Drug Design—Past, Present, Future


{kind=link}
{kind=link}
Abstract
1. Introduction
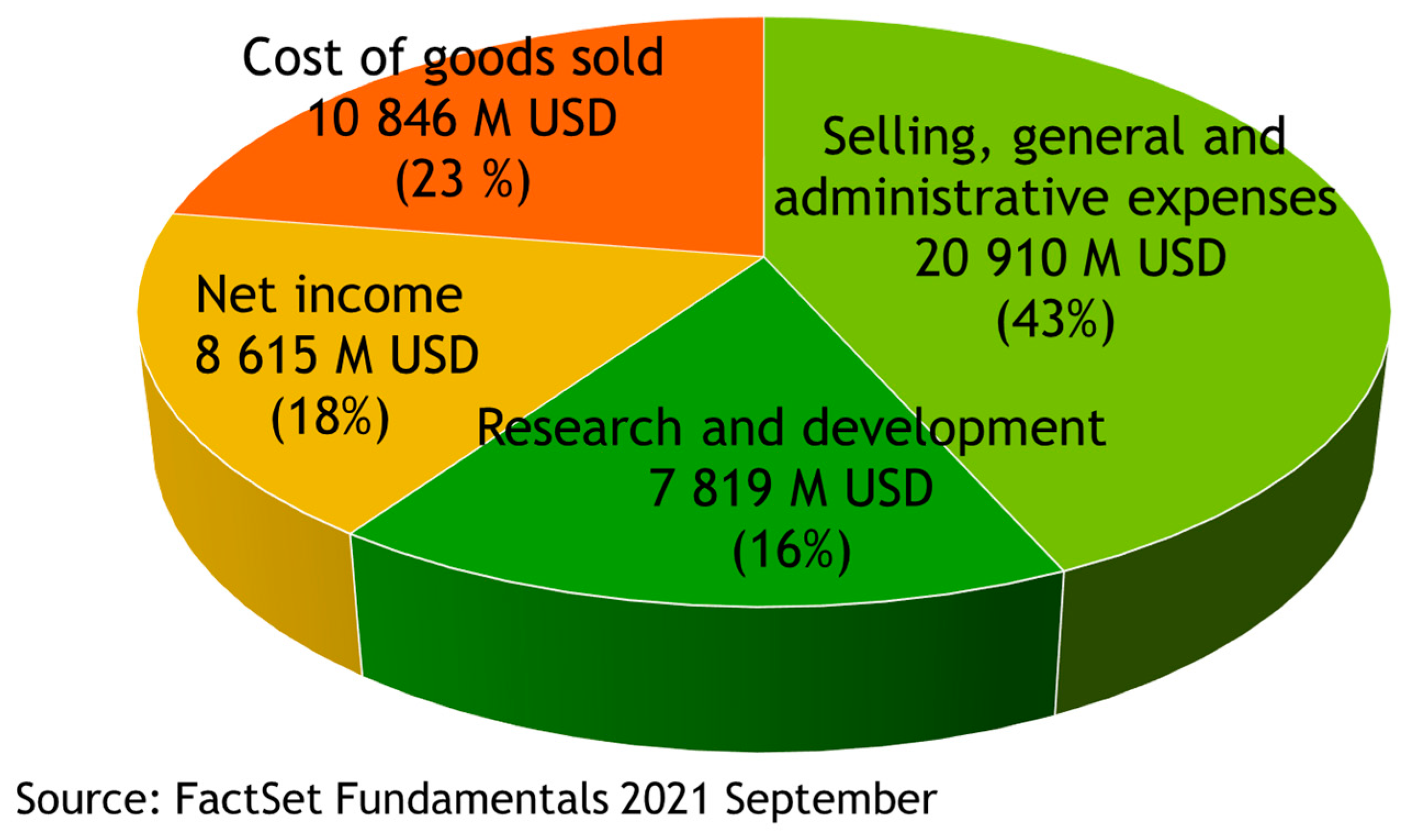
2. Drugs Are High Value-Added Products
3. Drug Discovery
4. Drug Design—Historical Notes
5. Current Methods for Drug Design
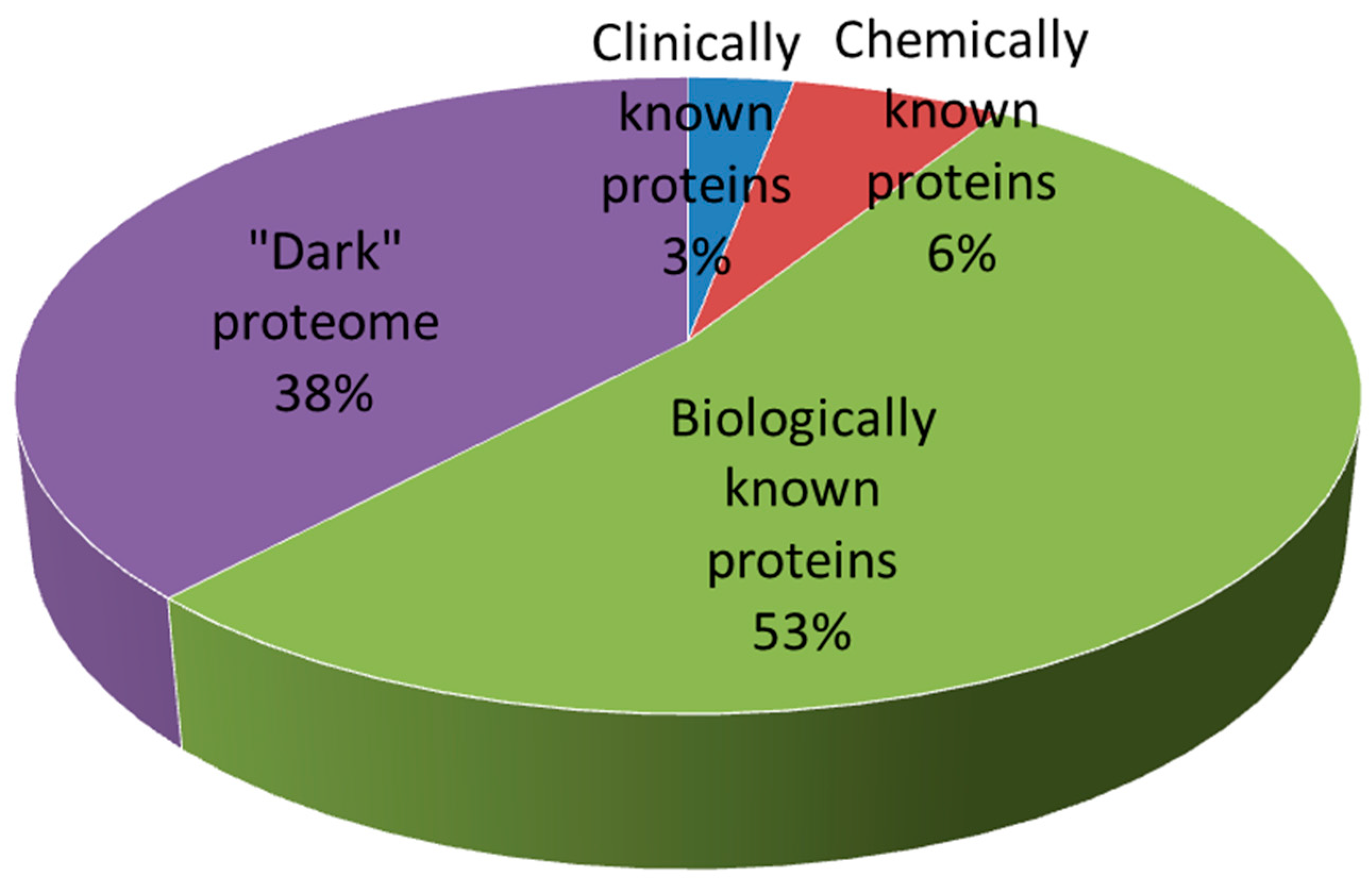
6. Future Trends in Drug Design
Acknowledgments
Conflicts of Interest
References
- Drugs@FDA Glossary of Terms; US Food & Drug Administration: Silver Spring, MD, USA, 2022.
- Molecular Conceptor Learning Series; Synergix Ltd.: Singapore, 2012.
- Young, D.C. (Ed.) Properties that make a molecule a good drug. In Computational Drug Design; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 9–39. [Google Scholar]
- Rowland, M.; Tozer, T.N. (Eds.) Fundamental concepts and terminology. In Clinical Pharmacokinetics and Pharmacodynamics, 4th ed.; Lippincott Williams & Wilkins: Baltimore, MD, USA, 2011; pp. 17–45. [Google Scholar]
- Sinha, S.; Vohora, D. (Eds.) Drug discovery and development: An overview. In Pharmaceutical Medicine and Translational Clinical Research; Elsevier Inc.: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- FactSet Fundamentals; FactSet Research Systems Inc.: Connecticut, CT, USA, 2021.
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [PubMed]
- McGrail, S. Key differences in small molecule, biologics drug development. Pharma News Intelligence. 20 August 2021. Available online: https://pharmanewsintel.com/news/key-differences-in-small-molecule-biologics-drug-development (accessed on 20 February 2022).
- Doytchinova, I.; Atanasova, M.; Valkova, I.; Stavrakov, G.; Philipova, I.; Zhivkova, Z.; Zheleva-Dimitrova, D.; Konstantinov, S.; Dimitrov, I. Novel hits for acetylcholinesterase inhibition derived by docking-based screening on ZINC database. J. Enzym. Inhib. Med. Chem. 2018, 33, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef]
- Fogel, D.B. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: A review. Contemp. Clin. Trials Commun. 2018, 11, 156–164. [Google Scholar] [CrossRef]
- Prentis, R.A.; Lis, Y.; Walker, S.R. Pharmaceutical innovation by the seven UK-owned pharmaceutical companies (1964–1985). Br. J. Clin. Pharmacol. 1988, 25, 387–396. [Google Scholar] [CrossRef]
- Ban, T.A. The role of serendipity in drug discovery. Dialogues Clin. Neurosci. 2006, 8, 335–344. [Google Scholar]
- Cheng, M. Hartmann Stahelin (1925–2011) and the contested history of cyclosporin A. Clin. Transplant. 2013, 27, 326–329. [Google Scholar] [CrossRef]
- Guo, Z. The modification of natural products for medical use. Acta Pharm. Sin. B 2017, 7, 119–136. [Google Scholar] [CrossRef]
- Montinari, M.R.; Minelli, S.; De Caterina, R. The first 3500 years of aspirin history from its roots—A concise summary. Vascul. Pharmacol. 2019, 113, 1–8. [Google Scholar] [CrossRef]
- Roberts, C.J. Clinical pharmacokinetics of ranitidine. Clin. Pharmacokinet. 1984, 9, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Meier, J. Pharmacokinetic comparison of pindolol with other beta-adrenoceptor-blocking agents. Am. Heart J. 1982, 104, 364–373. [Google Scholar] [CrossRef]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Butkiewicz, M.; Wang, Y.; Bryant, S.H.; Lowe, E.W., Jr.; Weaver, D.C.; Meiler, J. High-throughput screening assay datasets from the PubChem database. Chem. Inform. 2017, 3, 1. [Google Scholar] [CrossRef]
- Smith, A. Screening for drug discovery: The leading question. Nature 2002, 418, 453–455. [Google Scholar] [CrossRef]
- Bentley, R. Different roads to discovery; Prontosil (hence sulfa drugs) and penicillin (hence beta-lactams). J. Ind. Microbiol. Biotechnol. 2009, 36, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.E.; Wani, M.C. Camptothecin and taxol: Discovery to clinic—Thirteenth Bruce F. Cain Memorial Award Lecture. Cancer Res. 1995, 55, 753–760. [Google Scholar] [PubMed]
- Flower, D.R. (Ed.) Molecular informatics: Sharpening drug desisign’s cutting edge. In Drug Design: Cutting Edge Approaches; RSC: Cambridge, UK, 2002; pp. 1–52. [Google Scholar]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef]
- Oprea, T.I.; Bologa, C.G.; Brunak, S.; Campbell, A.; Gan, G.N.; Gaulton, A.; Gomez, S.M.; Guha, R.; Hersey, A.; Holmes, J.; et al. Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov. 2018, 17, 317–332. [Google Scholar] [CrossRef]
- Sheils, T.K.; Mathias, S.L.; Kelleher, K.J.; Siramshetty, V.B.; Nguyen, D.T.; Bologa, C.G.; Jensen, L.J.; Vidović, D.; Koleti, A.; Schürer, S.C.; et al. TCRD and Pharos 2021: Mining the human proteome for disease biology. Nucleic Acids Res. 2021, 49, D1334–D1346. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The worldwide Protein Data Bank (wwPDB): Ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007, 35, D301–D303. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Thakkar, A.; Mercado, R.; Engkvist, O. Molecular representations in AI-driven drug discovery: A review and practical guide. J. Cheminform. 2020, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Carracedo-Reboredo, P.; Liñares-Blanco, J.; Rodríguez-Fernández, N.; Cedrón, F.; Novoa, F.J.; Carballal, A.; Maojo, V.; Pazos, A.; Fernandez-Lozano, C. A review on machine learning approaches and trends in drug discovery. Comput. Struct. Biotechnol. J. 2021, 19, 4538–4558. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Steward, A.R. The use of substitutent constants in the analysis of the structure-activity relationship in penicillin derivatives. J. Med. Chem. 1964, 7, 691–694. [Google Scholar] [CrossRef]
- Martin, Y.; Stouch, T. In tribute to Corwin Hansch, father of QSAR. J. Comput. Aided Mol. Des. 2011, 25, 491. [Google Scholar] [CrossRef][Green Version]
- Wold, S.; Dunn III, W.J. Multivariate quantitative structure-activity relationships (QSAR): Conditions for their applicability. J. Chem. Inf. Comput. Sci. 1983, 23, 6–13. [Google Scholar] [CrossRef]
- Wold, S.; Gelada, P.; Esbensen, K.; Öhman, J. Multiway principal components and PLS-analysis. J. Chemometr. 1987, 1, 41–56. [Google Scholar] [CrossRef]
- Xu, J.; Hagler, A. Chemoinformatics and drug discovery. Molecules 2002, 7, 566–600. [Google Scholar] [CrossRef]
- Mitchell, J.B. Machine learning methods in chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 468–481. [Google Scholar] [CrossRef]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Hessler, G.; Baringhaus, K.-H. Artificial Intelligence in Drug Design. Molecules 2018, 23, 2520. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A., Jr.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 2020, 19, 353–364. [Google Scholar] [CrossRef]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2021, 26, 80–93. [Google Scholar] [CrossRef]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Bragina, M.E.; Daina, A.; Perez, M.A.S.; Michielin, O.; Zoete, V. SwissSimilarity 2021 web tool: Novel chemical libraries and additional methods for an enhanced ligand-based virtual screening experience. Int. J. Mol. Sci. 2022, 23, 811. [Google Scholar] [CrossRef]
- Cuizzo, A.; Daina, A.; Perez, M.A.S.; Michielin, O.; Zoete, V. SwissBioisostere 2021: Updated structural, bioactivity and physicochemical data delivered by a reshaped web interface. Nucleic Acids Res. 2022, 50, D1382–D1390. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Klucznik, T.; Mikulak-Klucznik, B.; McCormack, M.P.; Lima, H.; Szymkuć, S.; Bhowmick, M.; Molga, K.; Zhou, Y.; Rickershauser, L.; Gajewska, E.P.; et al. Efficient syntheses of diverse, medicinally relevant targets planned by computer and executed in the laboratory. Chem 2018, 4, 522–532. [Google Scholar] [CrossRef]
- MacKinnon, S.S.; Madani Tonekaboni, S.A.; Windemuth, A. Proteome-scale drug-target interaction predictions: Approaches and applications. Curr. Protoc. 2021, 1, e302. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Xing, J.; Li, X.; Liu, X.; Fu, Z.; Xiong, Z.; Lu, D.; Wu, X.; Zhao, J.; Tan, X.; et al. Artificial intelligence in drug design. Sci. China Life Sci. 2018, 61, 1191–1204. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Grisoni, F.; Weskamp, N.; Schneider, G. Artificial intelligence in drug discovery: Recent advances and future perspectives. Expert Opin. Drug Discov. 2021, 16, 949–959. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Di Filippo, J.I. Artificial intelligence in the early stages of drug discovery. Arch. Biochem. Biophys. 2021, 698, 108730. [Google Scholar] [CrossRef]
- Chan, H.; Shan, H.; Dahoun, T.; Vogel, H.; Yuan, S. Advancing drug discovery via artificial intelligence. Trends Pharmacol. Sci. 2019, 40, 592–604. [Google Scholar] [CrossRef]
- Zimmer, M. Bioorganic molecular mechanics. Chem. Rev. 1995, 95, 2629–2649. [Google Scholar] [CrossRef]
- Young, D.C. (Ed.) Molecular Mechanics. In Computational Drug Design; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 119–123. [Google Scholar]
- Ganesan, A.; Coote, M.L.; Barakat, K. Molecular dynamics-driven drug discovery: Leaping forward with confidence. Drug Discov. Today 2017, 22, 249–269. [Google Scholar] [CrossRef]
- Chen, Z.; Li, H.L.; Zhang, Q.J.; Bao, X.G.; Yu, K.Q.; Luo, X.M.; Zhu, W.L.; Jiang, H.L. Pharmacophore-based virtual screening versus docking-based virtual screening: A benchmark comparison against eight targets. Acta Pharmacol. Sin. 2009, 30, 1694–1708. [Google Scholar] [CrossRef]
- Muthas, D.; Sabnis, Y.A.; Lundborg, M.; Karlén, A. Is it possible to increase hit rates in structure-based virtual screening by pharmacophore filtering? An investigation of the advantages and pitfalls of post-filtering. J. Mol. Graph. Model. 2008, 26, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- King, R.D.; Whelan, K.E.; Jones, F.M.; Reiser, P.G.; Bryant, C.H.; Muggleton, S.H.; Kell, D.B.; Oliver, S.G. Functional genomic hypothesis generation and experimentation by a robot scientist. Nature 2004, 427, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Bilsland, E.; Sparkes, A.; Aubrey, W.; Young, M.; Soldatova, L.N.; De Grave, K.; Ramon, J.; de Clare, M.; Sirawaraporn, W.; et al. Cheaper faster drug development validated by the repositioning of drugs against neglected tropical diseases. J. R. Soc. Interface 2015, 12, 20141289. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doytchinova, I. Drug Design—Past, Present, Future. Molecules 2022, 27, 1496. https://doi.org/10.3390/molecules27051496
Doytchinova I. Drug Design—Past, Present, Future. Molecules. 2022; 27(5):1496. https://doi.org/10.3390/molecules27051496
Chicago/Turabian StyleDoytchinova, Irini. 2022. "Drug Design—Past, Present, Future" Molecules 27, no. 5: 1496. https://doi.org/10.3390/molecules27051496
APA StyleDoytchinova, I. (2022). Drug Design—Past, Present, Future. Molecules, 27(5), 1496. https://doi.org/10.3390/molecules27051496