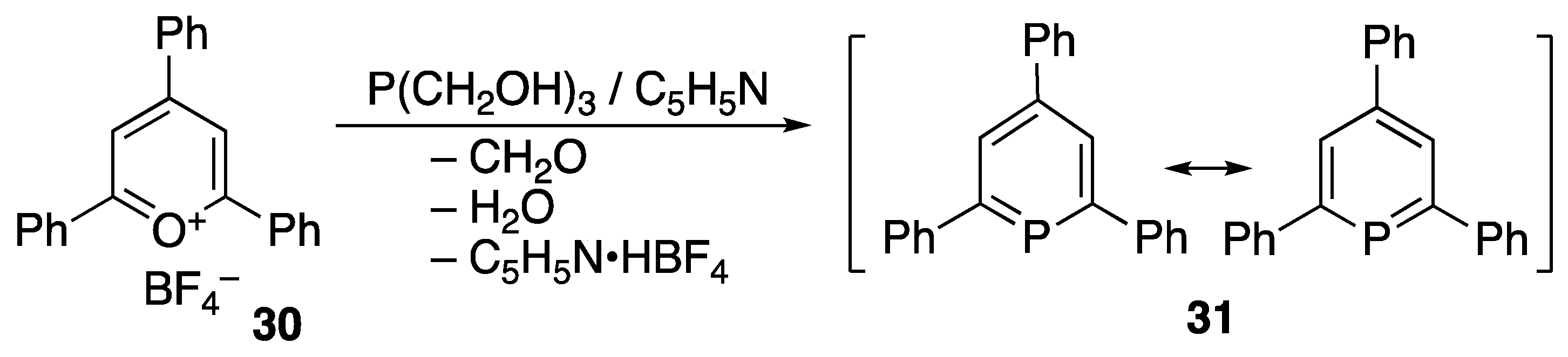
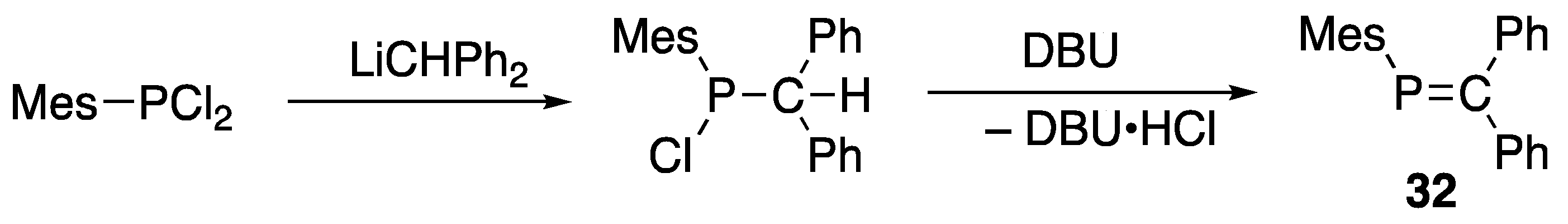
3.1. Stable Phosphaalkenes and the Related Compounds
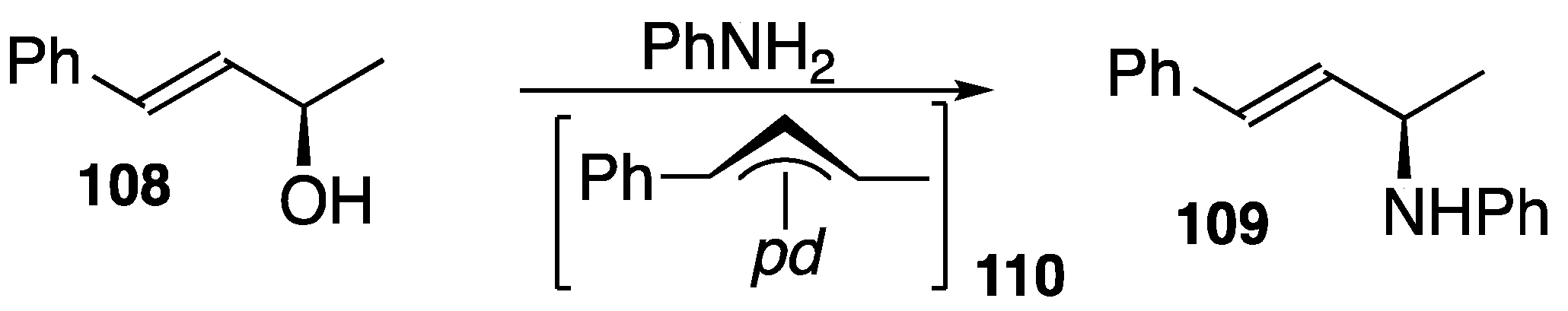
3.2. Some Reactions of Phosphaalkenes
3.3. Stable Phosphaalkynes
3.4. Singlet Biradicals from Phosphaalkyne
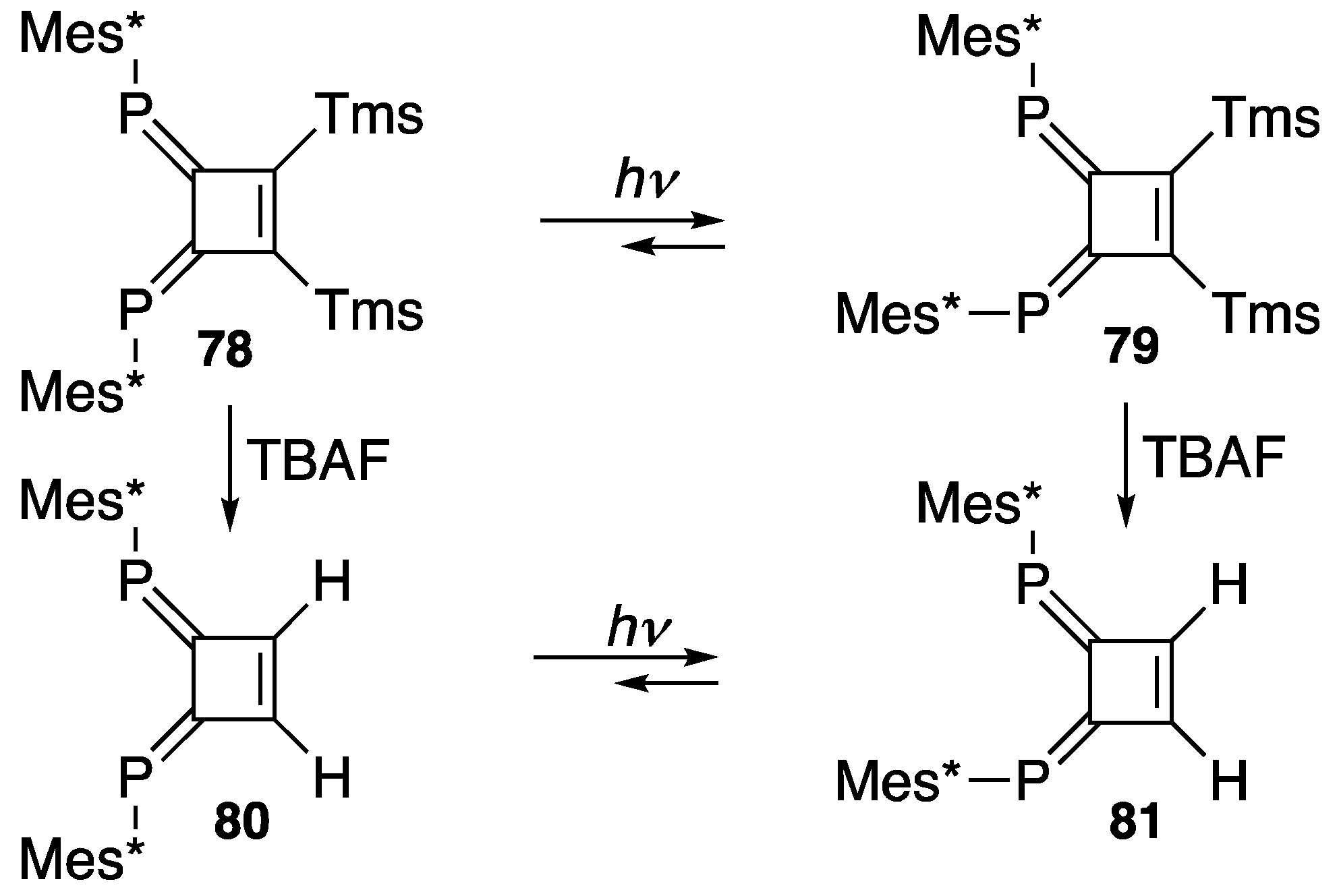
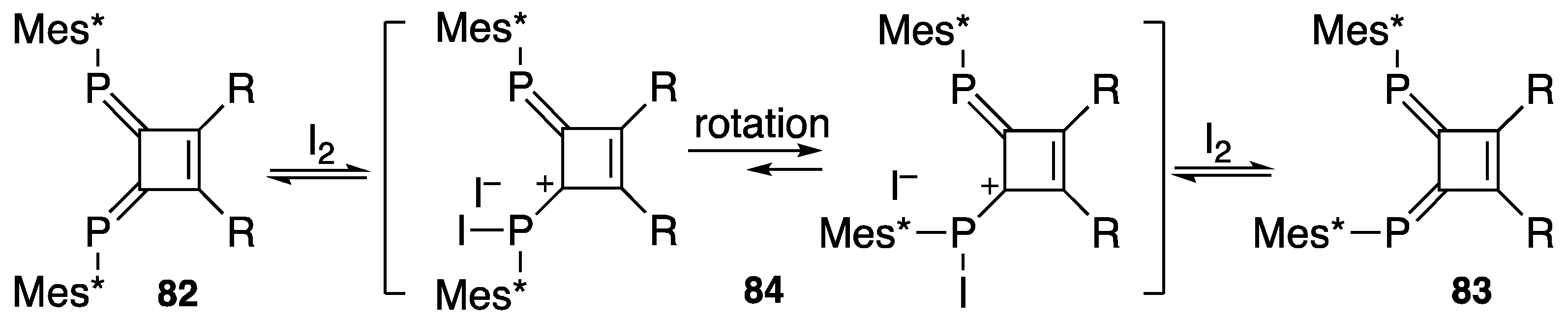
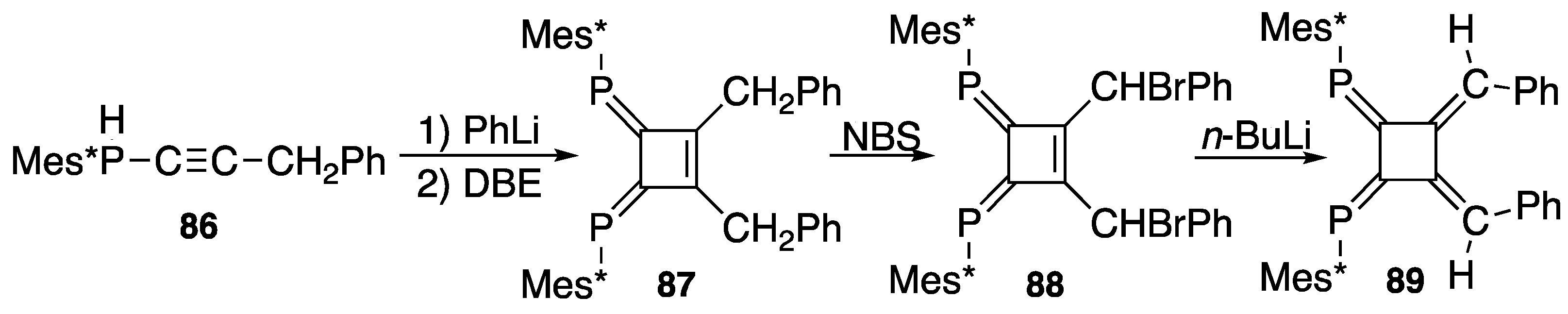
4. Diphosphinidenecyclobutenes
4.1. Preparation of Diphosphinidenecyclobutenes
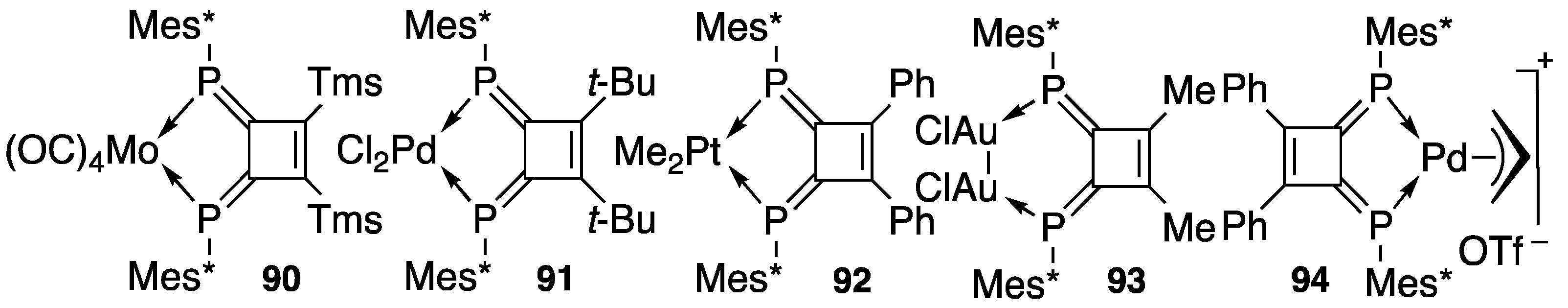
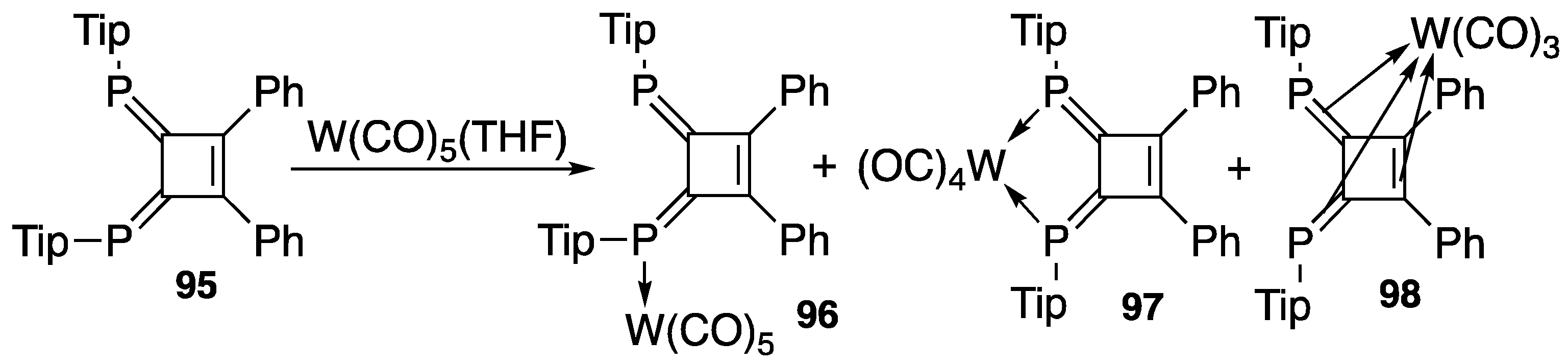
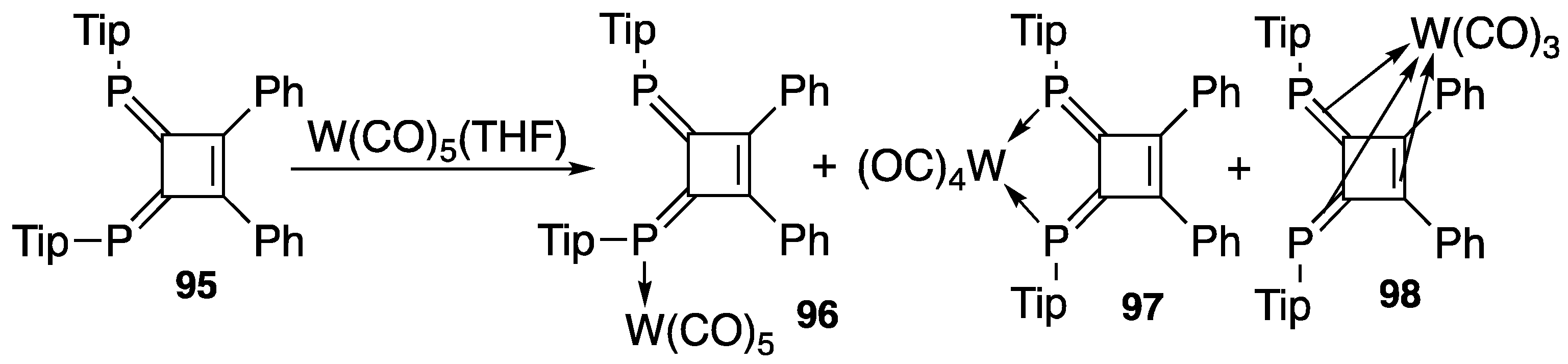
4.2. Transition Metal Complexes of Diphosphinidenecyclobutenes
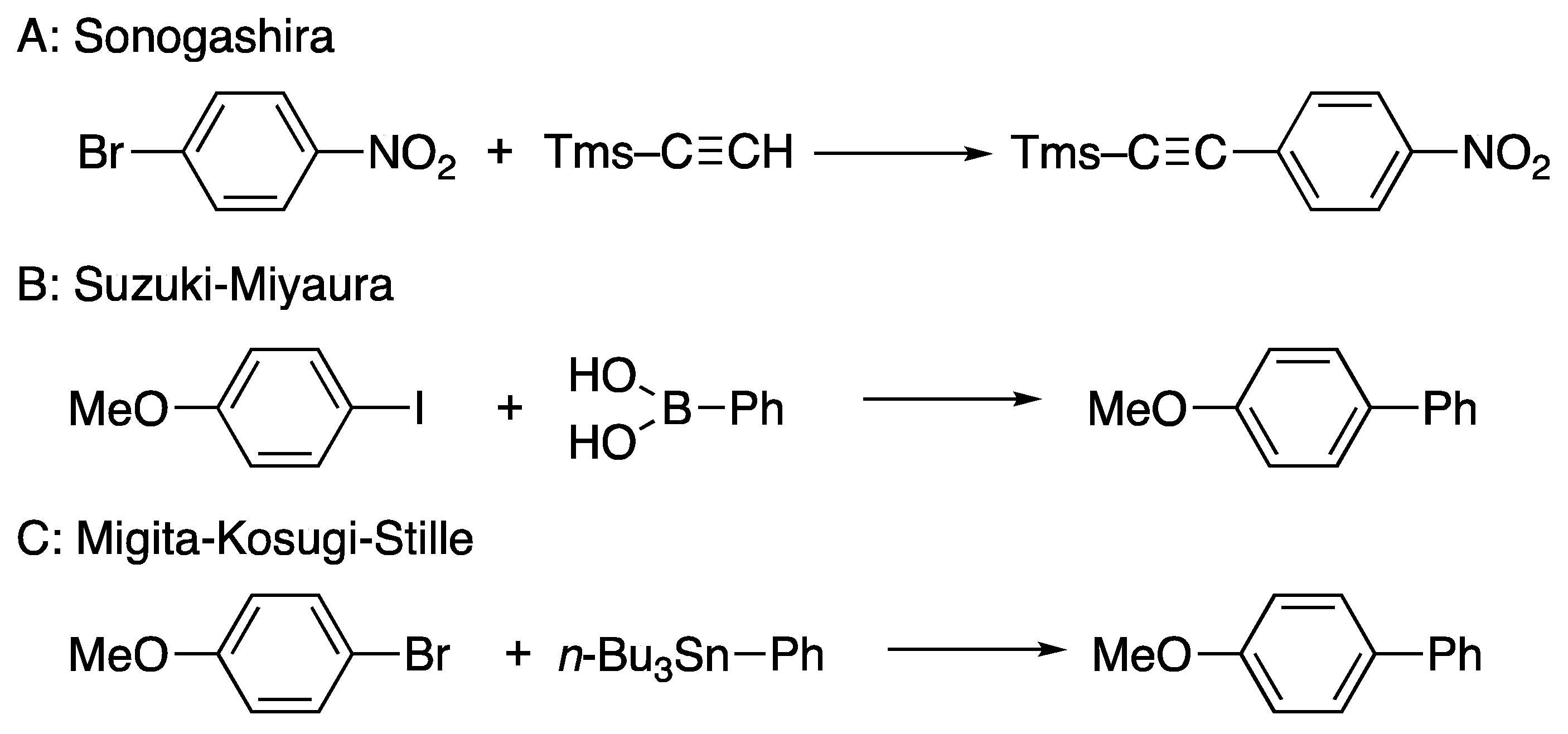
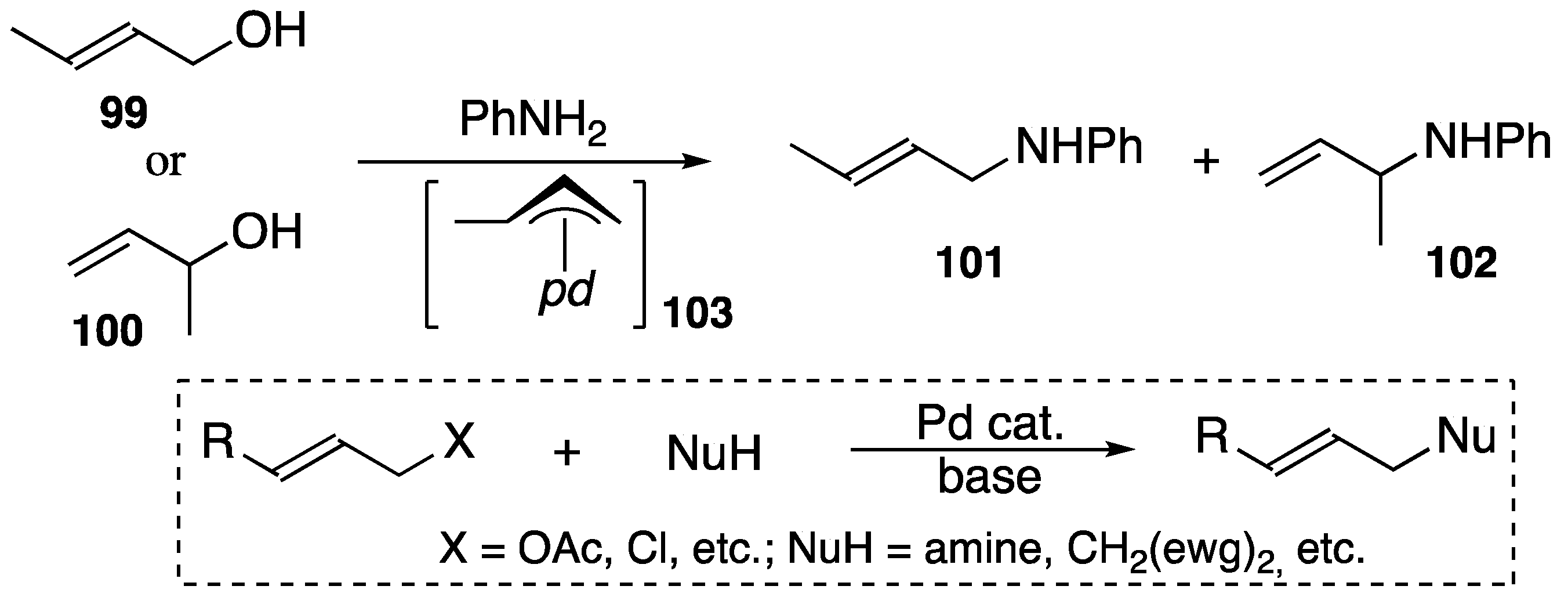
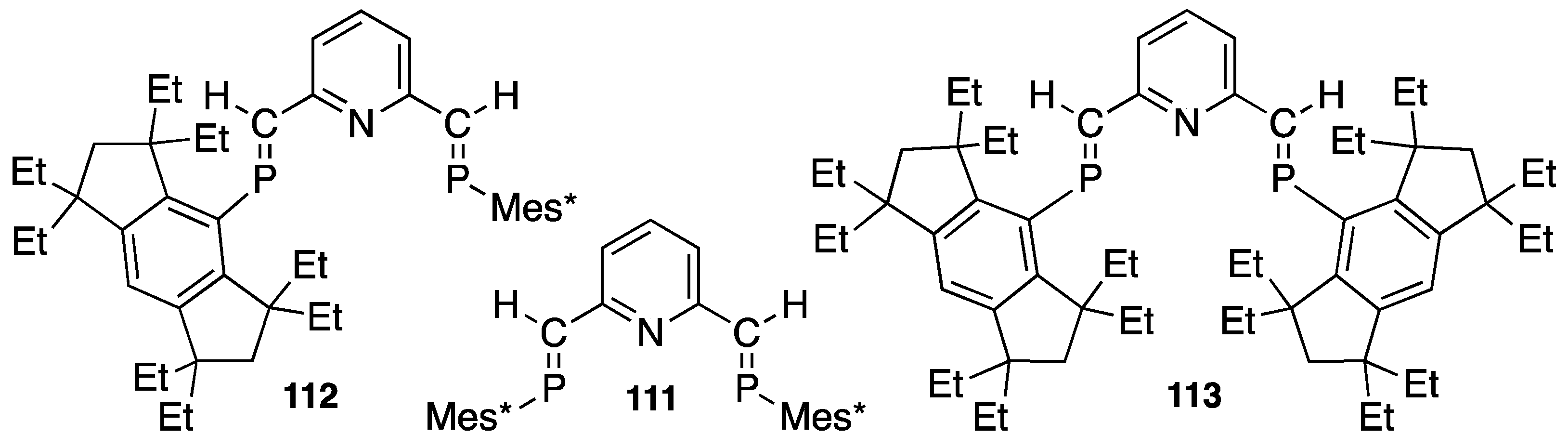
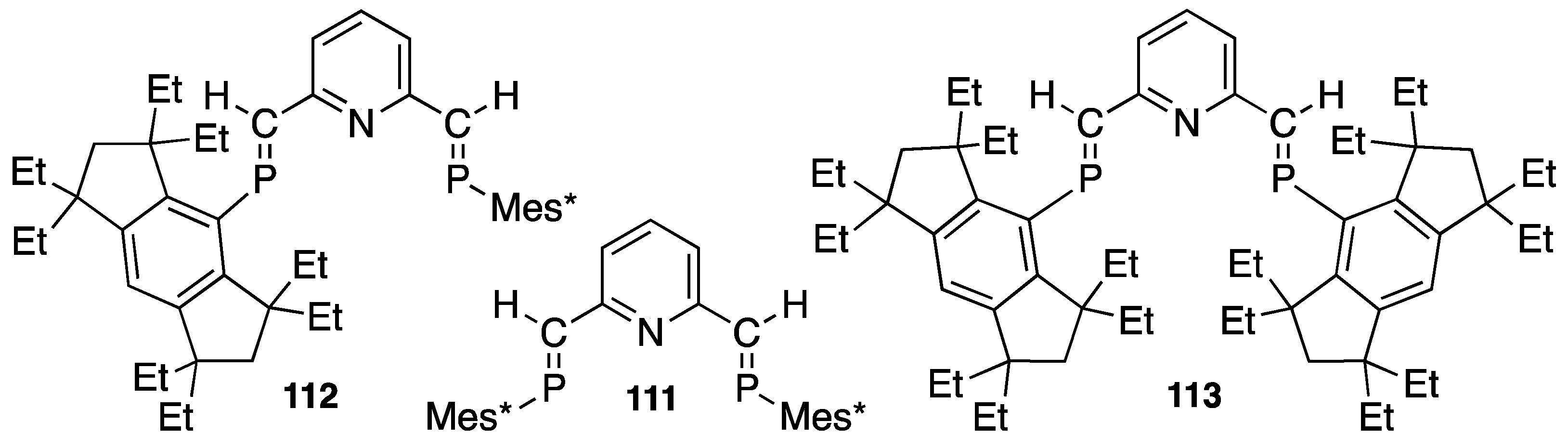
4.3. Synthetic Application to Catalytic Reactions of DPCB–Transition Metal Complexes
5. Closing Remarks
Funding
Acknowledgments
Conflicts of Interest
References
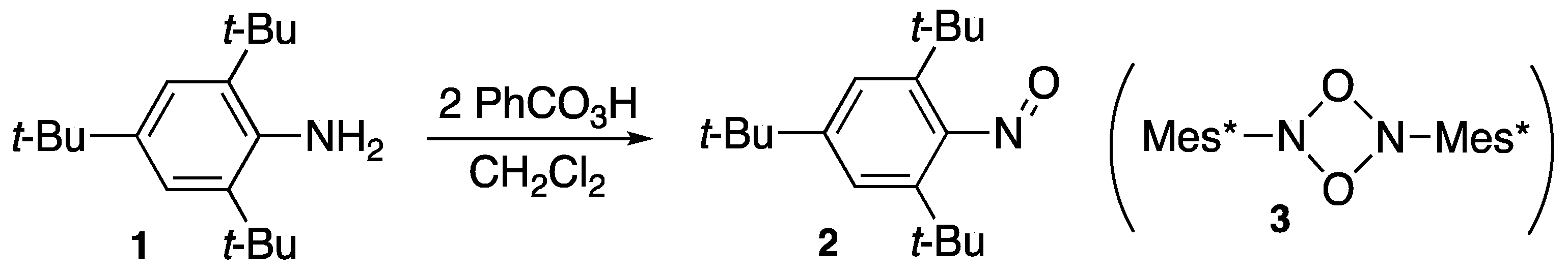
- Okazaki, R.; Hosogai, T.; Iwadare, E.; Hashimoto, M.; Inamoto, N. Preparation of sterically hindered nitrosobenzenes. Bull. Chem. Soc. Jpn. 1969, 42, 3611–3612. [Google Scholar] [CrossRef] [Green Version]
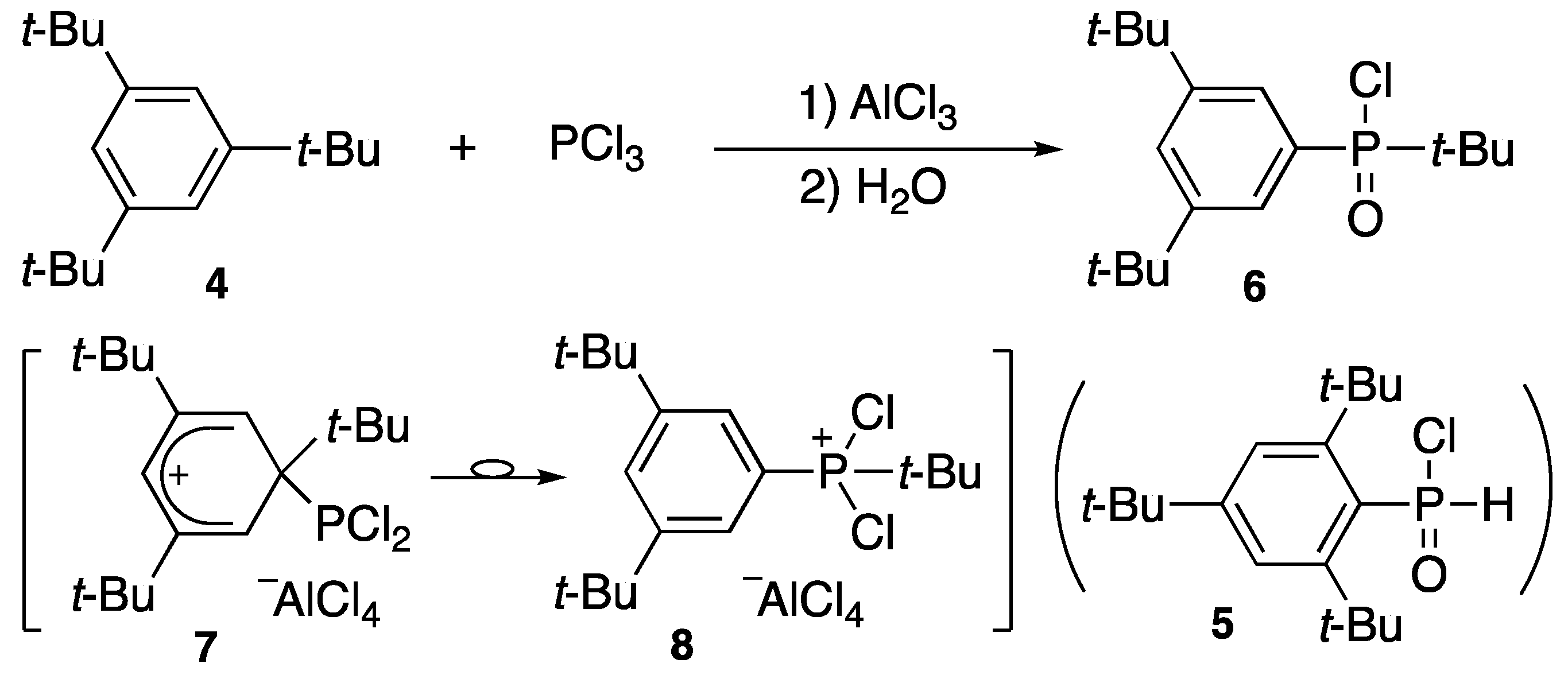
- Cook, A.G. Syntheses and reactions of some hindered organophosphorus compounds. J. Org. Chem. 1965, 30, 1262–1263. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Fujishima, I.; Okazaki, R.; Inamoto, N. Reaction of 1,3,5-tri-tert-butylbenzene with phosphorus trichloride: Revised structure of 2,4,6-tri-tert-butylphenylphosphinic chloride. Chem. Ind. (Lond.) 1970, 19, 625. [Google Scholar]
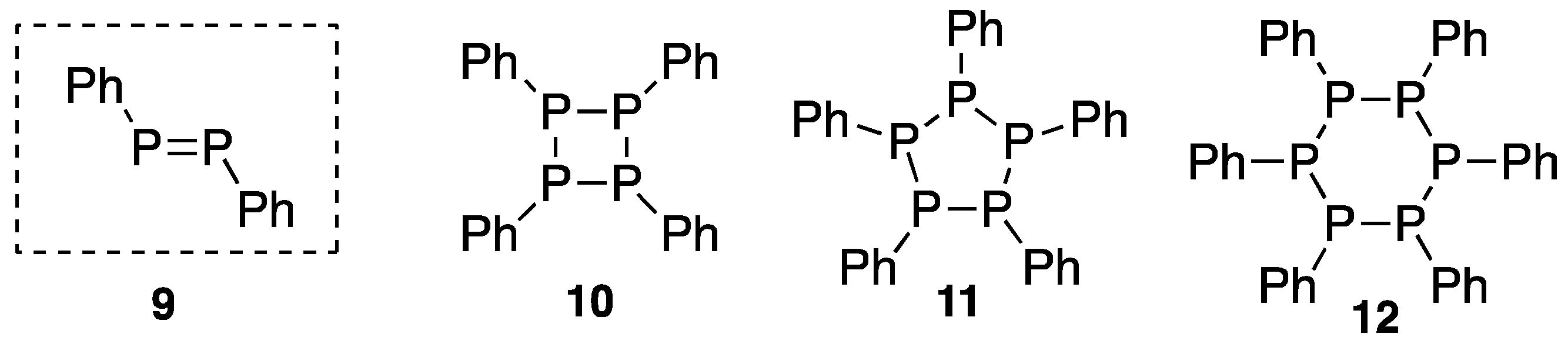
- Köhler, H.; Michaelis, A. Über Phenylphosphin und Phosphobenzol (Diphosphenyl). Ber. Dtsch. Chem. Ges. 1877, 10, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Kuchen, W.; Buchwald, H. Zur Kenntnis der Organophosphorverbindungen, 1. Das Tetraphenyl-cyclotetraphosphin. Chem. Ber. 1958, 91, 2296–2304. [Google Scholar] [CrossRef]
- Daly, J.J.; Maier, L. Molecular structure of phosphobenzene. Nature 1964, 203, 1167–1168. [Google Scholar] [CrossRef]
- Daly, J.J. The crystal and molecular structure of phosphobenezene A, (PC6H5)5. J. Chem. Soc. 1964, 6147–6166. [Google Scholar] [CrossRef]
- Daly, J.J.; Maier, L. Structure of phosphobenzene. Nature 1965, 208, 383–384. [Google Scholar] [CrossRef]
- Daly, J.J. The crystal and molecular structure of a trigonal form of phosphobenezene B, (PC6H5)6. J. Chem. Soc. 1965, 4789–4799. [Google Scholar] [CrossRef]
- Daly, J.J. The crystal and molecular structure of a triclinic form of phosphobenzene, (PC6H5)6, referred as B. J. Chem. Soc. A 1966, 428–439. [Google Scholar] [CrossRef]
- Yoshifuji, M. 2,4,6-Tri-tert.-butylphenylphosphane, (E)-1,2-bis(2,4,6-tri-tert.-butylphenyl)diphosphene, (E)-1-(2,4,6-tri-methylphenyl)-2-(2,4,6-tri-tert.-butylphenyl)diphosphene. In Synthetic Methods in Inorganic and Organometallic Chemistry: Phosphorus, Arsenic, Antimony, and Bismuth; Herrmann, W.A., Karsch, H.H., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 1996; Volume 3, pp. 118–125. [Google Scholar]
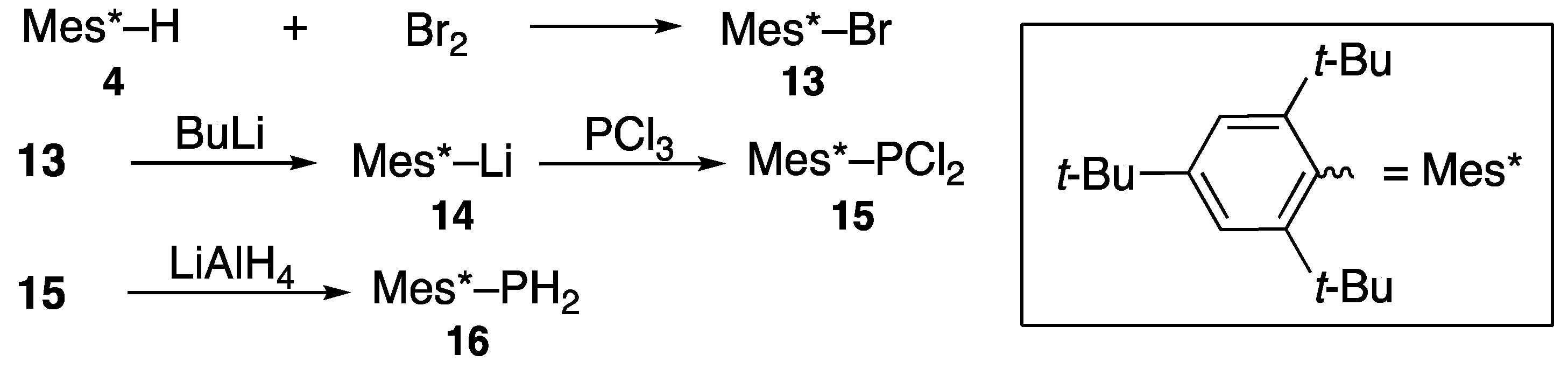
- Yoshifuji, M.; Shima, I.; Inamoto, N.; Hirotsu, K.; Higuchi, T. Synthesis and structure of bis(2,4,6-tri-tert-butylphenyl)diphosphene; Isolation of a true “phosphobenzene”. J. Am. Chem. Soc. 1981, 103, 4587–4589. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Shima, I.; Inamoto, N.; Hirotsu, K.; Higuchi, T. Synthesis and structure of bis(2,4,6-tri-tert-butylphenyl)diphosphene; Isolation of a true "phosphobenzene" (correction). J. Am. Chem. Soc. 1982, 104, 6167. [Google Scholar] [CrossRef]
- Hamaguchi, H.; Tasumi, M.; Yoshifuji, M.; Inamoto, N. The P=P stretching frequency observed in the resonance Raman spectrum of bis(2,4,6-tri-tert-butylphenyl)diphosphene. J. Am. Chem. Soc. 1984, 106, 508–509. [Google Scholar] [CrossRef]
- Cetinkaya, B.; Hudson, A.; Lappert, M.F.; Goldwhite, H. Generation and e.s.r. spectra of some new phosphorus-centred radicals Ṗ2Ar2X, Ṗ(Ar)X, Ṗ(OAr)2, ṖAr2(:O), ṖAr[N(SiMe3)2](:NSiMe3), and [P2Ar2]˙– derived from the bulky group C6H2But3-2,4,6 (= Ar). J. Chem. Soc. Chem. Commun. 1982, 609–610. [Google Scholar] [CrossRef]
- Cowley, A.H.; Kilduff, J.E.; Newman, T.H.; Pakulski, M. Diphosphenes (RP:PR). Synthesis and NMR characterization. J. Am. Chem. Soc. 1982, 104, 5820–5821. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Shibayama, K.; Inamoto, N.; Watanabe, T. Reduction of diphosphene; Formation of dl- and meso-diphosphanes. Chem. Lett. 1983, 12, 585–588. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Shibayama, K.; Inamoto, N.; Matsushita, T.; Nishimoto, K. Isolation of some sterically protected unsymmetrical diphosphenes; Nature of the phosphorus-phosphorus double bond. J. Am. Chem. Soc. 1983, 105, 2495–2497. [Google Scholar] [CrossRef]
- Yoshifuji, M. 31P NMR in studies of some one- and two-coordinate phosphorus compounds. In Phosphorus-31 NMR Spectral Properties in Compound Characterization and Structural Analysis; Quin, L.D., Verkade, J., Eds.; VCH Publishers: New York, NY, USA, 1994; pp. 175–187. [Google Scholar]
- Yoshifuji, M.; Sasaki, S.; Inamoto, N. The first preparation of an unsymmetrical 1,3-diphospha-allene carrying 2,4,6-tri-t-butylphenyl and 2,4,6-tri-t-pentylphenyl as protecting groups. J. Chem. Soc. Chem. Commun. 1989, 22, 1732–1733. [Google Scholar] [CrossRef]
- Smit, C.N.; van der Knaap, T.A.; Bickelhaupt, F. Stable unsymmetrical diphosphenes. Tetrahedron Lett. 1983, 24, 2031–2034. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Inamoto, N. Reaction of a diaryldiphosphene with hexacarbonylchromium(0)–Formation of (arene)tricarbonylchromium(0) complexes. Tetrahedron Lett. 1983, 24, 4855–4858. [Google Scholar] [CrossRef]
- Cowley, A.H.; Kilduff, J.E.; Mehrotra, S.K.; Norman, N.C.; Pakulski, M. A new approach to the formation of phosphorus–phosphorus double bonds. J. Chem. Soc. Chem. Commun. 1983, 9, 528–529. [Google Scholar] [CrossRef]
- Jutzi, P.; Meyer, U.; Krebs, B.; Dartmann, M. Bis(pentamethylcyclopentadienyl)diphosphene—A molecule with fluxional structure and reactive P≡C bond. Angew. Chem. Int. Ed. Engl. 1986, 25, 919–921. [Google Scholar] [CrossRef]
- Couret, C.; Escudié, J.; Satgé, J. Un nouveau diphosphene stable: Le bis[tris(trimethylsilyl)methyl]diphosphene. Tetrahedron Lett. 1982, 23, 4941–4942. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Sato, T.; Inamoto, N. Wavelength- and temperature-dependent photolysis of a diphosphene. Generation of 2,4,6-tri-t-butylphenylphosphinidene and E/Z isomerization. Chem. Lett. 1988, 17, 1735–1738. [Google Scholar] [CrossRef] [Green Version]
- Caminade, A.-M.; Verrier, M.; Ades, C.; Paillous, N.; Koenig, M. Laser irradiation of a diphosphene: Evidence for the first cis–trans isomerization. J. Chem. Soc. Chem. Commun. 1984, 13, 875–877. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Shinohara, N.; Toyota, K. Application of 1,2-bis(2-bromo-3,5-di-t-butylphenyl)ethane to preparation of compounds having two diphosphene units. Tetrahedron Lett. 1996, 37, 7815–7818. [Google Scholar] [CrossRef]
- Smith, R.C.; Shah, S.; Protasiewicz, J.D. A role for free phosphinidenes in the reaction of magnesium and sterically encumbered ArPCl2 in solution at room temperature. J. Organomet. Chem. 2002, 646, 255–261. [Google Scholar] [CrossRef]
- West, R.; Fink, M.J.; Michl, J. Tetramesityldisilene, a stable compound containing a silicon-silicon double bond. Science 1981, 214, 1343–1344. [Google Scholar] [CrossRef]
- Yoshifuji, M. Sterically protected organophosphorus compounds of unusual structures. Pure Appl. Chem. 2017, 89, 281–286. [Google Scholar] [CrossRef]
- Yoshifuji, M. Sterically protected organophosphorus compounds of unusual structures. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1452–1456. [Google Scholar] [CrossRef]
- Yoshifuji, M. Phosphinylidenephosphines (diphosphenes). In Multiple Bonds and Low Coordination in Phosphorus Chemistry; Regitz, M., Scherer, O.J., Eds.; Thieme: Stuttgart, Germany, 1990; pp. 321–337. [Google Scholar]
- Weber, L. The chemistry of diphosphenes and their heavy congeners: Synthesis, structure, and reactivity. Chem. Rev. 1992, 92, 1839–1905. [Google Scholar] [CrossRef]
- Dillon, K.B.; Mathey, F.; Nixon, J.F. Phosphorus: The Carbon Copy from Organophosphorus to Phospha-Organic Chemistry; John Wiley & Sons: Chichester, UK, 1998. [Google Scholar]
- Yoshifuji, M. Recent developments in the chemistry of low-coordinated organophosphorus compounds. Pure Appl. Chem. 2005, 77, 2011–2020. [Google Scholar] [CrossRef]
- Yoshifuji, M. Sterically protected diphosphenes. Eur. J. Inorg. Chem. 2016, 5, 607–615. [Google Scholar] [CrossRef]
- Yoshifuji, M. Protecting groups for stabilization of unusual organophosphorus compounds. Main Group Chem. News 1998, 6, 20–26. [Google Scholar]
- Yoshifuji, M. Protecting groups for stabilization of inter-element linkages. J. Organomet. Chem. 2000, 611, 210–216. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Shibayama, K.; Inamoto, N.; Hirotsu, K.; Higuchi, T. Reaction of the diphosphene ArP=PAr (Ar=2,4,6-But3C6H2) with sulphur: Isolation and X-ray structure of the diphosphene monosulphide. J. Chem. Soc. Chem. Commun. 1983, 862–863. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Ando, K.; Shibayama, K.; Inamoto, N.; Hirotsu, K.; Higuchi, T. The first X-ray structure analysis of a thiadiphosphirane. Angew. Chem. Int. Ed. Engl. 1983, 22, 418–419. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Hashida, T.; Inamoto, N.; Hirotsu, K.; Horiuchi, T.; Higuchi, T.; Ito, K.; Nagase, S. E/Z Photoisomerization of a diphosphene of carbonyl metal complexes (M = Cr, Mo, W). Angew. Chem. Int. Ed. Engl. 1985, 24, 211–212. [Google Scholar] [CrossRef]
- Märkl, G. 2,4,6-Triphenylphosphabenzene. Angew. Chem. Int. Ed. 1966, 5, 846–847. [Google Scholar] [CrossRef]
- Klebach, T.C.; Lourens, R.; Bickelhaupt, F. Synthesis of mesityldiphenylmethylenephosphine: A stable compound with a localized phosphorus:carbon bond. J. Am. Chem. Soc. 1978, 100, 4886–4888. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Inamoto, N. Photoisomerization of benzylidenephosphine containing phosphorus in low coordination state. Tetrahedron Lett. 1985, 26, 1727–1730. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Matsuda, I.; Niitsu, T.; Inamoto, N.; Hirotsu, K.; Higuchi, T. Synthesis and structures of E- and Z-phosphaethylenes. Tetrahedron 1988, 44, 1363–1367. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Okamoto, Y.; Asakura, T. Separation of an optically active phosphaallene of pseudo axial dissymmetry. Tetrahedron Lett. 1990, 31, 2311–2314. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Inamoto, N. Isolation and characterisation of the first stable diphospha-allene; P,P’-bis(2,4,6-tri-t-butylphenyl)-1,3-diphospha-allene. J. Chem. Soc. Chem. Commun. 1984, 689–690. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Yoshimura, H.; Hirotsu, K.; Okamoto, A. Reactions of dichlorocarbene with sterically protected phosphaallene and 1,3-diphosphaallene. J. Chem. Soc. Chem. Commun. 1991, 2, 124–125. [Google Scholar] [CrossRef]
- Märkl, G.; Sejpka, H.; Dietl, S.; Nuber, B.; Ziegler, M.L. 1-Phospha-1,2,3-butatriene. Angew. Chem. Int. Ed. 1986, 25, 1003–1004. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Yoshimura, H. Preparation of 1,4-diphosphabutatriene from 3-dichloromethylene-1,2-diphosphirane. Chem. Lett. 1991, 20, 491–492. [Google Scholar] [CrossRef]
- Märkl, G.; Kreitmeier, P. 1,4-Diphosphabutatriene. Angew. Chem. Int. Ed. 1988, 27, 1360–1361. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Shibayama, K.; Inamoto, N. Isolation and characterization of some very stable 1-phospha-allenes; sterically protected iminomethylene- and ethenylidenephosphines. Tetrahedron Lett. 1984, 25, 1809–1812. [Google Scholar] [CrossRef]
- Appel, R.; Paulen, W. The first stable phosphaketene. Angew. Chem. Int. Ed. Engl. 1983, 22, 785–786. [Google Scholar]
- Becker, G.; Gresser, G.; Uhl, W. Acyl- und Alkylidenphosphane, XV. 2,2-Dimethylpropylidinphosphan, eine stabile Verbindung mit einem Phosphoratom der Koordinationszahl 1. Z. Naturforsch. B 1981, 36, 16–19. [Google Scholar] [CrossRef]
- Märkl, G.; Sejpka, H. 2-(2,4,6-Tri-tert-butylphenyl)-1-phosphaethin, 1,4-bis-(trimethylsiloxy)-1,4-bis(2,4,6-tri-tert-butylphenyl)-2,3-diphosphabutadien. Tetrahedron Lett. 1986, 27, 171–174. [Google Scholar] [CrossRef]
- Appel, R.; Casser, C.; Immenkeppel, M.; Knoch, F. Easy synthesis of phosphaalkenes by a phosphorus-analogous isocyanide reaction and an atypical crystal structure of a tetracarbonyl(phosphaalkene)iron complex. Angew. Chem. Int. Ed. Engl. 1984, 23, 895–896. [Google Scholar] [CrossRef]
- Karsch, H.H.; Köhler, F.H.; Reisacher, H.-U. C-Phosphinosubstituierte Phosphaalkene und eine erstes Carbodiphosphan. Tetrahedron Lett. 1984, 25, 3687–3690. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Yoshimura, H.; Toyota, K. Reaction of dichlorocarbene with phosphaethylenes. Preparation of phosphaallene from phosphaethylene via dichlorophosphirane. Chem. Lett. 1990, 19, 827–830. [Google Scholar] [CrossRef]
- Makosza, M.; Wawrzyniewicz, M. Reactions of organic anions. XXIV. Catalytic method for preparation of dichlorocyclopropane derivatives in aqueous medium. Tetrahedron Lett. 1969, 10, 4659–4662. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Sasaki, S.; Niitsu, T.; Inamoto, N. A convenient new route from diphosphene to 1,3-diphospha-allene and dynamic NMR studies of the 2,4,6-tri-t-butylphenyl derivative. Tetrahedron Lett. 1989, 30, 187–188. [Google Scholar] [CrossRef]
- Karsch, H.H.; Reisacher, H.-U.; Müller, G. Molecular structure of a 1,3-diphosphaallene: (2,4,6-tBu3C6H2)P=C=P(2,4,6-tBu3C6H2), a phosphorus analogue of carbon disulfide. Angew. Chem. Int. Ed. 1984, 23, 618–619. [Google Scholar] [CrossRef]
- Ito, S.; Sekiguchi, S.; Yoshifuji, M. Formation and electrochemical properties of a 1,3-diphosphafulvene including formal dimerization of 1-phosphaallene. J. Org. Chem. 2004, 69, 4181–4184. [Google Scholar] [CrossRef]
- Ito, S.; Sekiguchi, S.; Freytag, M.; Yoshifuji, M. An efficient synthetic method of 1-(2,4,6-tri-t-butylphenyl)-3-phenyl-1-phosphaallenes [Mes*P=C=CR–Ph; R = H, SiMe3] from dibromophosphaethene [Mes*P=CBr2] (Mes* = 2,4,6-t-Bu3C6H2). Bull. Chem. Soc. Jpn. 2005, 78, 1142–1144. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Niitsu, T.; Inamoto, N.; Okamoto, Y.; Aburatani, R. The first separation of an optically active 1,3-diphospha-allene of axial dissymmetry. J. Chem. Soc. Chem. Commun. 1986, 20, 1550–1551. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Niitsu, T.; Inamoto, N. Formation of a phospha-alkyne via migration of the phenyl group from phosphorus to carbon. Chem. Lett. 1988, 17, 1733–1734. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Ito, S. Chemistry of phosphanylidene carbenoids. Top. Curr. Chem. 2003, 223, 67–89. [Google Scholar]
- Yoshifuji, M.; Kawanami, H.; Kawai, Y.; Toyota, K.; Yasunami, M.; Niitsu, T.; Inamoto, N. A new preparative route to phospha ethynes from dichlorophosphaethenes by lithiation, photoisomerization, and re-lithiation involving migration. Chem. Lett. 1992, 21, 1053–1056. [Google Scholar] [CrossRef]
- Köbrich, G.; Akhtar, A.; Ansari, F.; Breckoff, W.E.; Büttner, H.; Drischel, W.; Fischer, R.H.; Flory, K.; Fröhlich, H.; Goyert, W.; et al. Chemistry of stable α-halogenoorganolithium compounds and the mechanism of carbenoid reactions. Angew. Chem. Int. Ed. Engl. 1967, 6, 41–52. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Ito, S. Phosphorus version of the Fritsch-Buttenberg-Wiechell reaction. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 140–145. [Google Scholar] [CrossRef]
- Appel, R.; Immenkeppel, M. Niederkoordinierte Phosphorverbindungen, 58. Synthese und Reaktivität der 2,4,6-Tri-tert-butylphenyl(halogenmethylen)phosphane. Z. Anorg. Allg. Chem. 1987, 553, 7–14. [Google Scholar] [CrossRef]
- Ito, S.; Toyota, K.; Yoshifuji, M. Formation of 5,7-di-tert-butyl-1,1-dimethyl-4-phospha-1,2-dihydronaphthalene by the reaction of 2,2-dibromo-1-(2,4,6-tri-tert-butylphenyl)-1-phosphaethene with butyllithium. Chem. Commun. 1997, 1637–1638. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Ito, S. Theoretical studies of the intramolecular cyclization of 2,4,6-tBu3C6H2P=C: And effects of conjugation between the P=C and aromatic moieties. Beilstein J. Org. Chem. 2014, 10, 1032–1036. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.T.; Ha, T.K. Ab initio study of structures and relative stabilities of RCP (R = H, F) and their energetically higher-lying isomers RPC. J. Mol. Struct. (Theochem.) 1986, 139, 145–152. [Google Scholar] [CrossRef]
- Lehmann, K.K.; Ross, S.C.; Lohr, L.L. Experimental and ab initio determination of the bending potential of HCP. J. Chem. Phys. 1985, 82, 4460–4469. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, H.; Ito, S.; Yoshifuji, M. Synthesis of a 1,3-diphosphacyclobutane-2,4-diyl from Mes*C P (Mes* = 2,4,6-tBu3C6H2). Angew. Chem. Int. Ed. 2003, 42, 3802–3804. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H.; Ito, S.; Yoshifuji, M. Preparation and reactions of 1,3-diphosphacyclobutane-2,4-diyls featuring an amino substituent and/or a carbonyl group. Chem. Eur. J. 2004, 10, 2700–2706. [Google Scholar] [CrossRef] [PubMed]
- Niecke, E.; Fuchs, A.; Baumeister, F.; Nieger, M.; Schoeller, W.W. A P2C2-four-membered ring with unusual bonding–synthesis, structure, and ring opening of a 1,3-diphosphacyclobutane-2,4-diyl. Angew. Chem. Int. Ed. 1995, 34, 555–557. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Hirano, Y.; Schnakenburg, G.; Streubel, R.; Niecke, E.; Ito, S. New aspects of 1,3-diphosphacyclobutane-2,4-diyls. Helv. Chim. Acta 2012, 95, 1723–1729. [Google Scholar] [CrossRef]
- Ueta, Y.; Mikami, K.; Ito, S. Access to air-stable 1,3-diphosphacyclobutane-2,4-diyls by an arylation reaction with arynes. Angew. Chem. Int. Ed. 2016, 55, 7525–7529. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Arduengo, A.J., III; Konovalova, T.A.; Kispert, L.D.; Kikuchi, M.; Ito, S. Oxidation of 1,3-diphosphacyclobutane-2,4-diyl with ammoniumyl antimonate and EPR study of the corresponding cation radical. Chem. Lett. 2006, 35, 1136–1137. [Google Scholar] [CrossRef]
- Ito, S.; Kikuchi, M.; Sugiyama, H.; Yoshifuji, M. Synthesis and properties of air-stable 1,3-diphosphacyclobutane-2,4-diyls and the related compounds. J. Organomet. Chem. 2007, 692, 2761–2767. [Google Scholar] [CrossRef]
- Ito, S.; Miura, J.; Morita, N.; Yoshifuji, M.; Arduengo, A.J., III. Modeling the direct activation of dihydrogen by a P2C2-cyclic biradical: Formation of a cyclic bis(P-H λ5-phosphorane). Inorg. Chem. 2009, 48, 8063–8065. [Google Scholar] [CrossRef]
- Ito, S.; Miura, J.; Morita, N.; Yoshifuji, M.; Arduengo, A.J., III. Diverse reactions of sterically protected 1,3-diphosphacyclobutane-2,4-diyls with hydride. Dalton Trans. 2010, 39, 8281–8287. [Google Scholar] [CrossRef]
- Ito, S.; Ueta, Y.; Koshino, K.; Kojima, K.M.; McKenzie, I.; Mikami, K. Observation of a metastable P-heterocyclic radical by muonium addition to a 1,3-diphosphacyclobutane-2,4-diyl. Angew. Chem. Int. Ed. 2018, 57, 8608–8613. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Miura, J.; Morita, N.; Yoshifuji, M.; Arduengo, A.J., III. Poly(biradicals): Oligomers of 1,3-diphosphacyclobutane-2,4-diyl units. Angew. Chem. Int. Ed. 2008, 47, 6418–6421. [Google Scholar] [CrossRef] [PubMed]
- Ueta, Y.; Mikami, K.; Ito, S. Chemical detection of hydrogen fluoride by the phosphorus congener of cyclobutane-1,3-diyl. Inorg. Chem. 2015, 54, 8778–8785. [Google Scholar] [CrossRef]
- Ito, S.; Kikuchi, M.; Yoshifuji, M.; Arduengo, A.J., III; Konovalova, T.A.; Kispert, L.D. Preparation and characterization of an air-tolerant 1,3-diphosphacyclobuten-4-yl radical. Angew. Chem. Int. Ed. 2006, 45, 4341–4345. [Google Scholar] [CrossRef] [PubMed]
- Yoshifuji, M. Chemistry of diphosphinidenecyclobutenes. J. Synth. Org. Chem. Jpn. 2003, 61, 1116–1123. [Google Scholar] [CrossRef]
- Toyota, K.; Horikawa, K.; Jensen, R.S.; Omori, K.; Kawasaki, S.; Ito, S.; Yoshifuji, M.; Morita, N. Improved preparative method for 1,2-diaryl-3,4-diphosphinidenecyclobutenes and its application to the studies of 1,2-bis(arylthienyl)-3,4-diphosphinidenecyclobutenes. Bull. Chem. Soc. Jpn. 2007, 80, 1580–1586. [Google Scholar] [CrossRef]
- Appel, R.; Winkhaus, V.; Knoch, F. Niederkoordinierte Phosphorverbindungen, 56. Valenzisomerisierung eines 3,4-Diphospha-1,5-hexadiins zu einem 3,4-Bis(phosphamethylen)-1-cyclobuten. Chem. Ber. 1987, 120, 243–245. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Toyota, K.; Murayama, M.; Yoshimura, H.; Okamoto, A.; Hirotsu, K.; Nagase, S. Preparation of sterically protected 3,4-bis(phosphinidene)cyclobutenes and their isomerism. Chem. Lett. 1990, 2195–2198. [Google Scholar] [CrossRef]
- Toyota, K.; Tashiro, K.; Yoshifuji, M.; Nagase, S. Preparation and isomerization of 3,4-bis(2,4,6-tri-t-butylphenylphosphinidene)cyclobutenes. Bull. Chem. Soc. Jpn. 1992, 65, 2297–2299. [Google Scholar] [CrossRef]
- Toyota, K.; Tashiro, K.; Abe, T.; Yoshifuji, M. Iodine-induced E/Z isomerization of sterically protected 3,4-diphosphinidenecyclobutenes and 1-phosphaethenes. Heteroat. Chem. 1994, 5, 549–553. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Ichikawa, Y.; Toyota, K.; Kasashima, E.; Okamoto, Y. Rotational isomers of a 1,2-diphenyl-3,4-diphosphinidenecyclobutene. Chem. Lett. 1997, 26, 87–88. [Google Scholar] [CrossRef]
- Toyota, K.; Tashiro, K.; Yoshifuji, M. Synthesis and structure of diphospha[4]radialene. Angew. Chem. Int. Ed. Engl. 1993, 32, 1163–1165. [Google Scholar] [CrossRef]
- Ozawa, F.; Kawagishi, S.; Ishiyama, T.; Yoshifuji, M. Synthesis, Structures, and Reactions of Methylplatinum(II) and -palladium(II) Complexes Bearing Diphosphinidenecyclobutene Ligands (DPCB-Y). Organometallics 2004, 23, 1325–1332. [Google Scholar] [CrossRef]
- Toyota, K.; Tashiro, K.; Yoshifuji, M.; Miyahara, I.; Hayashi, A.; Hirotsu, K. X-Ray structure analyses of a diphosphinidenecyclobutene and its chelate type tetracarbonylmolybdenum(0) complex. J. Organometal. Chem. 1992, 431, C35–C41. [Google Scholar] [CrossRef]
- Toyota, K.; Masaki, K.; Abe, T.; Yoshifuji, M. Preparation and structure of dichloropalladium(II) complexes of sterically protected diphosphinidenecyclobutene and their synthetic application as homogeneous catalyst. Chem. Lett. 1995, 24, 221–222. [Google Scholar] [CrossRef]
- Ikeda, S.; Ohhata, F.; Miyoshi, M.; Tanaka, R.; Minami, T.; Ozawa, F.; Yoshifuji, M. Synthesis and reactions of palladium and platinum complexes bearing diphosphinidenecyclobutene ligands: A thermally stable catalyst for ethylene polymerization. Angew. Chem. Int. Ed. 2000, 39, 4512–4513. [Google Scholar] [CrossRef]
- Freytag, M.; Ito, S.; Yoshifuji, M. Coordination behavior of sterically protected phosphaalkenes on the AuCl moietyleading to catalytic 1,6-enyne cycloisomerization. Chem. Asian J. 2006, 1, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Freytag, M.; Yoshifuji, M. Structural and coordination properties of 1,2-bis(cyclopropyl)-3,4-bis(tri-t-butylphenyl)-3,4-diphosphinidenecyclobutene prepared by dehydrogenative homocoupling of 3-cyclopropyl-1-phosphaallene. J. Chem. Soc. Dalton 2006, 5, 710–713. [Google Scholar] [CrossRef]
- Minami, T.; Okamoto, H.; Ikeda, S.; Tanaka, R.; Ozawa, F.; Yoshifuji, M. (π-Allyl)palladium complexes bearing diphosphinidenecyclobutene ligands: A highly active catalyst for hydroamination of 1,3-dienes. Angew. Chem. Int. Ed. 2001, 40, 4501–4503. [Google Scholar] [CrossRef]
- Yoshifuji, M.; Ichikawa, Y.; Yamada, N.; Toyota, K. Preparation and X-ray analysis of novel carbonyltungsten(0) complexes of diphosphinidenecyclobutenes. Chem. Commun. 1998, 1, 27–28. [Google Scholar] [CrossRef]
- Ozawa, F.; Yoshifuji, M. Catalytic applications of transition metal complexes bearing diphosphinidenecyclobutenes (DPCB). Dalton Trans. 2006, 42, 4987–4995. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, F.; Yoshifuji, M. Synthesis and catalytic properties of diphosphinidenecyclobutene-coordinated palladium and platinum complexes. C. R. Chim. 2004, 7, 747–754. [Google Scholar] [CrossRef]
- Ozawa, F.; Katayama, H.; Toyota, K.; Yoshifuji, M. Catalytic applications of transition metal complexes bearing sp2 hybridized phosphorus compounds. Catal. Catal. 2005, 47, 544–549. [Google Scholar]
- Gajare, A.S.; Jensen, R.S.; Toyota, K.; Yoshifuji, M.; Ozawa, F. Low coordinated diphosphinidene ligands: A new entry for Stille cross-coupling of aryl bromides. Synlett 2005, 144–148. [Google Scholar] [CrossRef]
- Gajare, A.S.; Toyota, K.; Yoshifuji, M.; Ozawa, F. Solvent free amination reactions of aryl bromides at room temperature catalyzed by a (π-allyl)palladium complex bearing a diphosphinidenecyclobutene ligand. J. Org. Chem. 2004, 69, 6504–6506. [Google Scholar] [CrossRef]
- Gajare, A.S.; Toyota, K.; Yoshifuji, M.; Ozawa, F. Application of a diphosphinidenecyclobutene ligand in solvent free Cu-catalysed amination reactions of aryl halides. Chem. Commun. 2004, 17, 1994–1995. [Google Scholar] [CrossRef]
- Jensen, R.S.; Gajare, A.S.; Toyota, K.; Yoshifuji, M.; Ozawa, F. A convenient procedure for palladium catalyzed cyanation using a unique bidentate phosphorus ligand. Tetrahedron Lett. 2005, 46, 8645–8647. [Google Scholar] [CrossRef]
- Ozawa, F.; Okamoto, H.; Kawagishi, S.; Yamamoto, S.; Minami, T.; Yoshifuji, M. (π-Allyl)palladium complexes bearing diphosphinidenecyclobutene ligands (DPCB): Highly active catalysts for direct conversion of allylic alcohols. J. Am. Chem. Soc. 2002, 124, 10968–10969. [Google Scholar] [CrossRef]
- Taguchi, H.; Takeuchi, K.; Tsujimoto, S.; Matsuo, T.; Tanaka, H.; Yoshizawa, K.; Ozawa, F. Unsymmetrical PNP-pincer type phosphaalkene ligands protected by a fused-ring bulky Eind group: Synthesis and applications to Rh(I) and Ir(I) complexes. Organometallics 2016, 35, 1526–1533. [Google Scholar] [CrossRef]
- Takeuchi, K.; Tanaka, Y.; Tanigawa, I.; Ozawa, F. Cu(I) complex bearing a PNP-pincer-type phosphaalkene ligand with a bulky fused-ring Eind group: Properties and applications to FLP-type bond activation and catalytic CO2 reduction. Dalton Trans. 2020, 49, 3630–3637. [Google Scholar] [CrossRef]
- Yoshifuji, M. Low coordinate phosphorus compounds for 40 years in memory of the late Professor François Mathey, École Polytechnique. Phosphorus Sulfur Silicon Relat. Elem. 2021, 1–6. [Google Scholar] [CrossRef]


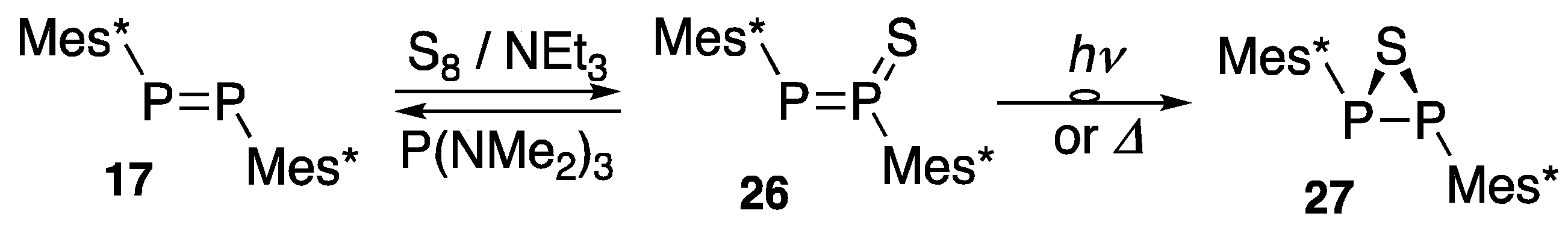



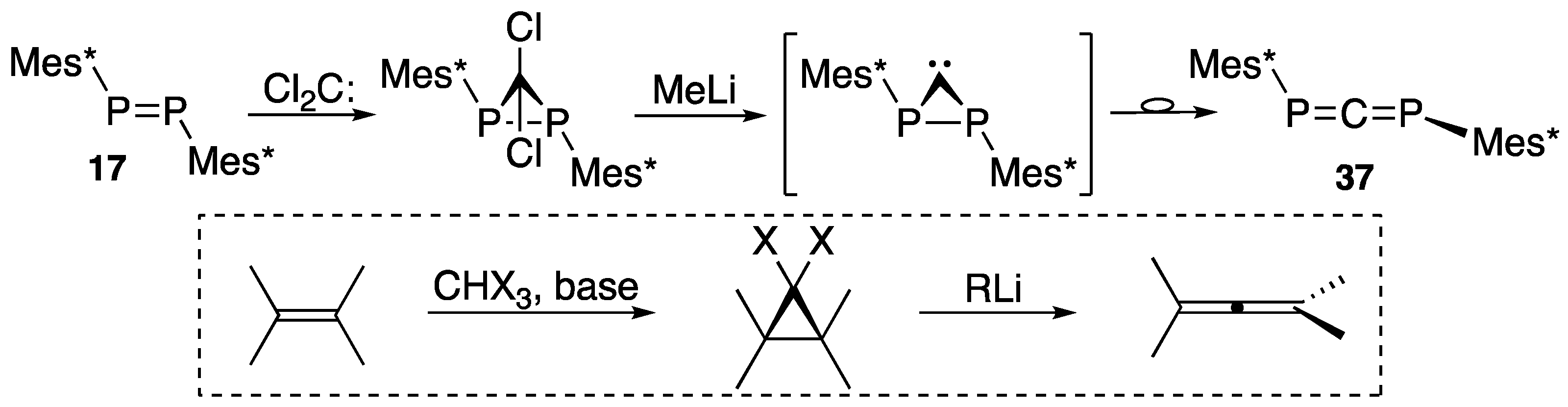

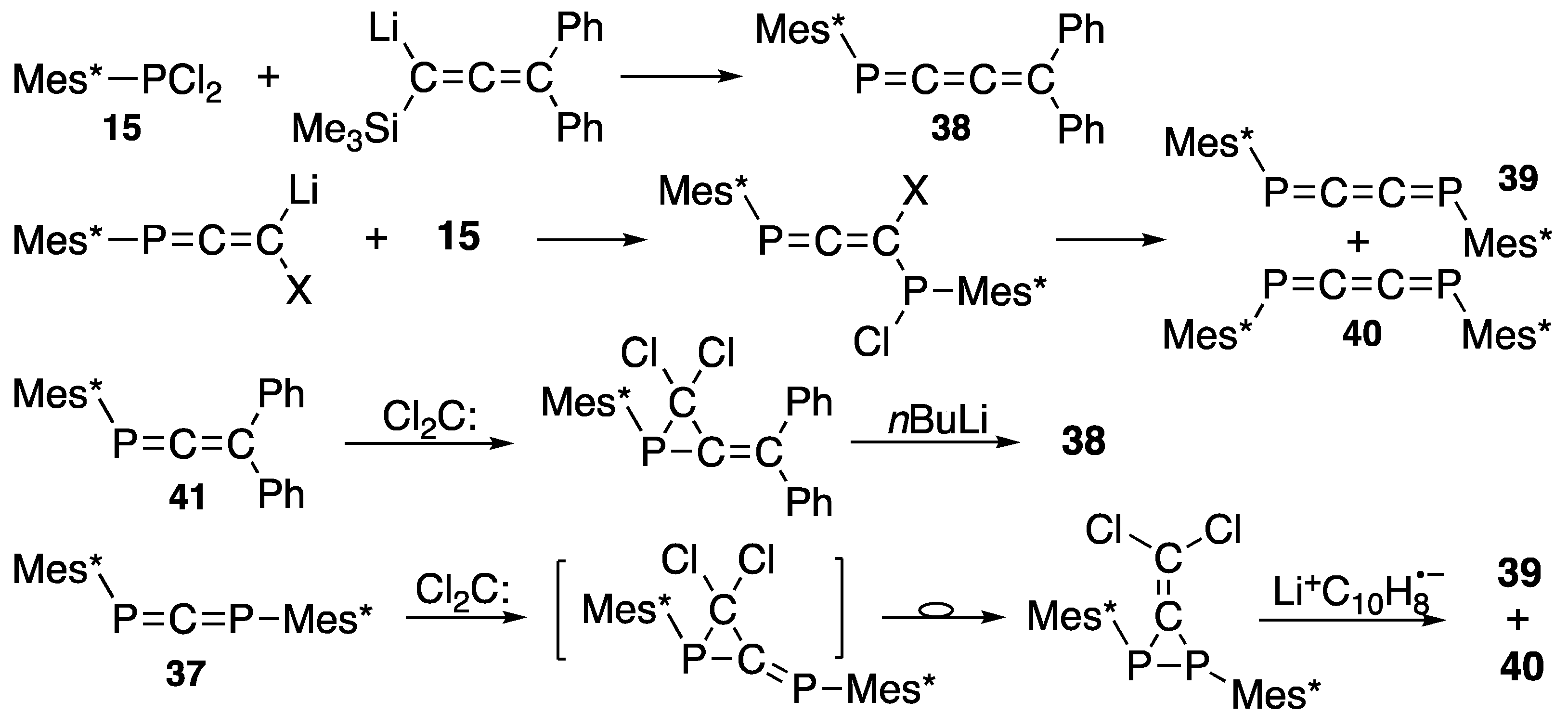
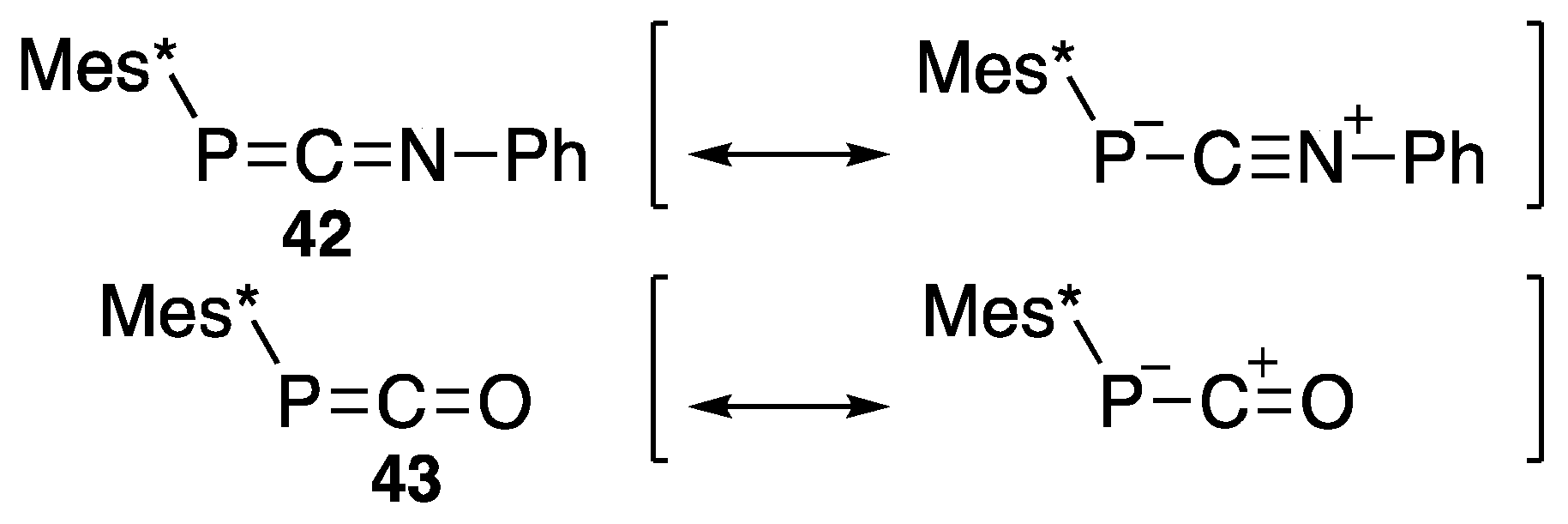
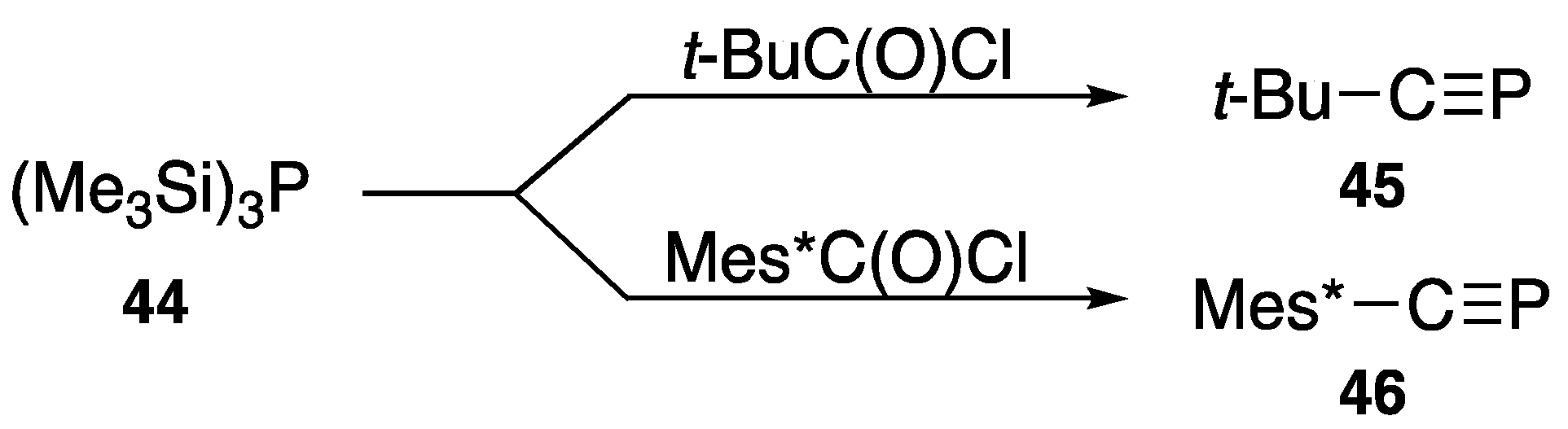
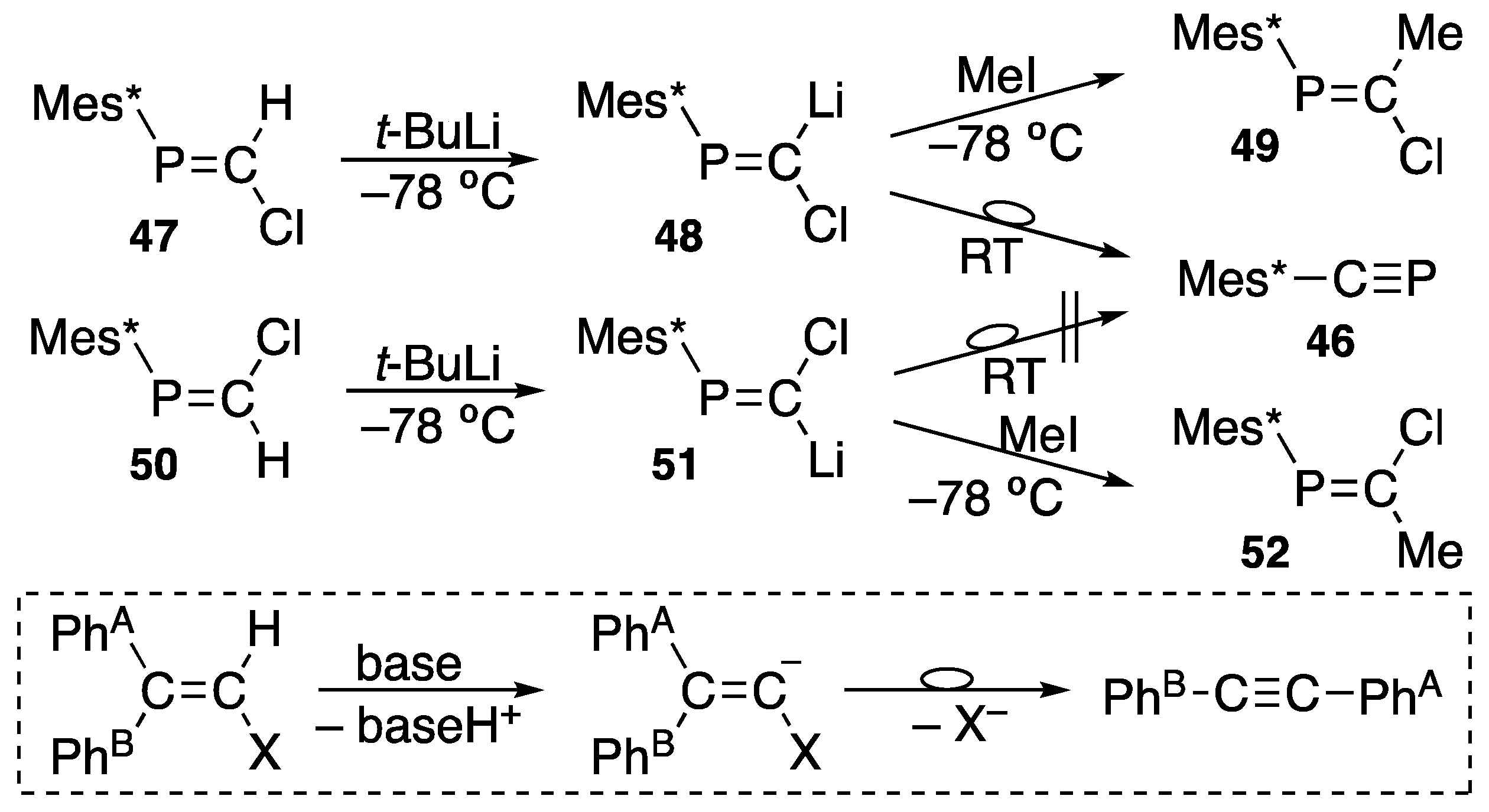
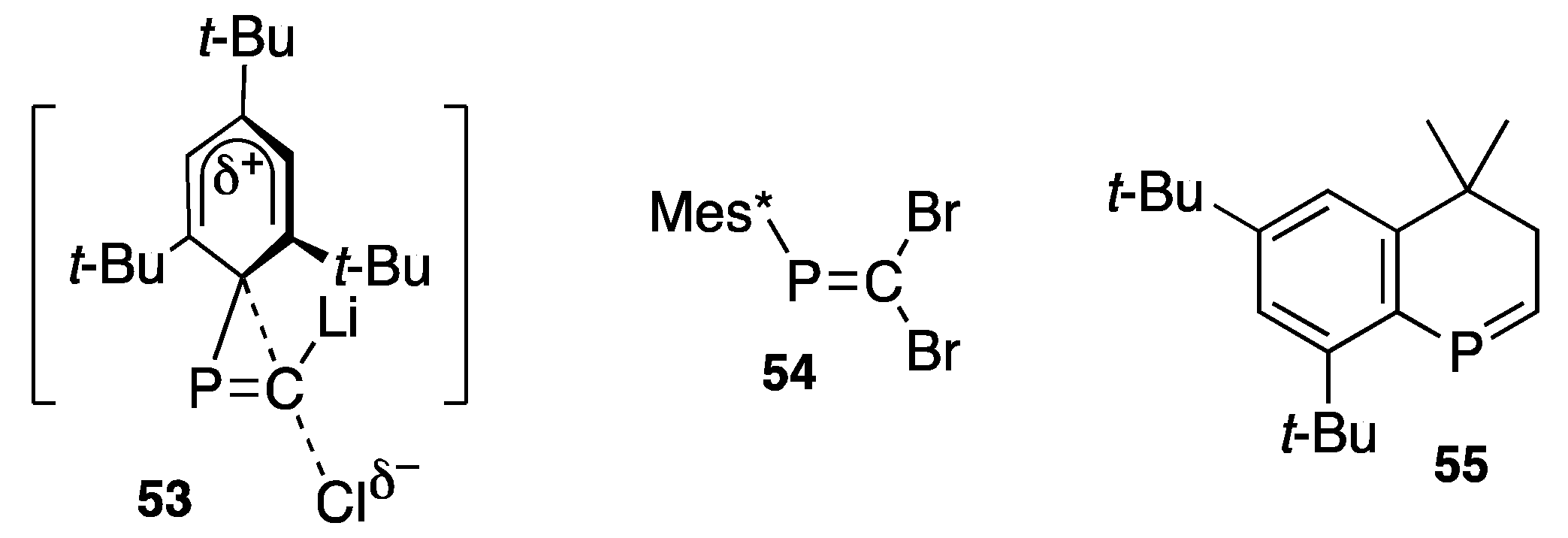
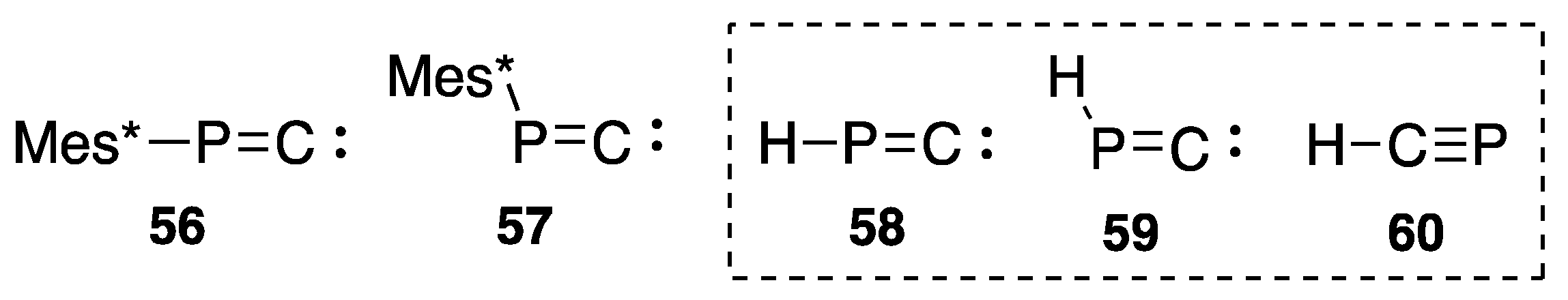
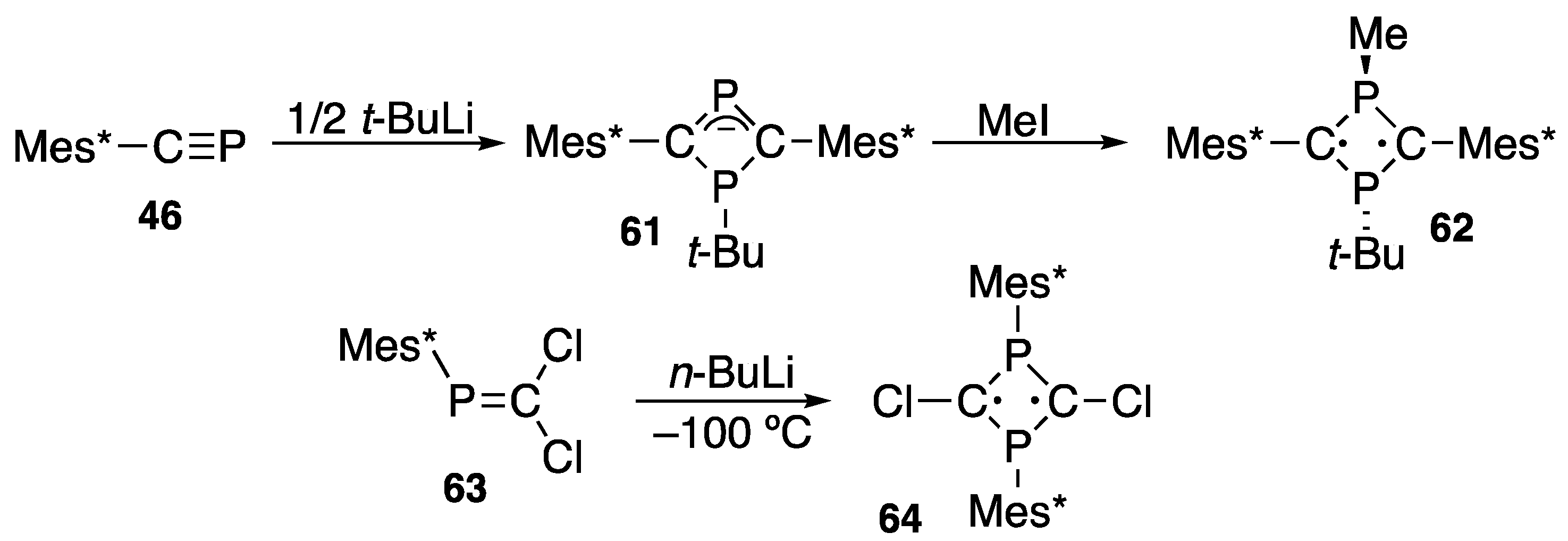

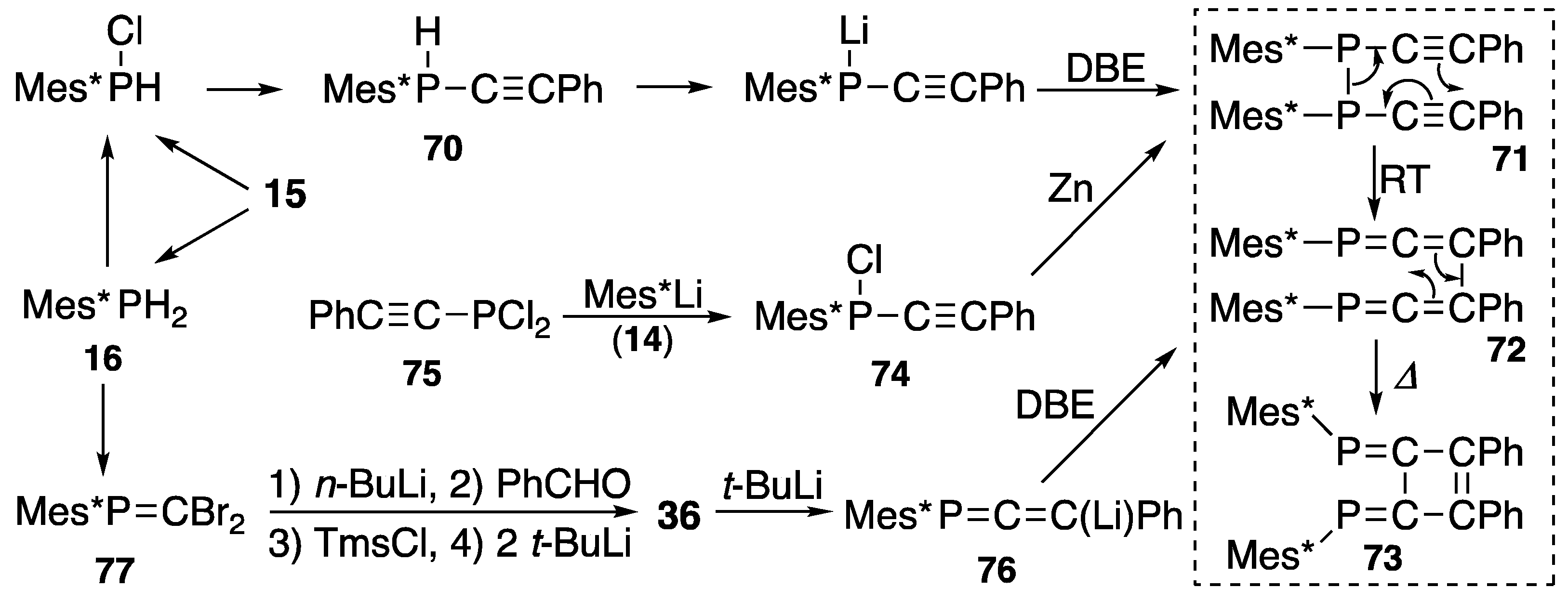
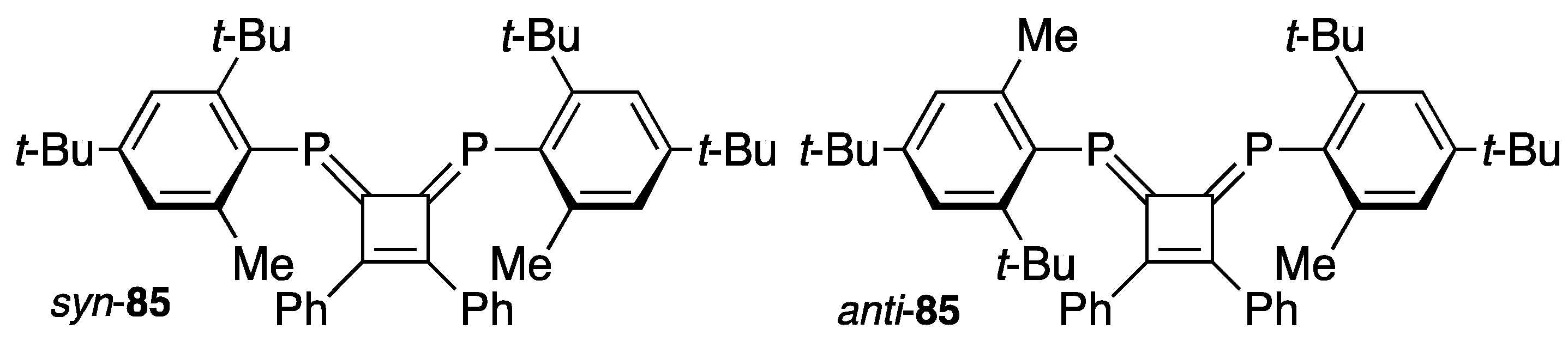


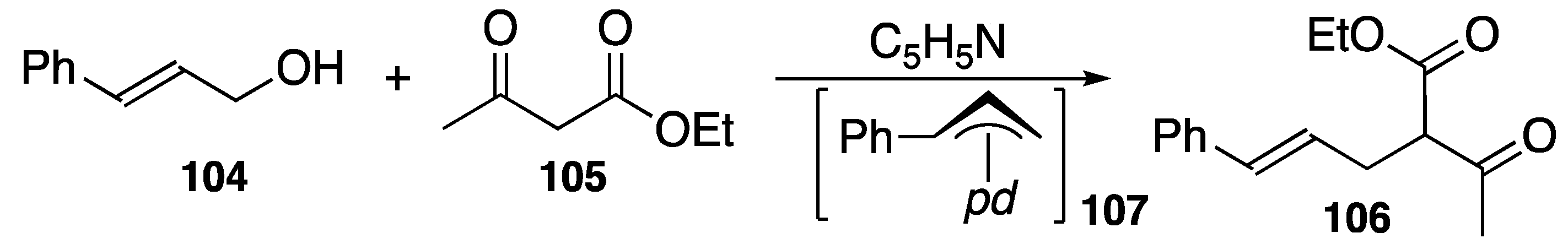

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
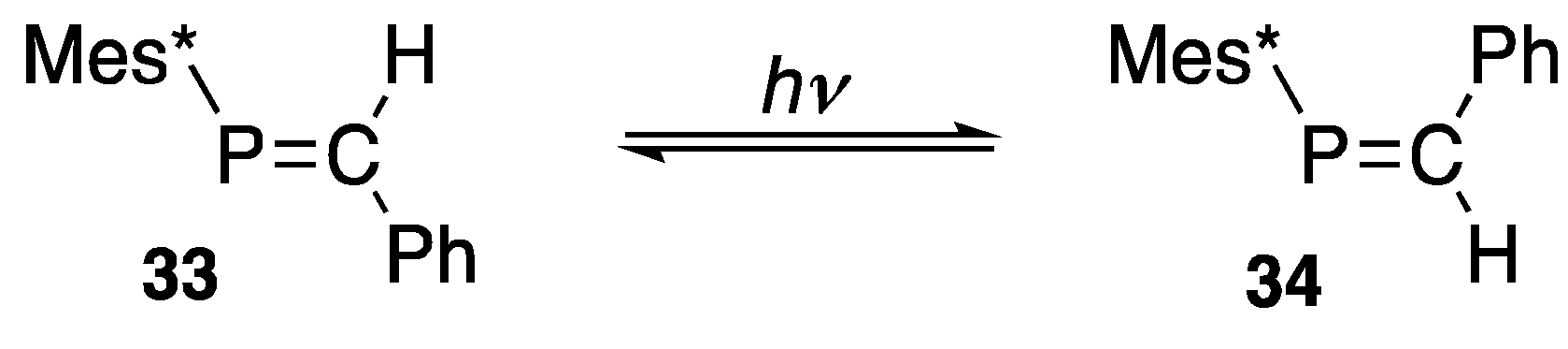{kind=link}
{kind=link}
{kind=link}
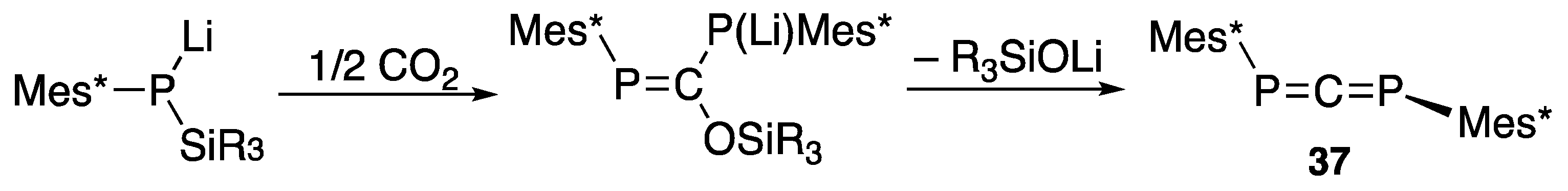{kind=link}
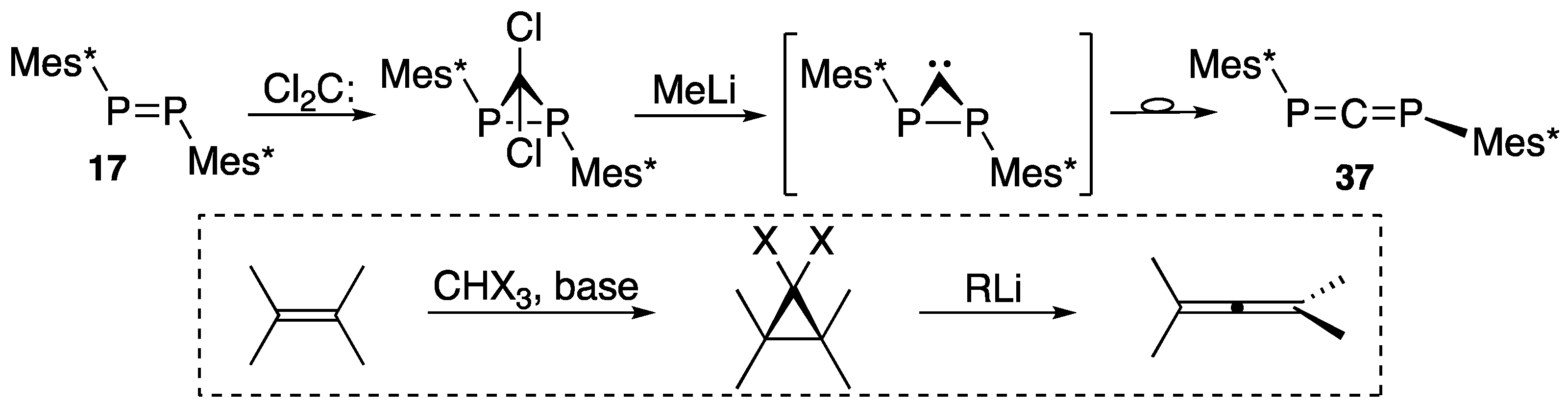{kind=link}
{kind=link}
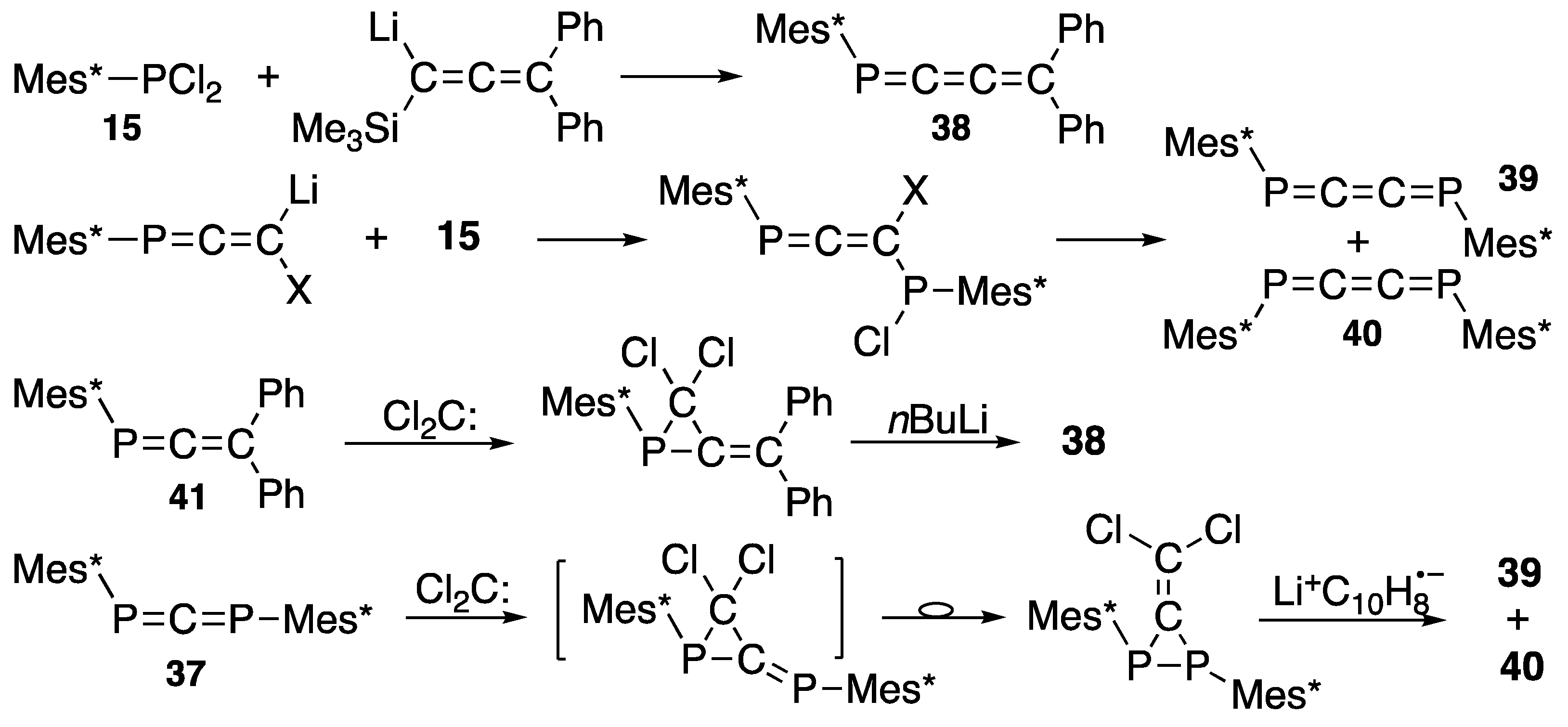{kind=link}
{kind=link}
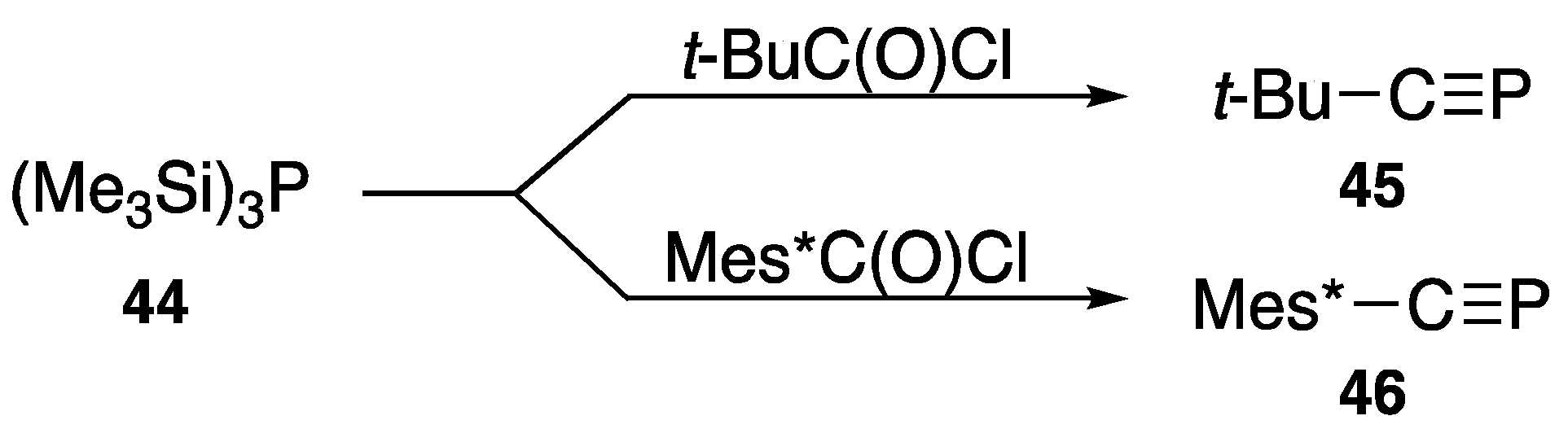{kind=link}
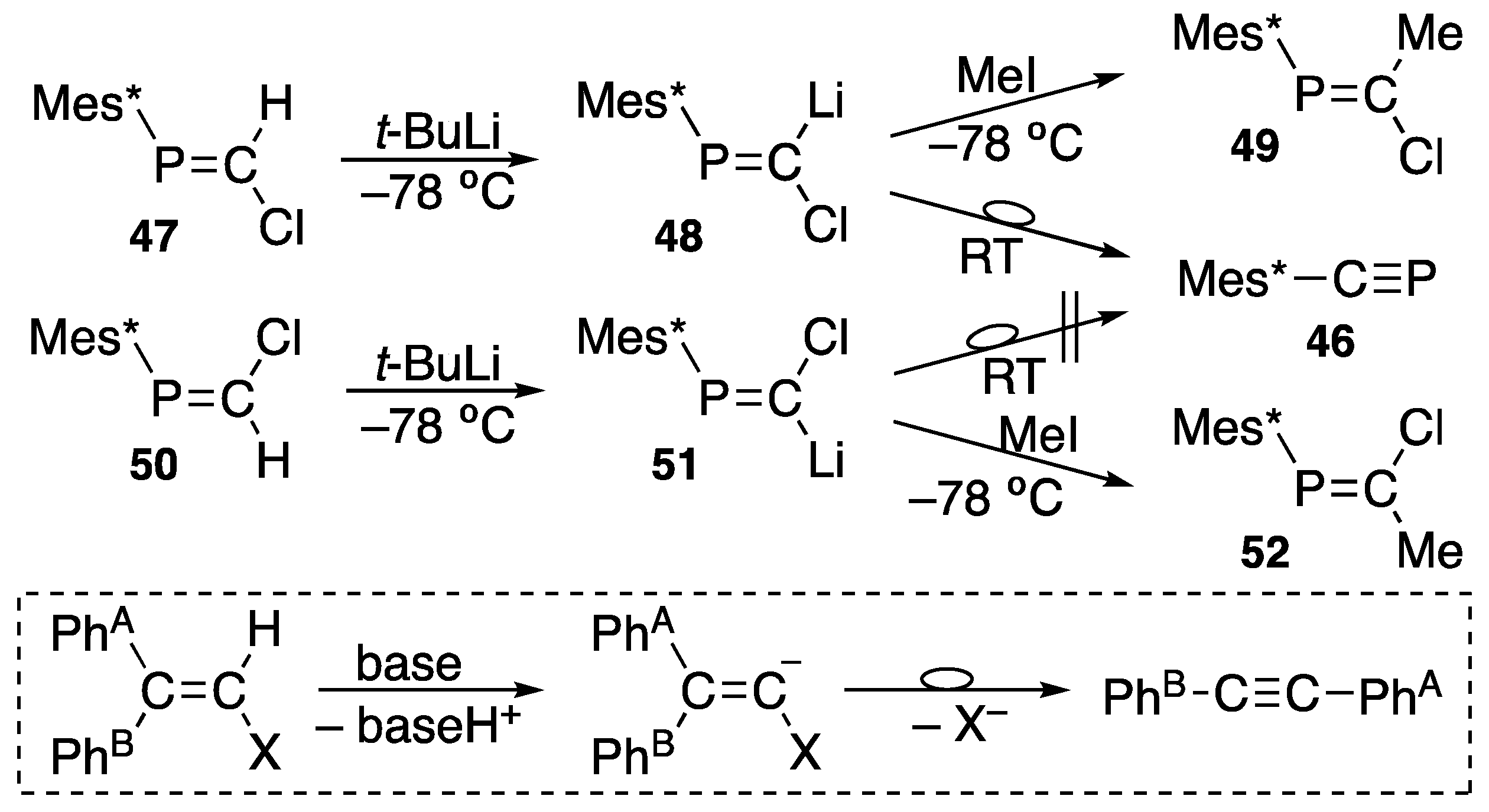{kind=link}
{kind=link}
{kind=link}
{kind=link}
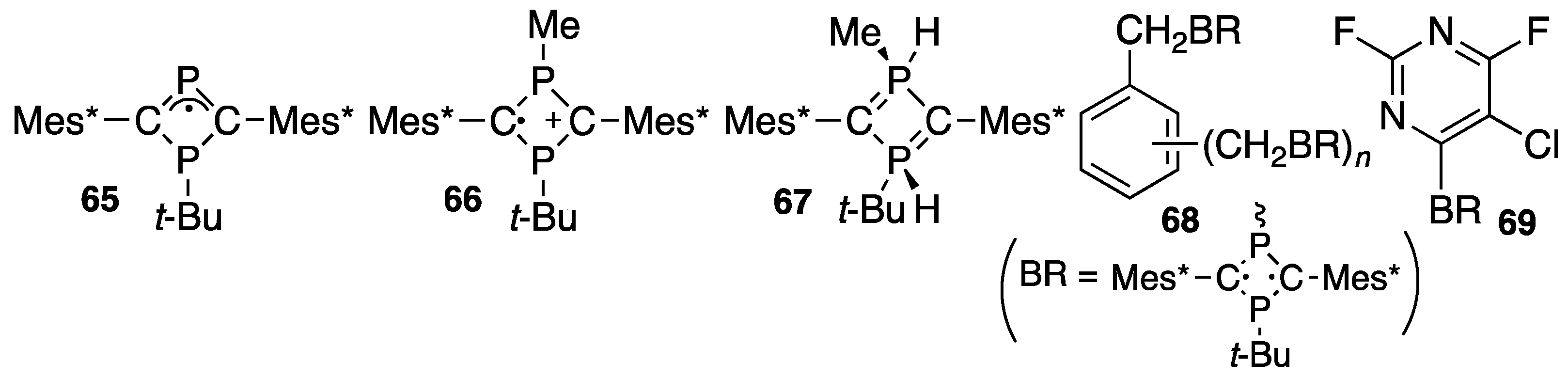{kind=link}
{kind=link}
{kind=link}
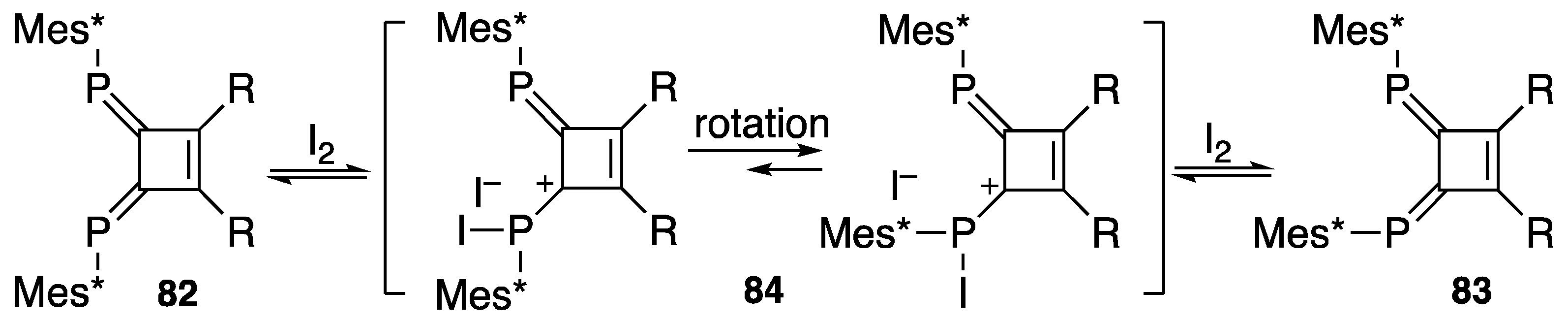{kind=link}
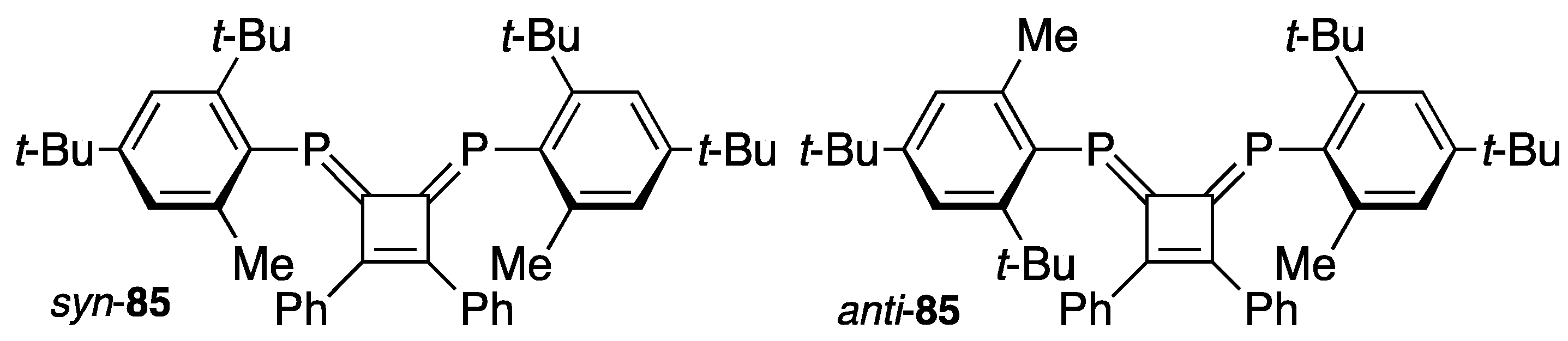{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Detailed Description | Ref. |
|---|---|---|
| MS | Found: 552.4012; Calcd: 552.4012 | [12] |
| Mol. Wt. (C6H6) | 552.1 | [12] |
| UV (CH2Cl2) | λmax (ε) 284 (15,660), 340 (7690), 460 (1360) nm | [12] |
| 1H NMR (CCl4) | δ 7.30 (s, 4H, arom.), 1.45 (s, 36H, o-t-Bu), 1.35 (18H, p-t-Bu) | [12] |
| 31P NMR (C6D6) | δP 492.4 ppm | [13] |
| Raman | ν(P=P) 610 cm–1 | [14] |
| X-ray | P=P 2.034(2), P–C 1.862(2) Å; P–C–C 102.8(1), C–P–P–C 172.2(1)° | [12] |
| Diphosphene | E/Z | Chemical Shift δP | Coupling Constant 1JPP/Hz | Ref. | |
|---|---|---|---|---|---|
| Mes*–P=P–Mes* | (17) | E | 492.4 | ––– | [12,13] |
| Mes*–P=P–2,4-t-Bu2-6-MeC6H2 | (18A) | E | 517.0; 480.1 | 583.5 | [18] |
| Mes*–P=P–Mes | (18B) | E | 540.4; 467.6 | 573.7 | [18] |
| Mes*–P=P–Ph | (18C) | E | 525.5; 455.5 | 548.7 | [18] |
| Mes*–P=P–2,4,6-t-Pn3C6H2 | (18D) | E | 491.2; 489.9 | 582.9 | [20] |
| Mes*–P=P–2,4,6-i-Pr3C6H2 | (18E) | E | 535; 470 | 572 | [21] |
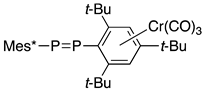 | (18F) | E | 503.2; 475.6 | 590.8 | [22] |
| Mes*–P=P–C(SiMe3)3 | (18G) | E | 533.1; 530.0 | 619.7 | [23] |
| Cp*–P=P–Cp* | (19) | E | 504.0 | ––– | [24] |
| (Me3Si)3C–P=P–C(SiMe3)3 | (20) | E | 599.6 | ––– | [16,25] |
| Mes*–P=P–Mes* | (21) | Z | 368 | ––– | [26,27] |
 | (22) | Z | 394 | ––– | [28] |
| Compound | δP/ppm | Ref. | |
|---|---|---|---|
 | (31) | 178.2 | [43] |
 | (32) | 233.0 | [44] |
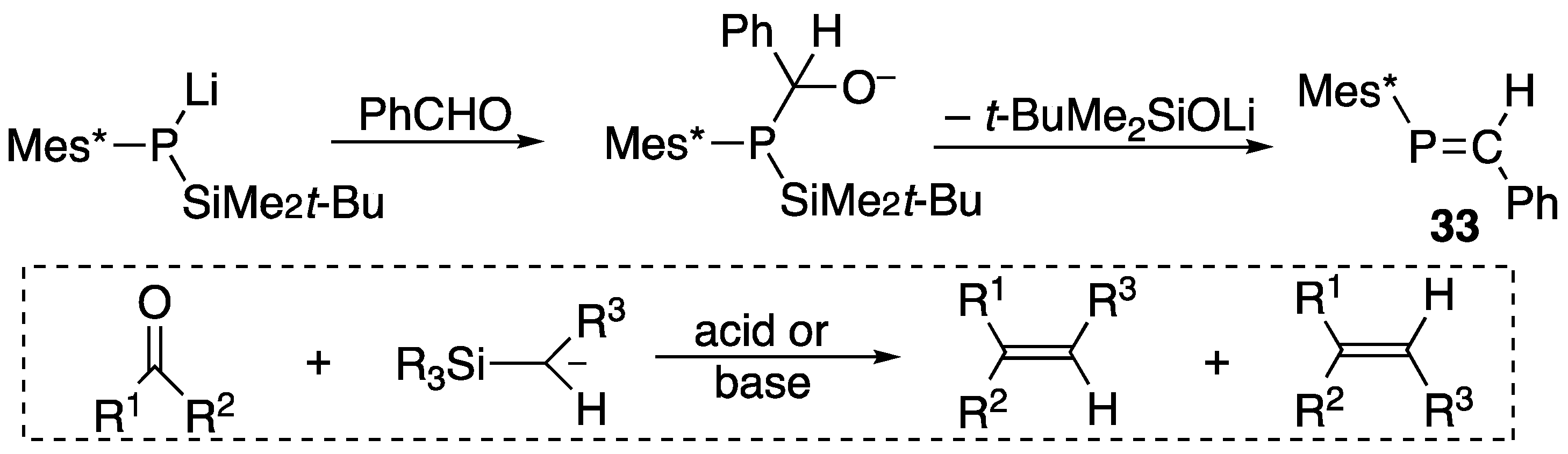
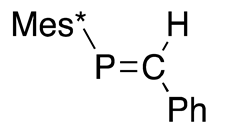 | (33) | 259.3 | [45] |
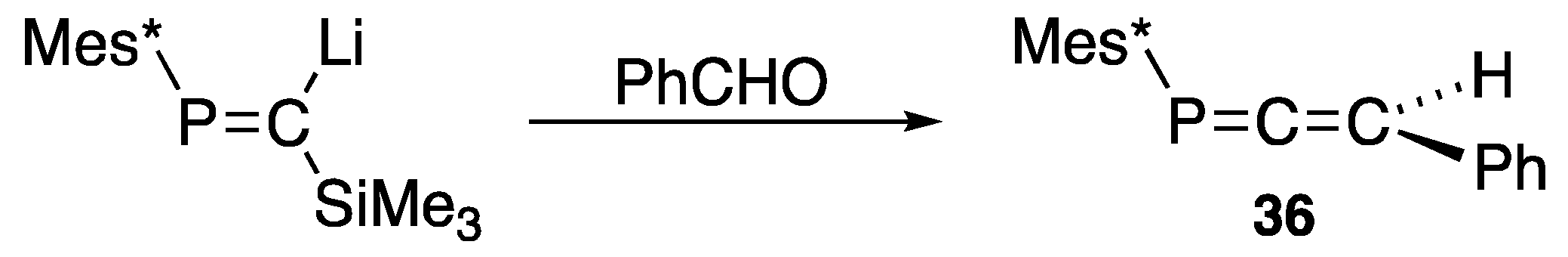
 | (34) | 241.6 | [45] |
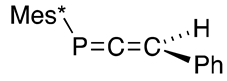 | (36) | 75.2 | [47] |
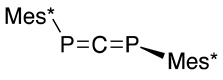 | (37) | 142.4 | [48] |
 | (38) | 156.7 | [49,50] |
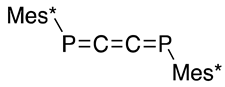 | (39) | 180.6 | [51,52] |
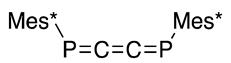 | (40) | 170.0 | [51,52] |
| Mes*–P=C=NPh | (42) | −106.2 | [53] |
| Mes*–P=C=O | (43) | −207.4 | [54] |
| t-Bu–C≡P | (45) | −69.2 | [55] |
| Mes*–C≡P | (46) | 34.4 | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshifuji, M. Chemistry of Several Sterically Bulky Molecules with P=P, P=C, and C≡P Bond. Molecules 2022, 27, 1557. https://doi.org/10.3390/molecules27051557
Yoshifuji M. Chemistry of Several Sterically Bulky Molecules with P=P, P=C, and C≡P Bond. Molecules. 2022; 27(5):1557. https://doi.org/10.3390/molecules27051557
Chicago/Turabian StyleYoshifuji, Masaaki. 2022. "Chemistry of Several Sterically Bulky Molecules with P=P, P=C, and C≡P Bond" Molecules 27, no. 5: 1557. https://doi.org/10.3390/molecules27051557
APA StyleYoshifuji, M. (2022). Chemistry of Several Sterically Bulky Molecules with P=P, P=C, and C≡P Bond. Molecules, 27(5), 1557. https://doi.org/10.3390/molecules27051557