
Stable Carbenes as Structural Components of Partially Saturated Sulfur-Containing Heterocycles

 , , , and
, , , and
Abstract
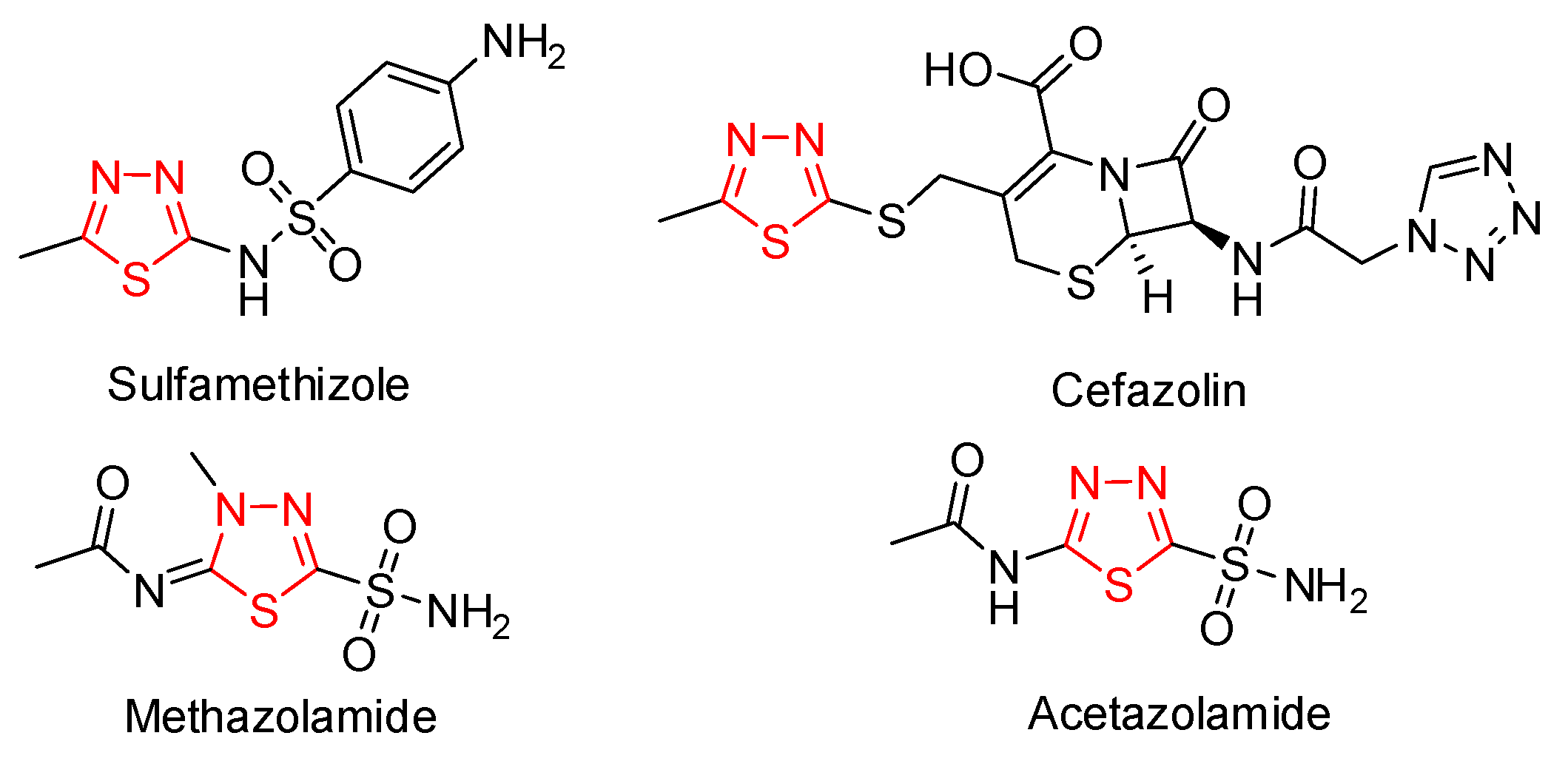
:1. Introduction
2. Results and Discussion
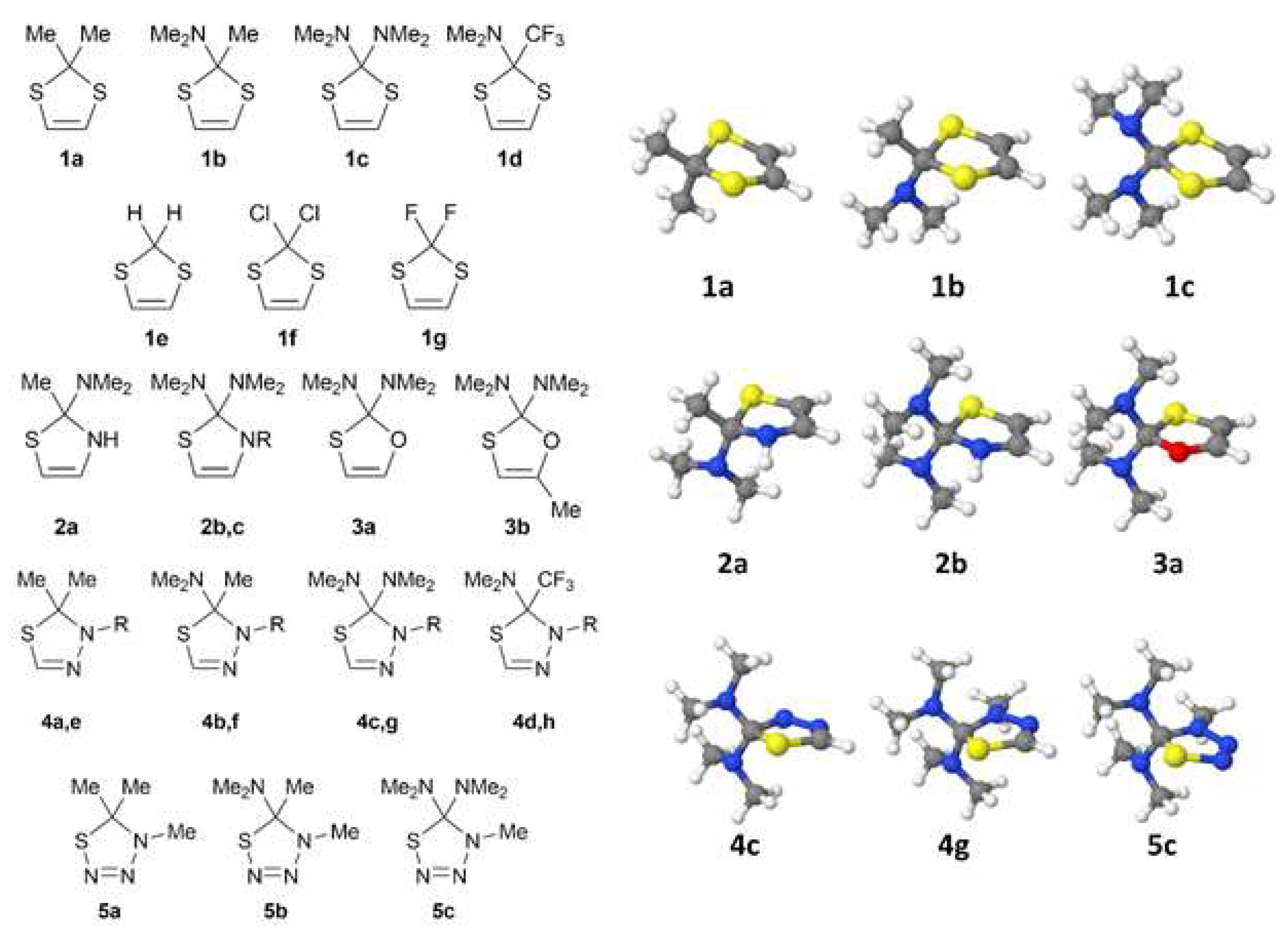
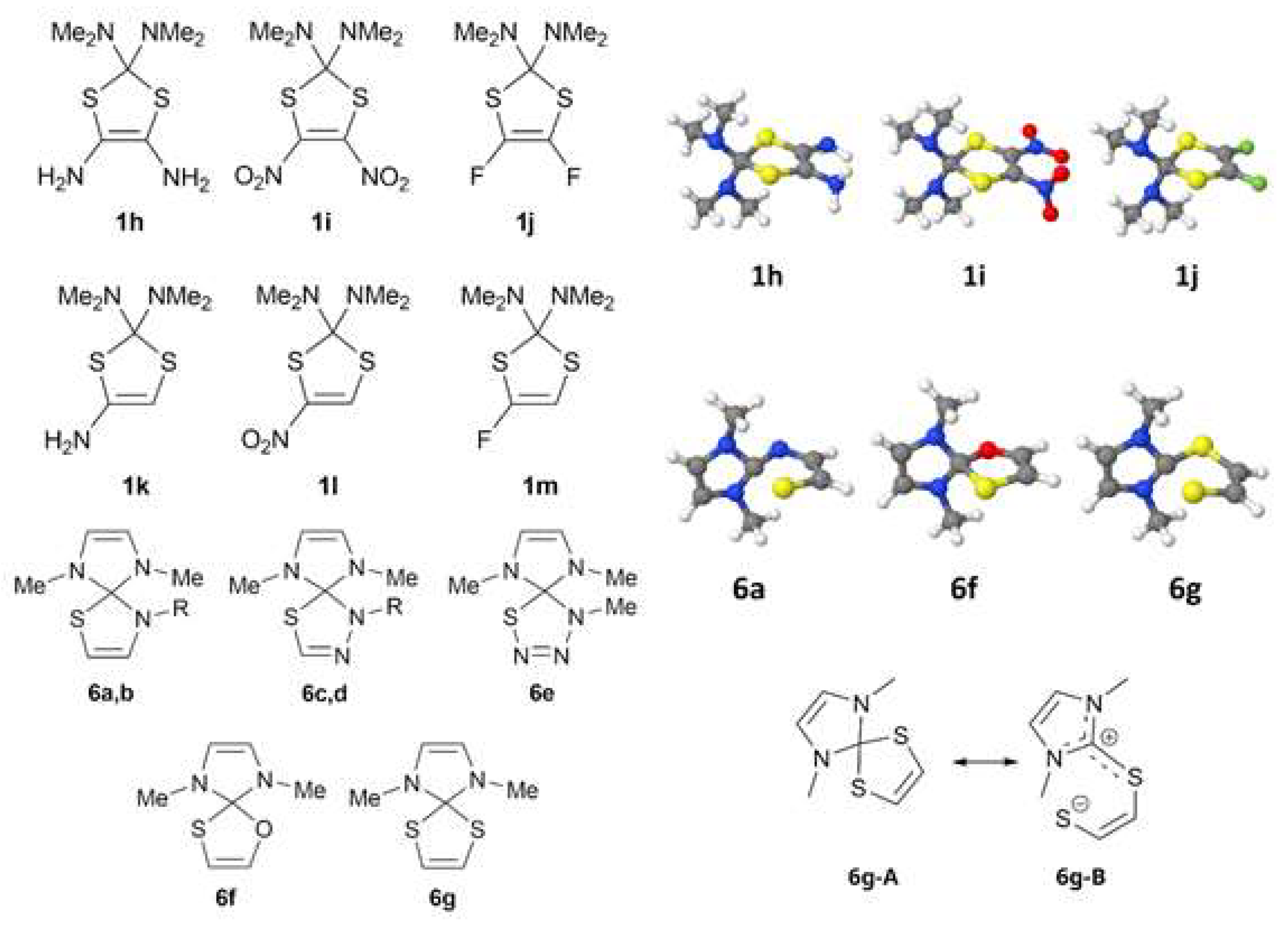
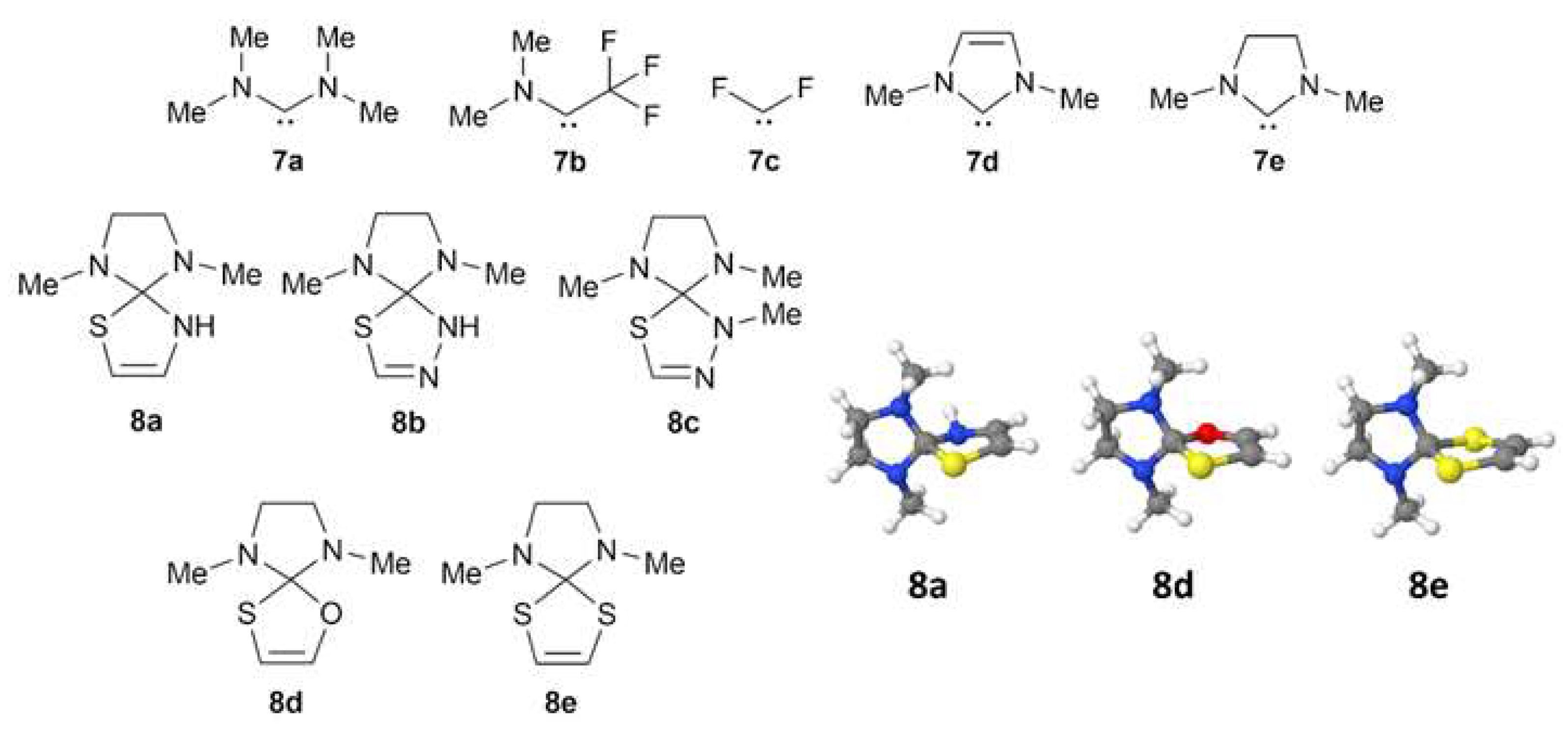
2.1. Quantum Chemical Modeling of Sulfur-Containing Heterocycles
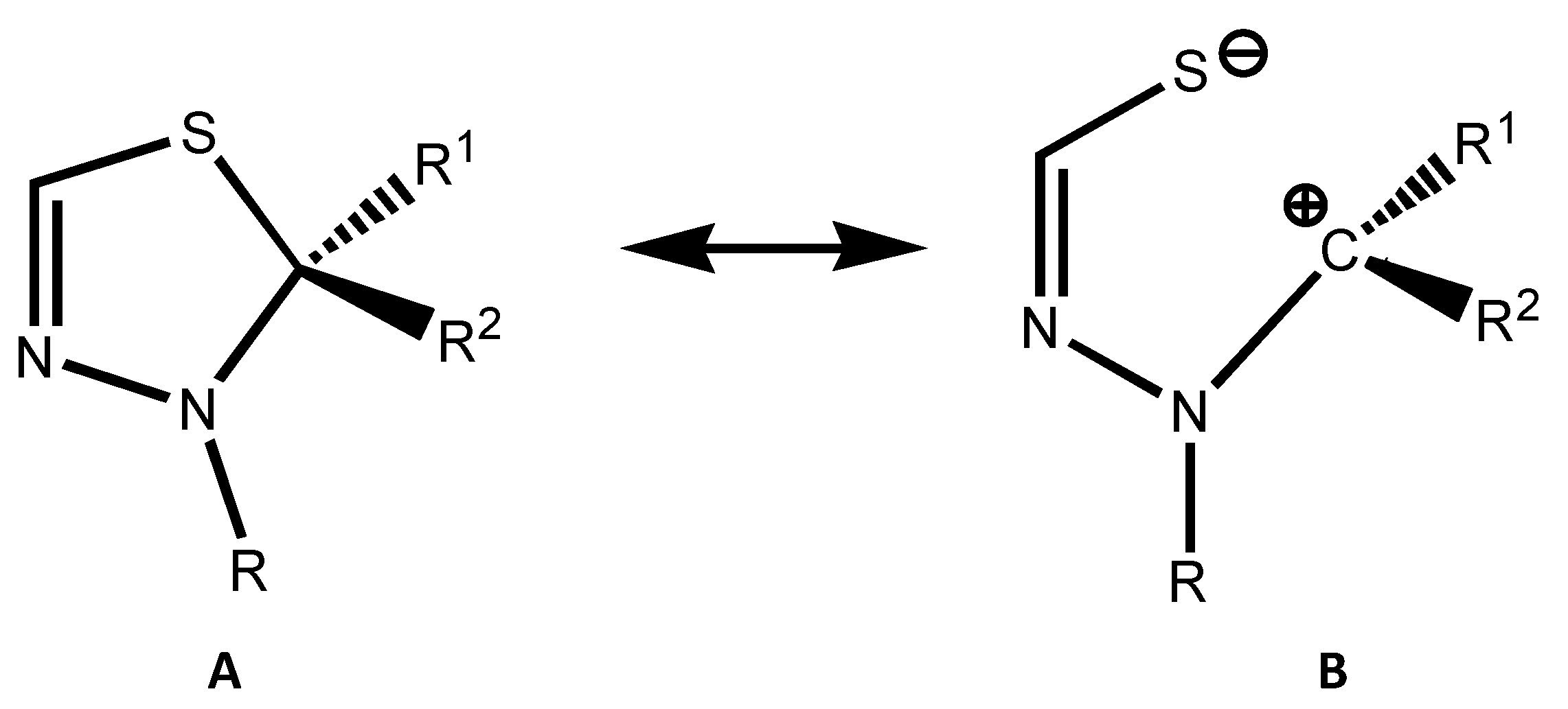
2.2. Arduengo Carbene-Based Thioheterocycles
2.3. Isodesmic Reactions of the Heterocycles with a Carbene Molecule
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Taylor, R.D.; MacCoss, M.; Lawson, A.D.G. Rings in Drugs. J. Med. Chem. 2014, 57, 5845–5859. [Google Scholar] [CrossRef] [PubMed]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Kurita, M.; Nikaido, H.; Mera, M.; Konishi, N.; Nakagawa, R. 3,7-Disubstituted Cephalosporin Compounds and Preparation Thereof. U.S. Patent 3,516,997, 23 June 1970. [Google Scholar]
- World Health Organization. World Health Organization Model List of Essential Medicines: 21st List 2019; WHO: Geneva, Switzerland, 2019; Volume 21. [Google Scholar]
- Clapp, J.W.; Roblin, R.O.J. Heterocyclic Sulfonamides and Methods of Preparation Thereof. U.S. Patent 2,554,816, 29 May 1951. [Google Scholar]
- Camargo, J.d.N.A.; Pianoski, K.E.; dos Santos, M.G.; Lazarin-Bidóia, D.; Volpato, H.; Moura, S.; Nakamura, C.V.; Rosa, F.A. Antiparasitic Behavior of Trifluoromethylated Pyrazole 2-Amino-1,3,4-thiadiazole Hybrids and Their Analogues: Synthesis and Structure-Activity Relationship. Front. Pharmacol. 2020, 11, 591570. [Google Scholar] [CrossRef] [PubMed]
- Almandil, N.B.; Taha, M.; Rahim, F.; Wadood, A.; Imran, S.; Alqahtani, M.A.; Bamarouf, Y.A.; Ibrahim, M.; Mosaddik, A.; Gollapalli, M. Synthesis of Novel Quinoline-Based Thiadiazole, Evaluation of Their Antileishmanial Potential and Molecular Docking Studies. Bioorg. Chem. 2019, 85, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Sadat-Ebrahimi, S.E.; Mirmohammadi, M.; Tabatabaei, Z.M.; Arani, M.A.; Jafari-Ashtiani, S.; Hashemian, M.; Foroumadi, P.; Yahya-Meymandi, A.; Moghimi, S.; Moshafi, M.H.; et al. Novel 5-(nitrothiophene-2-yl)-1,3,4-Thiadiazole Derivatives: Synthesis and Antileishmanial Activity against Promastigote Stage of Leishmania Major. Iran. J. Pharm. Res. 2019, 18, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Mykhaylychenko, S.S.; Pikun, N.V.; Rusanov, E.B.; Rozhenko, A.B.; Shermolovich, Y.G. Synthesis of 2-Polyfluoroalkyl-2,3-dihydro-1,3,4-thiadiazoles via Regioselective [3+2] Cycloaddition of Nitrile Imines to Polyfluoroalkanethioamides. Chem. Heterocycl. Compd. 2017, 53, 1268–1276. [Google Scholar] [CrossRef]
- Utecht-Jarzyńska, G.; Mykhaylychenko, S.S.; Rusanov, E.B.; Shermolovich, Y.G.; Jasiński, M.; Mlostoń, G. Highly Fluorinated 2,3-Dihydro-1,3,4-Thiadiazole Derivatives via [3+2]-Cycloadditions of Tertiary Thioamides with Nitrile Imines Derived from Trifluoroacetonitrile. J. Fluor. Chem. 2021, 242, 109702. [Google Scholar] [CrossRef]
- Handbook of Chemistry and Physic, 84th ed.; Lide, D.R. (Ed.) CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Grimme, S. Improved Second-Order Møller–Plesset Perturbation Theory by Separate Scaling of Parallel- and Antiparallel-Spin Pair Correlation Energies. J. Chem. Phys. 2003, 118, 9095–9102. [Google Scholar] [CrossRef]
- Fink, R.F. Spin-Component-Scaled Møller-Plesset (SCS-MP) Perturbation Theory: A Generalization of the MP Approach with Improved Properties. J. Chem. Phys. 2010, 133, 174113. [Google Scholar] [CrossRef]
- Pitoňák, M.; Neogrády, P.; Černý, I.; Grimme, S.; Hobza, P. Scaled MP3 Non-Covalent Interaction Energies Agree Closely with Accurate CCSD(T) Benchmark Data. ChemPhysChem 2009, 10, 282–289. [Google Scholar] [CrossRef]
- Řezáč, J.; Hobza, P. Describing Noncovalent Interactions beyond the Common Approximations: How Accurate Is the “Gold Standard,” CCSD(T) at the Complete Basis Set Limit? J. Chem. Theory Comput. 2013, 9, 2151–2155. [Google Scholar] [CrossRef] [PubMed]
- Rozhenko, A.B.; Schoeller, W.W.; Leszczynski, J. On the Stability of Perfluoroalkyl-Substituted Singlet Carbenes: A Coupled-Cluster Quantum Chemical Study. J. Phys. Chem. A 2014, 118, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- TURBOMOLE. University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007; TURBOMOLE V6.4 2012; TURBOMOLE GmbH: Karlsruhe, Germany, 2007; Available online: http://www.turbomole.com (accessed on 1 December 2021).
- Furche, F.; Ahlrichs, R.; Hättig, C.; Klopper, W.; Sierka, M.; Weigend, F. Turbomole. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 91–100. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M. RI-MP2: First Derivatives and Global Consistency. Theor. Chem. Acc. 1997, 97, 331–340. [Google Scholar] [CrossRef]
- Haase, F.; Ahlrichs, R. Semidirect MP2 Gradient Evaluation on Workstation Computers: The MPGRAD Program. J. Comput. Chem. 1993, 14, 907–912. [Google Scholar] [CrossRef]
- Ghahremanpour, M.M.; van Maaren, P.J.; Ditz, J.C.; Lindh, R.; van der Spoel, D. Large-Scale Calculations of Gas Phase Thermochemistry: Enthalpy of Formation, Standard Entropy, and Heat Capacity. J. Chem. Phys. 2016, 145, 114305. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized Auxiliary Basis Sets and Demonstration of Efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Hättig, C.; Schmitz, G.; Koßmann, J. Auxiliary Basis Sets for Density-Fitted Correlated Wavefunction Calculations: Weighted Core-Valence and ECP Basis Sets for Post-d Elements. Phys. Chem. Chem. Phys. 2012, 14, 6549–6555. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.P.; Weinhold, F. Natural Hybrid Orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural Bond Orbital Methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://jmol.sourceforge.net/ (accessed on 1 December 2021).
- Canepa, P.; Hanson, R.M.; Ugliengo, P.; Alfredsson, M. J-ICE: A New Jmol Interface for Handling and Visualizing Crystallographic and Electronic Properties. J. Appl. Crystallogr. 2011, 44, 225–229. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
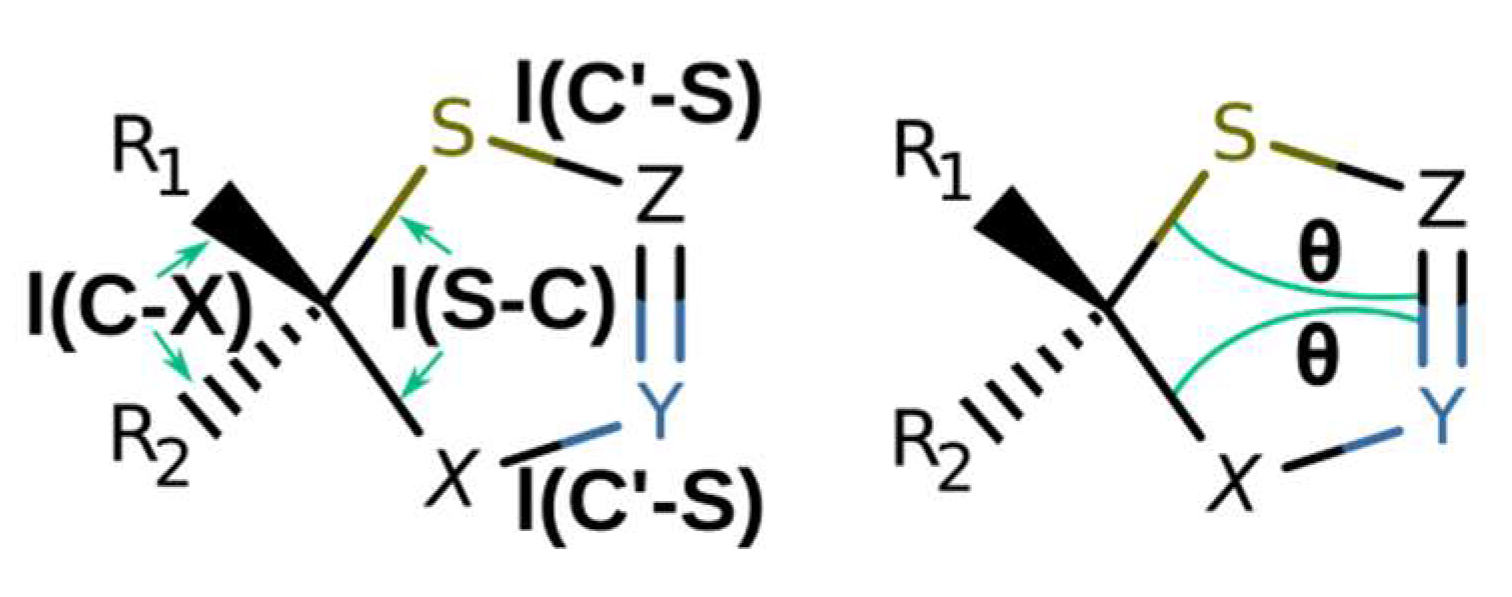
| Structure | l(S-C), Å | l(C′-S), Å | l(C-X), Å | θ, Degrees |
|---|---|---|---|---|
| 1a | 1.821 | 1.779 | 1.544 | 19.4, −19.4 |
| 1b | 1.852, 1.887 | 1.752 | 1.444 (C-N), 1.532 (C-C) | 15.8, −13.5 |
| 1c | 1.893 | 1.747 | 1.446 | 8.0, −5.0 |
| 1d | 1.852, 1.882 | 1.750, 1.751 | 1.439 (C-N), 1.552 (C-C) | 12.2, −8.9 |
| 1e | 1.823 | 1.763 | 1.087, 1.088 | 19.3, −19.3 |
| 1f | 1.822 | 1.755 | 1.779, 1.808 | 19.4, −19.4 |
| 1g | 1.826 | 1.750 | 1.353, 1.357 | 7.9, −7.9 |
| 1h | 1.882, 1.891 | 1.757, 1.773 | 1.445, 1.447 | 15.0, −12.4 |
| 1i | 1.897, 1.919 | 1.728, 1.744 | 1.426, 1.435 | 14.8, −12.6 |
| 1j | 1.903, 1.903 | 1.752, 1.752 | 1.436, 1.438 | 13.8, −11.7 |
| 1k | 1.886, 1.890 | 1.755, 1.761 | 1.444, 1.447 | 13.2, −13.2 |
| 1l | 1.898, 1.907 | 1.727, 1.744 | 1.437, 1.439 | 9.5, -8.1 |
| 1m | 1.897, 1.899 | 1.750, 1.751 | 1.439, 1.442 | 11.6, −8.8 |
| 2a | 1.897, 1.467 (C-N) | 1.765, 1.400 (C-N) | 1.452 (C-N), 1.527 (C-C) | 15.9, −9.9 |
| 2b | 1.898, 1.467 (C-N) | 1.761, 1.398 (C-N) | 1.450, 1.459 | 11.3, −5.4 |
| 2c | 1.906, 1.462 (C-N) | 1.760, 1.383 (C-N) | 1.447, 1.457 | 6.0, −12.6 |
| 3a | 1.908, 1.446 (C-O) | 1.755, 1.366 (C-O) | 1.434, 1.437 | 0.5, −2.8 |
| 3b | 1.905, 1.442 (C-O) | 1.756, 1.372 (C-O) | 1.435, 1.439 | 0.4, −2.5 |
| 4a | 1.846, 1.479 (C-N) | 1.764, 1.401 (N-N) | 1.522, 1.529 | 15.6, −28.5 |
| 4b | 1.888, 1.474 (C-N) | 1.756, 1.391 (N-N) | 1.444 (C-N), 1.527 (C-C) | 22.8, −11.6 |
| 4c | 1.898, 1.475 (C-N) | 1.751, 1.384 (N-N) | 1.438, 1.458 | 7.5, −20.0 |
| 4d | 1.853, 1.470 (C-N) | 1.756, 1.391 (N-N) | 1.446 (C-N), 1.551 (C-C) | −0.5, −8.0 |
| 4e | 1.847, 1.482 (C-N) | 1.760, 1.396 (N-N) | 1.523, 1.532 | 27.7, −16.1 |
| 4f | 1.888, 1.471 (C-N) | 1.755, 1.382 (N-N) | 1.444 (C-N), 1.529 (C-C) | 22.9, −12.7 |
| 4g | 1.902, 1.474 (C-N) | 1.749, 1.370 (N-N) | 1.441, 1.451 | 9.4, −20.0 |
| 4h | 1.858, 1.465 (C-N) | 1.754, 1.380 (N-N) | 1.459 (C-N), 1.559 (C-C) | 0.8, −5.8 |
| 5a | 1.829, 1.476 (C-N) | 1.761(S-N), 1.368 (N-N) | 1.525, 1.531 | 14.8, −24.3 |
| 5b | 1.857, 1.466 (C-N) | 1.752(S-N), 1.358 (N-N) | 1.450 (C-N), 1.530 (C-C) | 20.1, −12.9 |
| 5c | 1.868, 1.471 (C-N) | 1.739(S-N), 1.346 (N-N) | 1.447, 1.451 | 8.0, −15.7 |
| 6a | 2.272, 1.425 (C-N) | 1.763, 1.414 (C-N) | 1.409, 1.413 | 5.0, −13.2 |
| 6b | 1.990, 1.440 (C-N) | 1.758, 1.396 (C-N) | 1.429, 1.436 | 9.0, −16.7 |
| 6c | 2.064, 1.445 (C-N) | 1.740(S-N), 1.395 (N-N) | 1.408, 1.412 | 25.6, −11.3 |
| 6d | 2.045, 1.445 (C-N) | 1.740(S-N), 1.389 (N-N) | 1.412, 1.418 | 25.2, −11.6 |
| 6e | 1.919, 1.448 (C-N) | 1.739(S-N), 1.356 (N-N) | 1.437, 1.439 | 8.4, −16.8 |
| 6f | 2.034, 1.409 (C-O) | 1.750, 1.376 (C-O) | 1.411, 1.411 | 0.0, 0.0 |
| 6g | 2.155, 1.817 | 1.737, 1.755 | 1.399, 1.401 | 12.4, −14.7 |
| 8a | 1.890, 1.467 (C-N) | 1.760(C-S), 1.401 (C-N) | 1.448, 1.450 | 14.1, −9.2 |
| 8b | 1.886, 1.457 (C-O) | 1.756(C-S), 1.353 (C-O) | 1.425, 1.431 | 4.6, −6.0 |
| 8c | 1.885, 1.891 | 1.747, 1.747 | 1.432, 1.438 | 7.6, −12.7 |
| 8d | 1.894, 1.472 (C-N) | 1.748(C-S), 1.388 (N-N) | 1.448, 1.450 | 18.8, −9.5 |
| 8e | 1.895, 1.472 (C-N) | 1.747(C-S), 1.380 (N-N) | 1.436, 1.443 | 20.6, −11.3 |
| Item | Reaction | ΔH | ΔG |
|---|---|---|---|
| 1 | 1c + 7b → 1d + 7a | –5.2 | (–3.3) |
| 2 | 1c + 7c → 1g + 7a | –8.8 | –10.0 |
| 3 | 2b + 7d → 6a + 7a | 23.2 | 22.2 |
| 4 | 2c + 7d → 6b + 7a | 20.6 | 19.8 |
| 5 | 4c + 7d → 6c + 7a | 21.8 | 20.7 |
| 6 | 4g + 7d → 6d + 7a | 19.6 | 18.6 |
| 7 | 5c + 7d → 6e + 7a | 22.3 | 21.3 |
| 8 | 3a + 7d → 6f + 7a | 26.8 | 25.1 |
| 9 | 1c + 7d → 6g + 7a | 24.2 | 22.6 |
| 10 | 2b + 7e → 8a + 7a | 8.9 | 9.1 |
| 11 | 4c + 7e → 8b + 7a | 8.4 | 8.5 |
| 12 | 4g + 7e → 8c + 7a | 7.1 | 7.2 |
| 13 | 3a + 7e → 8d + 7a | 11.1 | 11.2 |
| 14 | 1c + 7e → 8e + 7a | 9.8 | 10.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozhenko, A.B.; Horbenko, Y.S.; Kyrylchuk, A.A.; Zarudnitskii, E.V.; Mykhaylychenko, S.S.; Shermolovich, Y.G.; Grafov, A.V. Stable Carbenes as Structural Components of Partially Saturated Sulfur-Containing Heterocycles. Molecules 2022, 27, 1458. https://doi.org/10.3390/molecules27051458
Rozhenko AB, Horbenko YS, Kyrylchuk AA, Zarudnitskii EV, Mykhaylychenko SS, Shermolovich YG, Grafov AV. Stable Carbenes as Structural Components of Partially Saturated Sulfur-Containing Heterocycles. Molecules. 2022; 27(5):1458. https://doi.org/10.3390/molecules27051458
Chicago/Turabian StyleRozhenko, Alexander B., Yuliia S. Horbenko, Andrii A. Kyrylchuk, Evgenij V. Zarudnitskii, Sergiy S. Mykhaylychenko, Yuriy G. Shermolovich, and Andriy V. Grafov. 2022. "Stable Carbenes as Structural Components of Partially Saturated Sulfur-Containing Heterocycles" Molecules 27, no. 5: 1458. https://doi.org/10.3390/molecules27051458
APA StyleRozhenko, A. B., Horbenko, Y. S., Kyrylchuk, A. A., Zarudnitskii, E. V., Mykhaylychenko, S. S., Shermolovich, Y. G., & Grafov, A. V. (2022). Stable Carbenes as Structural Components of Partially Saturated Sulfur-Containing Heterocycles. Molecules, 27(5), 1458. https://doi.org/10.3390/molecules27051458