
Two-Photon Absorption Cooperative Effects within Multi-Dipolar Ruthenium Complexes: The Decisive Influence of Charge Transfers



 and
and
Abstract
:1. Introduction
2. Results
2.1. Computational Results
2.1.1. Geometry Optimization
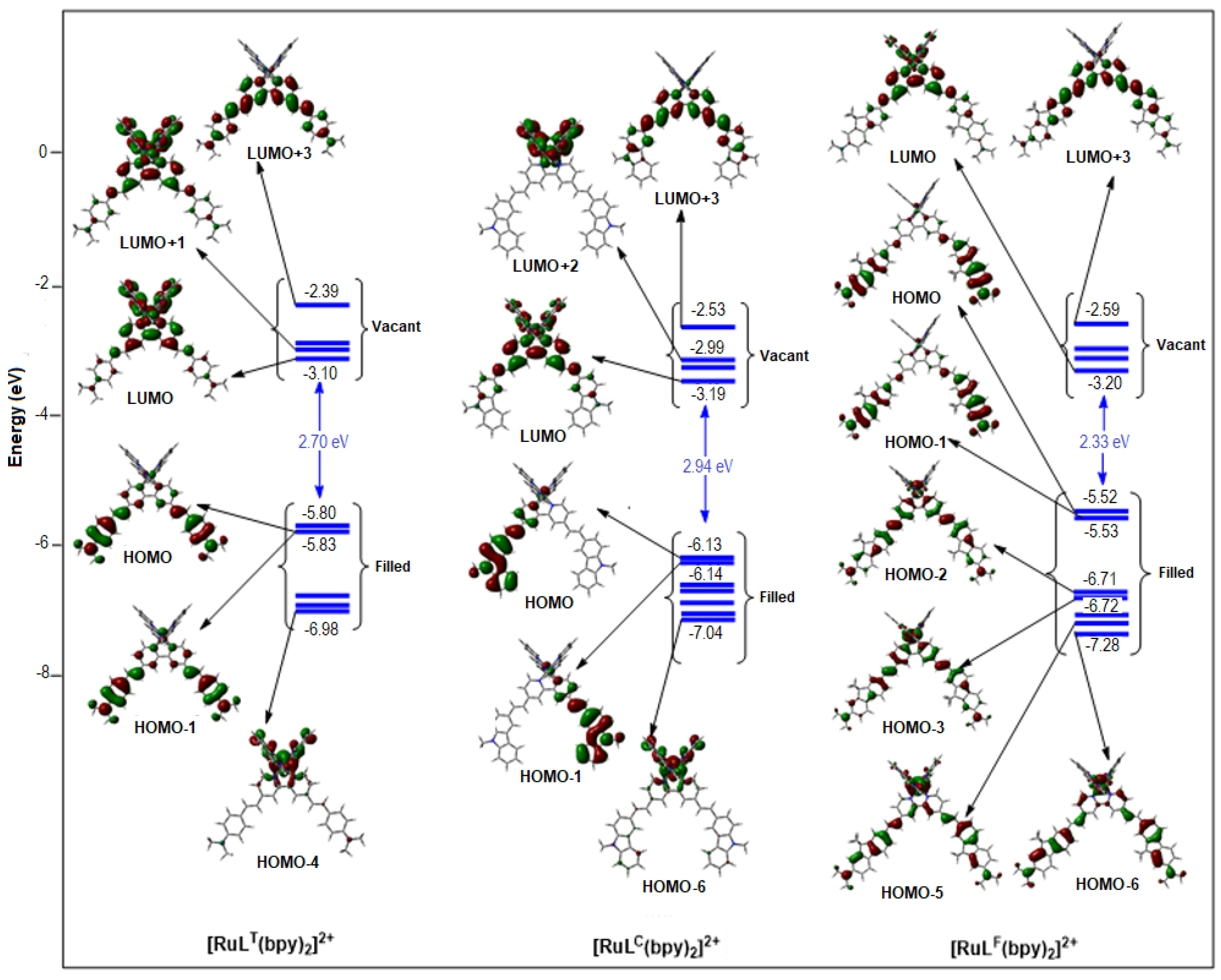
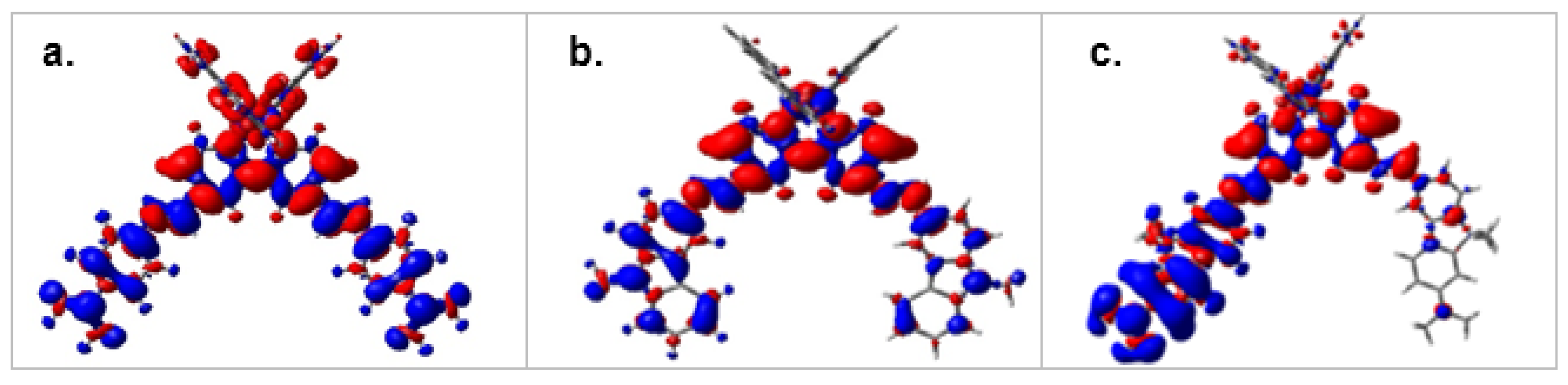
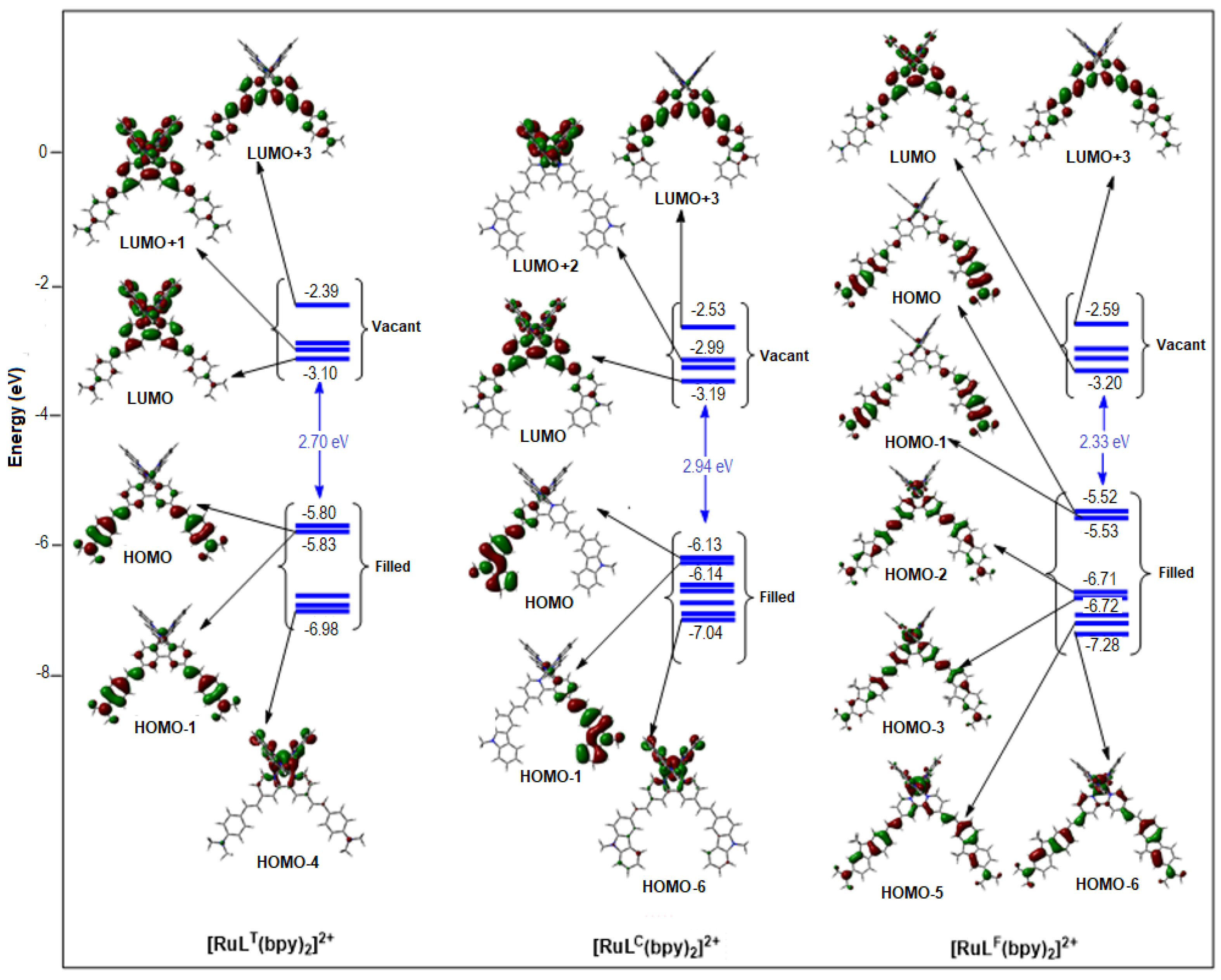
2.1.2. Molecular Orbitals
2.1.3. TD-DFT Calculations of the Electronic Absorption Spectra
3. Discussion
4. Materials and Methods
4.1. Synthesis and Photochemical Measurements
4.2. Theoretical Computations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
References
- Belfield, K.D.; Yao, S.; Bondar, M.V. Two-Photon Absorbing Photonic Materials: From Fundamentals to Applications; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2008; Volume 2008, pp. 97–156. [Google Scholar]
- Rumi, M.; Barlow, S.; Wang, J.; Perry, J.W.; Marder, S.R. Two-Photon Absorbing Materials and Two-Photon-Induced Chemistry; Springer International Publishing: Berlin/Heidelberg, Germany, 2008; Volume 2008, pp. 1–95. [Google Scholar]
- Pawlicki, M.; Collins, H.A.; Denning, R.G.; Anderson, H.L. Two-Photon Absorption and the Design of Two-Photon Dyes. Angew. Chem. Int. Ed. 2009, 48, 3244–3266. [Google Scholar] [CrossRef]
- Lee, W.-H.; Lee, H.; Kim, J.-A.; Choi, J.-H.; Cho, M.; Jeon, S.-J.; Cho, B.R. Two-Photon Absorption and Nonlinear Optical Properties of Octupolar Molecules. J. Am. Chem. Soc. 2001, 123, 10658–10667. [Google Scholar] [CrossRef]
- Kim, H.M.; Cho, B.R. Two-photon materials with large two-photon cross sections. Structure–property relationship. Chem. Commun. 2009, 2, 153–164. [Google Scholar] [CrossRef]
- Maruo, S.; Nakamura, O.; Kawata, S. Three-dimensional microfabrication with two-photon-absorbed photopolymerization. Opt. Lett. 1997, 22, 132–134. [Google Scholar] [CrossRef] [Green Version]
- Selimis, A.; Mironov, V.; Farsari, M. Direct laser writing: Principles and materials for scaffold 3D printing. Microelectron. Eng. 2015, 132, 83–89. [Google Scholar] [CrossRef]
- Tumbleston, J.R.; Shirvanyants, D.; Ermoshkin, N.; Janusziewicz, R.; Johnson, A.R.; Kelly, D.; Chen, K.; Pinschmidt, R.; Rolland, J.P.; Ermoshkin, A.; et al. Continuous liquid interface production of 3D objects. Science 2015, 347, 1349–1352. [Google Scholar] [CrossRef]
- Schwarz, C.M.; Grabill, C.N.; Digaum, J.L.; Williams, H.E.; Kuebler, S.M. Multiphoton Processing of Composite Materials and Functionalization of 3D Structures. In Multiphoton Lithography; Wiley-VCH Verlag GmbH & Co.: Hoboken, NJ, USA, 2016; pp. 221–264. [Google Scholar]
- Barner-Kowollik, C.; Bastmeyer, M.; Blasco, E.; Delaittre, G.; Müller, P.; Richter, B.; Wegener, M. 3D Laser Micro- and Nanoprinting: Challenges for Chemistry. Angew. Chem. Int. Ed. 2017, 56, 15828–15845. [Google Scholar] [CrossRef]
- Chang, J.; He, J.; Mao, M.; Zhou, W.; Lei, Q.; Li, X.; Li, D.; Chua, C.K.; Zhao, X. Advanced Material Strategies for Next-Generation Additive Manufacturing. Materials 2018, 11, 166. [Google Scholar] [CrossRef] [Green Version]
- Kawata, S.; Sun, H.; Tanaka, T.; Takada, K. Finer features for functional microdevices. Nature 2001, 412, 697–698. [Google Scholar] [CrossRef]
- Parthenopoulos, D.A.; Rentzepis, P.M. Three-Dimensional Optical Storage Memory. Science 1989, 245, 843–845. [Google Scholar] [CrossRef]
- Corredor, C.C.; Huang, Z.-L.; Belfield, K.D. Two-Photon 3D Optical Data Storage via Fluorescence Modulation of an Efficient Fluorene Dye by a Photochromic Diarylethene. Adv. Mater. 2006, 18, 2910–2914. [Google Scholar] [CrossRef]
- Dvornikov, A.S.; Walker, E.P.; Rentzepis, P.M. Two-Photon Three-Dimensional Optical Storage Memory. J. Phys. Chem. A 2009, 113, 13633–13644. [Google Scholar] [CrossRef]
- Lott, J.; Ryan, C.; Valle, B.; Johnson, J.R.; Schiraldi, D.A.; Shan, J.; Singer, K.D.; Weder, C. Two-Photon 3D Optical Data Storage via Aggregate Switching of Excimer-Forming Dyes. Adv. Mater. 2011, 23, 2425–2429. [Google Scholar] [CrossRef] [Green Version]
- Kallepalli, D.L.N.; AlShehri, A.M.; Marquez, D.T.; Andrzejewski, L.; Scaiano, J.C.; Bhardwaj, R. Ultra-high density optical data storage in common transparent plastics. Sci. Rep. 2016, 6, 26163. [Google Scholar] [CrossRef]
- Imhof, M.; Rhinow, D.; Hampp, N. Two-photon polarization data storage in bacteriorhodopsin films and its potential use in security applications. Appl. Phys. Lett. 2014, 104, 81921. [Google Scholar] [CrossRef]
- Kim, D.; Ryu, H.G.; Ahn, K.H. Recent development of two-photon fluorescent probes for bioimaging. Org. Biomol. Chem. 2014, 12, 4550–4566. [Google Scholar] [CrossRef] [Green Version]
- Bhawalkar, J.; Kumar, N.; Zhao, C.-F.; Prasad, P. Two-Photon Photodynamic Therapy. J. Clin. Laser Med. Surg. 1997, 15, 201–204. [Google Scholar] [CrossRef]
- Shen, Y.; Shuhendler, A.J.; Ye, D.; Xu, J.-J.; Chen, H.-Y. Two-photon excitation nanoparticles for photodynamic therapy. Chem. Soc. Rev. 2016, 45, 6725–6741. [Google Scholar] [CrossRef]
- Bolze, F.; Jenni, S.; Sour, A.; Heitz, V. Molecular photosensitisers for two-photon photodynamic therapy. Chem. Commun. 2017, 53, 12857–12877. [Google Scholar] [CrossRef]
- Gu, B.; Wu, W.; Xu, G.; Feng, G.; Yin, F.; Chong, P.H.J.; Qu, J.; Yong, K.-T.; Liu, B. Precise Two-Photon Photodynamic Therapy using an Efficient Photosensitizer with Aggregation-Induced Emission Characteristics. Adv. Mater. 2017, 29, 1701076. [Google Scholar] [CrossRef]
- Rumi, M.; Ehrlich, J.E.; Heikal, A.A.; Perry, J.W.; Barlow, S.; Hu, Z.; McCord-Maughon, D.; Parker, T.C.; Röckel, H.; Thayumanavan, S.; et al. Structure−Property Relationships for Two-Photon Absorbing Chromophores: Bis-Donor Diphenylpolyene and Bis(styryl)benzene Derivatives. J. Am. Chem. Soc. 2000, 122, 9500–9510. [Google Scholar] [CrossRef] [Green Version]
- Beljonne, D.; Wenseleers, W.; Zojer, E.; Shuai, Z.; Vogel, H.; Pond, S.J.; Brédas, J.L. Role of dimensionality on the two-photon absorption response of conjugated molecules: The case of octupolar compounds. Adv. Funct. Mater. 2002, 12, 631–641. [Google Scholar] [CrossRef]
- Katan, C.; Tretiak, S.; Werts, M.; Bain, A.J.; Marsh, R.; Leonczek, N.; Nicolaou, N.; Badaeva, E.; Mongin, A.O.; Blanchard-Desce, M. Two-Photon Transitions in Quadrupolar and Branched Chromophores: Experiment and Theory. J. Phys. Chem. B 2007, 111, 9468–9483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Chen, T.; Liu, B.; Huang, Z.-L.; Huang, T.; Li, S.; Xu, Y.; Qin, J. Two-photon absorption of a series of V-shape molecules: The influence of acceptor’s strength on two-photon absorption in a noncentrosymmetric D–π–A–π–D system. J. Mater. Chem. 2007, 17, 4685–4689. [Google Scholar] [CrossRef]
- Mongin, O.; Porrès, L.; Charlot, M.; Katan, C.; Blanchard-Desce, M. Synthesis, Fluorescence, and Two-Photon Absorption of a Series of Elongated Rodlike and Banana-Shaped Quadrupolar Fluorophores: A Comprehensive Study of Structure–Property Relationships. Chem. A Eur. J. 2007, 13, 1481–1498. [Google Scholar] [CrossRef]
- Andraud, C.; Fortrie, R.; Barsu, C.; Stéphan, O.; Chermette, H.; Baldeck, P.L. Excitonically coupled oligomers and dendrimers for two-photon absorption. In Photoresponsive Polymers II; Springer: Berlin/Heidelberg, Germany, 2008; pp. 149–203. [Google Scholar]
- Terenziani, F.; Katan, C.; Badaeva, E.; Tretiak, S.; Blanchard-Desce, M. Enhanced Two-Photon Absorption of Organic Chromophores: Theoretical and Experimental Assessments. Adv. Mater. 2008, 20, 4641–4678. [Google Scholar] [CrossRef] [Green Version]
- Albota, M.; Beljonne, D.; Brédas, J.-L.; Ehrlich, J.E.; Fu, J.-Y.; Heikal, A.A.; Hess, S.E.; Kogej, T.; Levin, M.D.; Marder, S.R.; et al. Design of Organic Molecules with Large Two-Photon Absorption Cross Sections. Science 1998, 281, 1653–1656. [Google Scholar] [CrossRef] [Green Version]
- Le Droumaguet, C.; Mongin, O.; Werts, M.H.V.; Blanchard-Desce, M. Towards “smart” multiphoton fluorophores: Strongly solvatochromic probes for two-photon sensing of micropolarity. Chem. Commun. 2005, 22, 2802–2804. [Google Scholar] [CrossRef]
- Katan, C.; Terenziani, F.; Mongin, O.; Werts, M.H.V.; Porrès, L.; Pons, T.; Mertz, J.; Tretiak, S.; Blanchard-Desce, M. Effects of (Multi)branching of Dipolar Chromophores on Photophysical Properties and Two-Photon Absorption. J. Phys. Chem. A 2005, 109, 3024–3037. [Google Scholar] [CrossRef] [Green Version]
- Grisanti, L.; Sissa, C.; Terenziani, F.; Painelli, A.; Roberto, D.; Tessore, F.; Ugo, R.; Quici, S.; Fortunati, I.; Garbin, E.; et al. Enhancing the efficiency of two-photon absorption by metal coordination. Phys. Chem. Chem. Phys. 2009, 11, 9450–9457. [Google Scholar] [CrossRef]
- Collini, E. Cooperative effects to enhance two-photon absorption efficiency: Intra- versus inter-molecular approach. Phys. Chem. Chem. Phys. 2012, 14, 3725–3736. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.D.; Vivas, M.G.; Silva, D.L.; Eucat, G.; Bretonnière, Y.; Andraud, C.; Mendonca, C.R.; De Boni, L. Intramolecular Cooperative and Anti-Cooperative Effect on the Two-Photon Absorption Cross Section in Triphenylamine Derivatives. J. Phys. Chem. Lett. 2019, 10, 2214–2219. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, S. Second-order nonlinear optical properties of transition metal complexes. Chem. Soc. Rev. 2001, 30, 355–366. [Google Scholar] [CrossRef]
- De Silva, I.C.; de Silva, R.M.; de Silva, K.N. Investigations of nonlinear optical (NLO) properties of Fe, Ru and Os organometallic complexes using high accuracy density functional theory (DFT) calculations. J. Mol. Struct. THEOCHEM 2005, 728, 141–145. [Google Scholar] [CrossRef]
- Di Bella, S.; Dragonetti, C.; Pizzotti, M.; Roberto, D.; Tessore, F.; Ugo, R. Coordination and Organometallic Complexes as Second-Order Nonlinear Optical Molecular Materials. In Molecular Organometallic Material for Optics; Bozec, H., Guerchais, V., Eds.; Springer: Heidelberg, Germany, 2010; pp. 1–55. [Google Scholar] [CrossRef]
- Coe, B.J. Developing iron and ruthenium complexes for potential nonlinear optical applications. Co-ord. Chem. Rev. 2013, 257, 1438–1458. [Google Scholar] [CrossRef]
- Coe, B.J.; Samoc, M.; Samoc, A.; Zhu, L.; Yi, Y.; Shuai, Z. Two-Photon Absorption Properties of Iron(II) and Ruthenium(II) Trischelate Complexes of 2,2′:4,4″:4′,4‴-Quaterpyridinium Ligands. J. Phys. Chem. A 2007, 111, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Coe, B.J.; Fielden, J.; Foxon, S.P.; Brunschwig, B.S.; Asselberghs, I.; Clays, K.; Samoc, A.; Samoc, M. Combining Very Large Quadratic and Cubic Nonlinear Optical Responses in Extended, Tris-Chelate Metallochromophores with Six π-Conjugated Pyridinium Substituents. J. Am. Chem. Soc. 2010, 132, 3496–3513. [Google Scholar] [CrossRef] [Green Version]
- Maury, O.; Le Bozec, H. Molecular Engineering of Octupolar NLO Molecules and Materials Based on Bipyridyl Metal Complexes. Accounts Chem. Res. 2005, 38, 691–704. [Google Scholar] [CrossRef]
- Feuvrie, C.; Maury, O.; Le Bozec, H.; Ledoux, I.; Morrall, J.P.; Dalton, G.T.; Samoc, A.M.; Humphrey, M.G. Nonlinear Optical and Two-Photon Absorption Properties of Octupolar Tris(bipyridyl)metal Complexes. J. Phys. Chem. A 2007, 111, 8980–8985. [Google Scholar] [CrossRef]
- Girardot, C.; Cao, B.; Mulatier, J.-C.; Baldeck, P.L.; Chauvin, J.; Riehl, D.; Delaire, J.A.; Andraud, C.; Lemercier, G. Ruthenium(II) Complexes for Two-Photon Absorption-Based Optical Power Limiting. ChemPhysChem 2008, 9, 1531–1535. [Google Scholar] [CrossRef]
- Rebane, A.K.; Stark, C.W.; Pahapill, J.; Mikhaylov, A.; Rammo, M. Probing metal-to-ligand charge transfer transitions in ruthenium complexes by quantitative two-photon absorption spectroscopy. Org. Photonic Mater. Dev. XX 2018, 10529, 105291D. [Google Scholar] [CrossRef]
- Durand, N.; Mhanna, R.; Savel, P.; Akdas-Kiliç, H.; Malval, J.-P.; Soppera, O.; Fillaut, J.-L. Unexpected disruption of the dimensionality-driven two-photon absorption enhancement within a multipolar polypyridyl ruthenium complex series. Chem. Commun. 2020, 56, 12801–12804. [Google Scholar] [CrossRef] [PubMed]
- Kuzyk, M. Fundamental limits on two-photon absorption cross sections. J. Chem. Phys. 2003, 119, 8327–8334. [Google Scholar] [CrossRef]
- Moreno, J.P.; Kuzyk, M. Fundamental limits of the dispersion of the two-photon absorption cross section. J. Chem. Phys. 2005, 123, 194101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfield, K.D.; Hagan, D.; Van Stryland, E.W.; Schafer, A.K.J.; Negres, R.A. New Two-Photon Absorbing Fluorene Derivatives: Synthesis and Nonlinear Optical Characterization. Org. Lett. 1999, 1, 1575–1578. [Google Scholar] [CrossRef]
- Yurchenko, O.; Freytag, D.; Borg, L.Z.; Zentel, R.; Heinze, J.; Ludwigs, S. Electrochemically Induced Reversible and Irreversible Coupling of Triarylamines. J. Phys. Chem. B 2011, 116, 30–39. [Google Scholar] [CrossRef]
- Kapturkiewicz, A.; Herbich, J.; Karpiuk, J.; Nowacki, J. Intramolecular Radiative and Radiationless Charge Recombination Processes in Donor−Acceptor Carbazole Derivatives. J. Phys. Chem. A 1997, 101, 2332–2344. [Google Scholar] [CrossRef]
- Bodapati, R.; Dey, G.R.; Ramteke, G.R.; Krishnakanth, K.N.; Rao, S.V.; Jose, K.V.J.; Das, S.K. Carbazole-based π-conjugated 2,2′-Bipyridines, a new class of organic chromophores: Photophysical, ultrafast nonlinear optical and computational studies. Dyes Pigments 2021, 185, 108932. [Google Scholar] [CrossRef]
- Song, H.; Wang, X.; Yang, W.; He, G.; Kuang, Z.; Li, Y.; Xia, A.; Zhong, Y.-W.; Kong, F. Ultrafast relaxation dynamics of amine-substituted bipyridyl ruthenium(ii) complexes. Chem. Phys. Lett. 2017, 683, 322–328. [Google Scholar] [CrossRef]
- Verma, S.; Kar, P.; Das, A.; Ghosh, H.N. Photophysical properties of ligand localized excited state in ruthenium(ii) polypyridyl complexes: A combined effect of electron donor–acceptor ligand. Dalton Trans. 2011, 40, 9765–9773. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee, KS, USA, 2007. [Google Scholar]
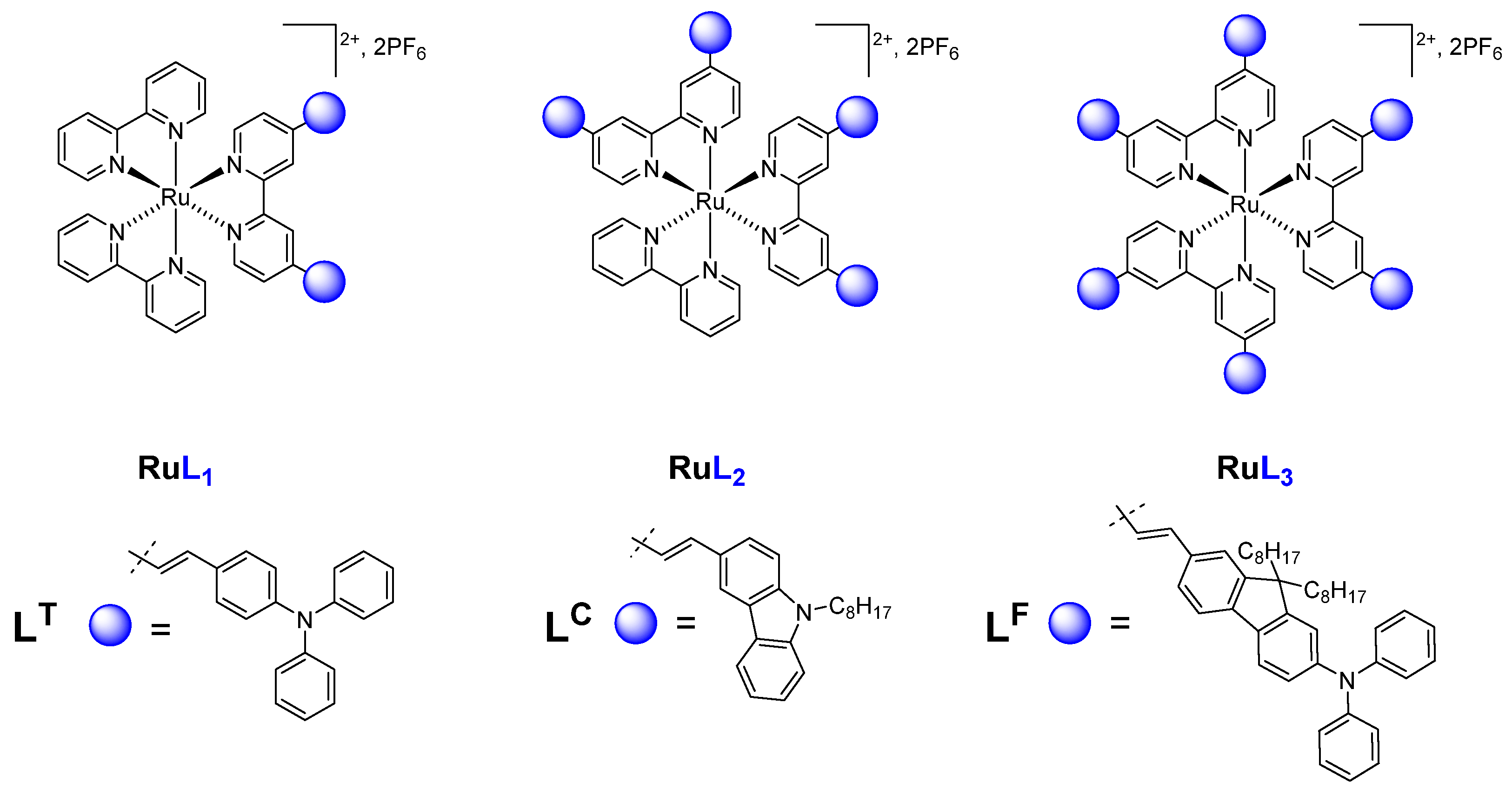
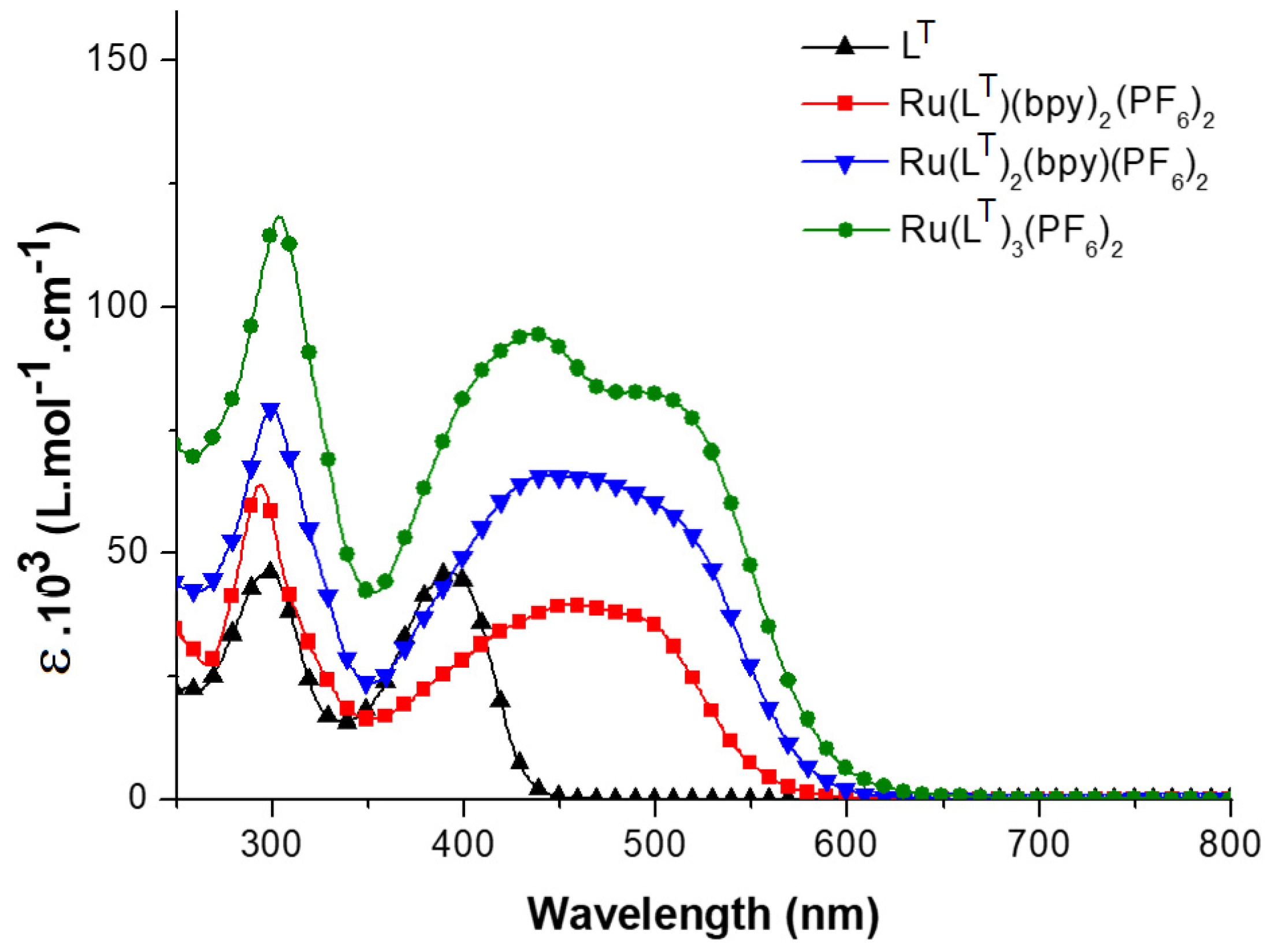
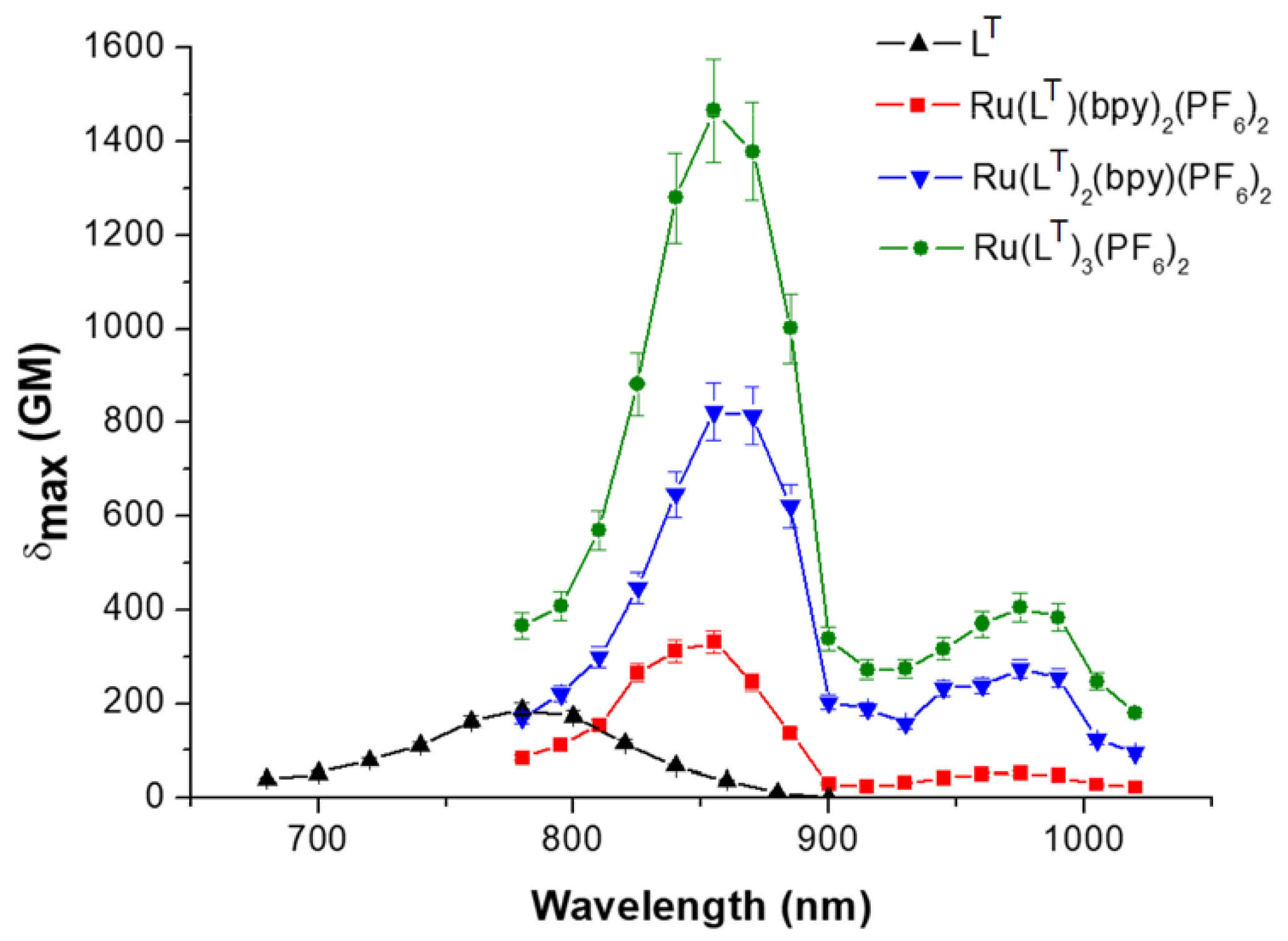
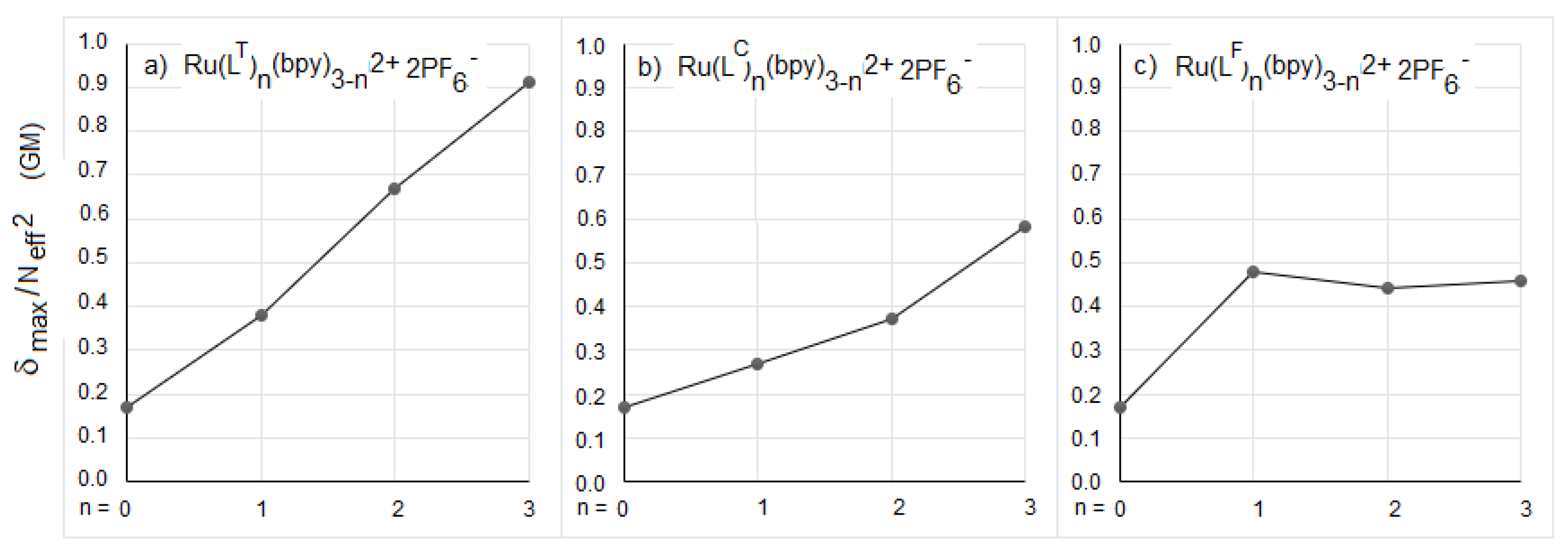

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λabs (nm) | ε.103 (L·mol−1·cm−1) | λem (nm) | Φem (N2 atm.) | |
|---|---|---|---|---|
| LT | 299, 392 | 46, 46 | 481 | 0.80 |
| [Ru(LT)(bpy)2]2+ | 294, 455, 489 | 64, 40, 37 | 695 | 0.0013 (0.014) |
| [Ru(LT)2(bpy)]2+ | 300, 445, 505 | 79, 66, 59 | 700 | 0.0021 (0.031) |
| [Ru(LT)3]2+ | 303, 437, 504 | 119, 94, 82 | 705 | 0.0032 (0.041) |
| LC | 300, 362 | 44, 51 | 433 | 0.11 |
| [Ru(LC)(bpy)2]2+ | 294, 415, 469 | 71, 34, 30 | 675 | 0.0034 (0.017) |
| [Ru(LC)2(bpy)]2+ | 300, 415, 494 | 100, 80, 55 | 685 | 0.0057 (0.047) |
| [Ru(LC)3]2+ | 303, 420, 503 | 147, 141, 86 | 695 | 0.0063 (0.056) |
| LF | 309, 395 | 57, 76 | 498 | 0.78 |
| [Ru(LF)(bpy)2]2+ | 294, 450, 493 | 101, 66, 62 | 695 | 0.0012 (0.017) |
| [Ru(LF)2(bpy)]2+ | 300, 426, 507 | 110, 100, 88 | 700 | 0.0018 (0.017) |
| [Ru(LF)3]2+ | 309, 429, 502 | 146, 139, 122 | 705 | 0.0028 (0.029) |
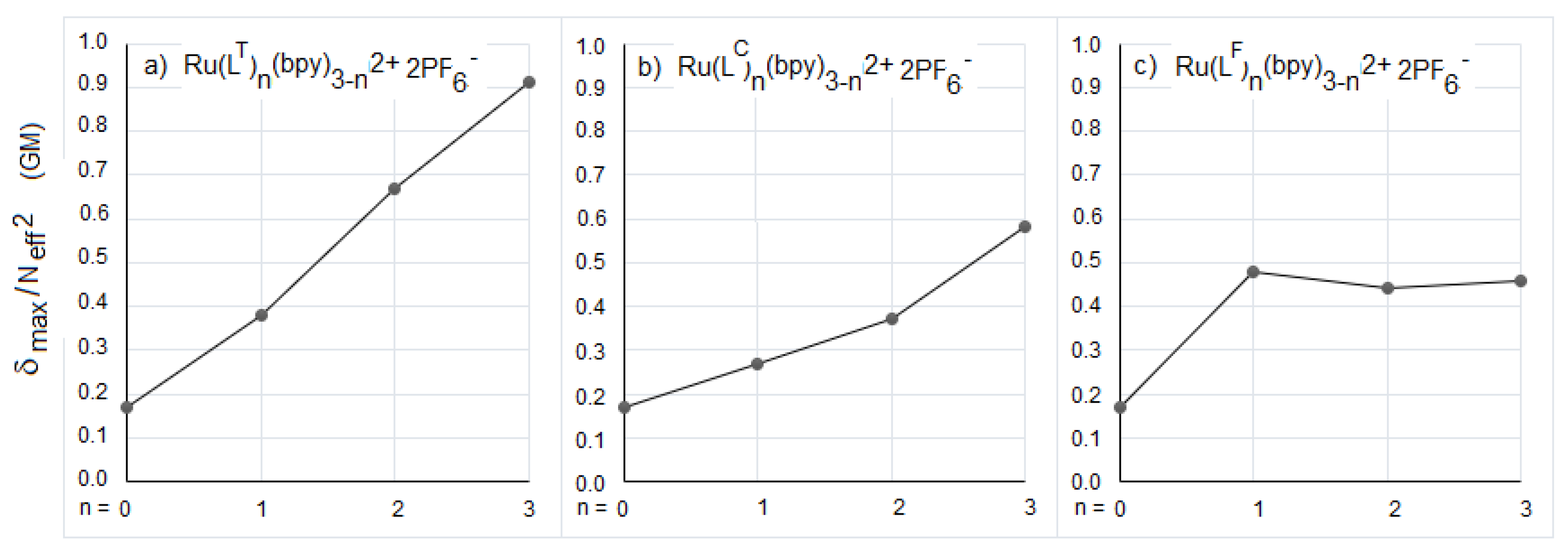
| λabs (nm) OPA | λabs (nm) TPA | δmax (GM) | Neff | δmax/Neff2 (GM) | |
|---|---|---|---|---|---|
| LT | 299, 392 | 780 | 187 | 23.1 | 0.35 |
| [Ru(LT)(bpy)2]2+ | 294, 455, 489 | 855 | 311 | 28.6 | 0.38 |
| [Ru(LT)2(bpy)]2+ | 300, 445, 505 | 855 | 822 | 35 | 0.67 |
| [Ru(LT)3]2+ | 303, 437, 504 | 855 | 1465 | 40.1 | 0.91 |
| LC | 300, 362 | 705 | 200 | 28.3 | 0.25 |
| [Ru(LC)(bpy)2]2+ | 294, 415, 469 | 795 | 395 | 38.3 | 0.27 |
| [Ru(LC)2(bpy)]2+ | 300, 415, 494 | 840 | 655 | 42.1 | 0.37 |
| [Ru(LC)3]2+ | 303, 420, 503 | 855 | 1381 | 48.8 | 0.58 |
| LF | 309, 395 | 800 | 208 | 30.7 | 0.22 |
| [Ru(LF)(bpy)2]2+ | 294, 450, 493 | 855 | 596 | 35.2 | 0.48 |
| [Ru(LF)2(bpy)]2+ | 300, 426, 507 | 870 | 889 | 44.9 | 0.44 |
| [Ru(LF)3]2+ | 309, 429, 502 | 870 | 1315 | 53.5 | 0.46 |
| HOMO-2 | HOMO-1 | HOMO | LUMO | LUMO+1 | LUMO+2 | |
|---|---|---|---|---|---|---|
| [Ru(LT)(bpy)2]2+ (% Ru) | −6.81 eV (66) | −5.83 eV (9) | −5.80 eV (18) | −3.10 eV (7) | −2.98 eV (20) | −2.95 eV (11) |
| Ru(LC)(bpy)2]2+ (% Ru) | −6.71 eV (7) | −6.14 eV (4) | −6.13 eV (5) | −3.19 eV (2) | −3.04 eV (4) | −2.99 eV (6) |
| [Ru(LF)(bpy)2]2+ (% Ru) | −6.71 eV (43) | −5.53 eV (1) | −5.52 eV (1) | −3.20 eV (2) | −3.04 eV (4) | −2.98 eV (6) |
| [Ru(LT)3]2+ (% Ru) | −5.68 eV (10) | −5.67 eV (11) | −5.67 eV (11) | −2.92 eV (1) | −2.78 eV (6) | −2.72 eV (5) |
| [Ru(LC)3]2+ (% Ru) | −6.14 eV (1) | −6.07 eV (10) | −6.04 eV (12) | −3.20 eV (1) | −3.07 eV (6) | −3.07 eV (6) |
| [Ru(LF)3]2+ (% Ru) | −5.51 eV (0) | −5.49 eV (0) | −5.48 eV (0) | −3.16 eV (0) | −3.04 eV (6) | −3.03 eV (6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durand, N.; Amar, A.; Mhanna, R.; Akdas-Kiliç, H.; Soppera, O.; Malval, J.-P.; Boucekkine, A.; Fillaut, J.-L. Two-Photon Absorption Cooperative Effects within Multi-Dipolar Ruthenium Complexes: The Decisive Influence of Charge Transfers. Molecules 2022, 27, 1493. https://doi.org/10.3390/molecules27051493
Durand N, Amar A, Mhanna R, Akdas-Kiliç H, Soppera O, Malval J-P, Boucekkine A, Fillaut J-L. Two-Photon Absorption Cooperative Effects within Multi-Dipolar Ruthenium Complexes: The Decisive Influence of Charge Transfers. Molecules. 2022; 27(5):1493. https://doi.org/10.3390/molecules27051493
Chicago/Turabian StyleDurand, Nicolas, Anissa Amar, Rana Mhanna, Huriye Akdas-Kiliç, Olivier Soppera, Jean-Pierre Malval, Abdou Boucekkine, and Jean-Luc Fillaut. 2022. "Two-Photon Absorption Cooperative Effects within Multi-Dipolar Ruthenium Complexes: The Decisive Influence of Charge Transfers" Molecules 27, no. 5: 1493. https://doi.org/10.3390/molecules27051493
APA StyleDurand, N., Amar, A., Mhanna, R., Akdas-Kiliç, H., Soppera, O., Malval, J.-P., Boucekkine, A., & Fillaut, J.-L. (2022). Two-Photon Absorption Cooperative Effects within Multi-Dipolar Ruthenium Complexes: The Decisive Influence of Charge Transfers. Molecules, 27(5), 1493. https://doi.org/10.3390/molecules27051493