
Hydrogen Bonding and Polymorphism of Amino Alcohol Salts with Quinaldinate: Structural Study



Abstract
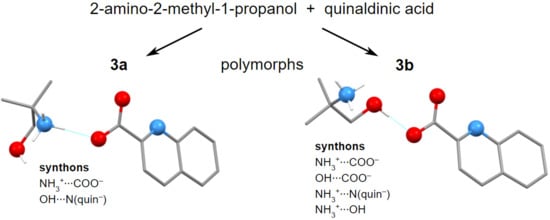
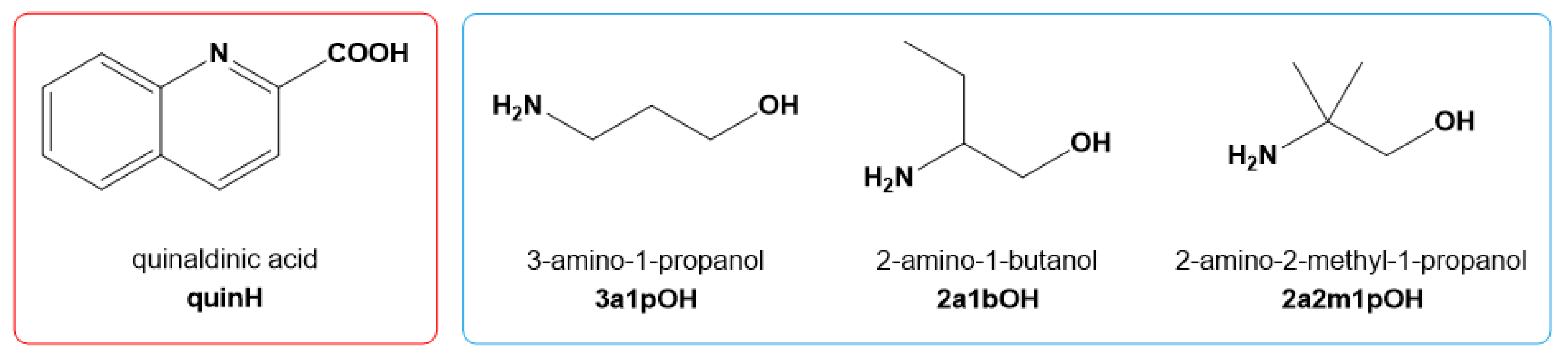
:
1. Introduction
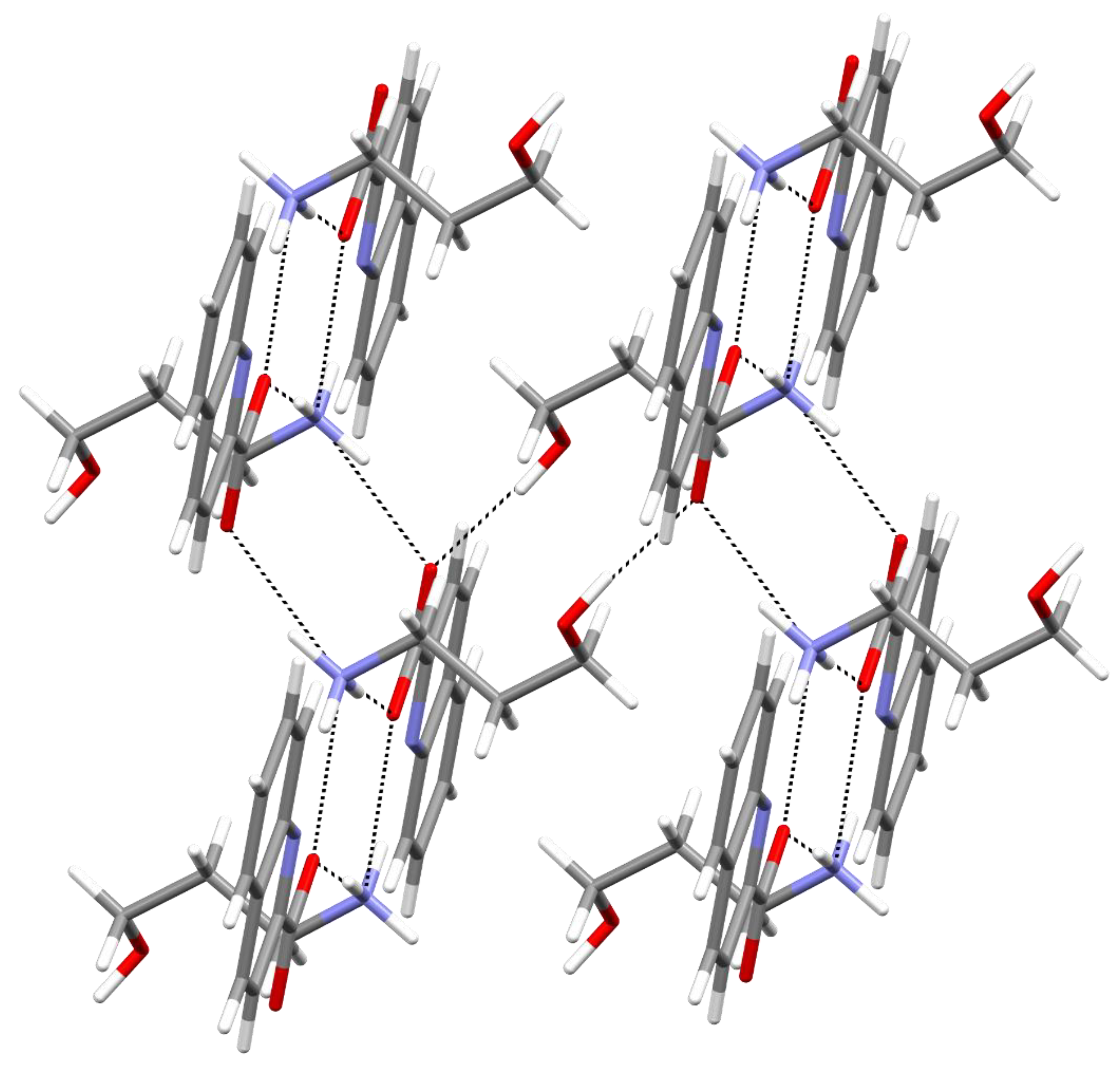
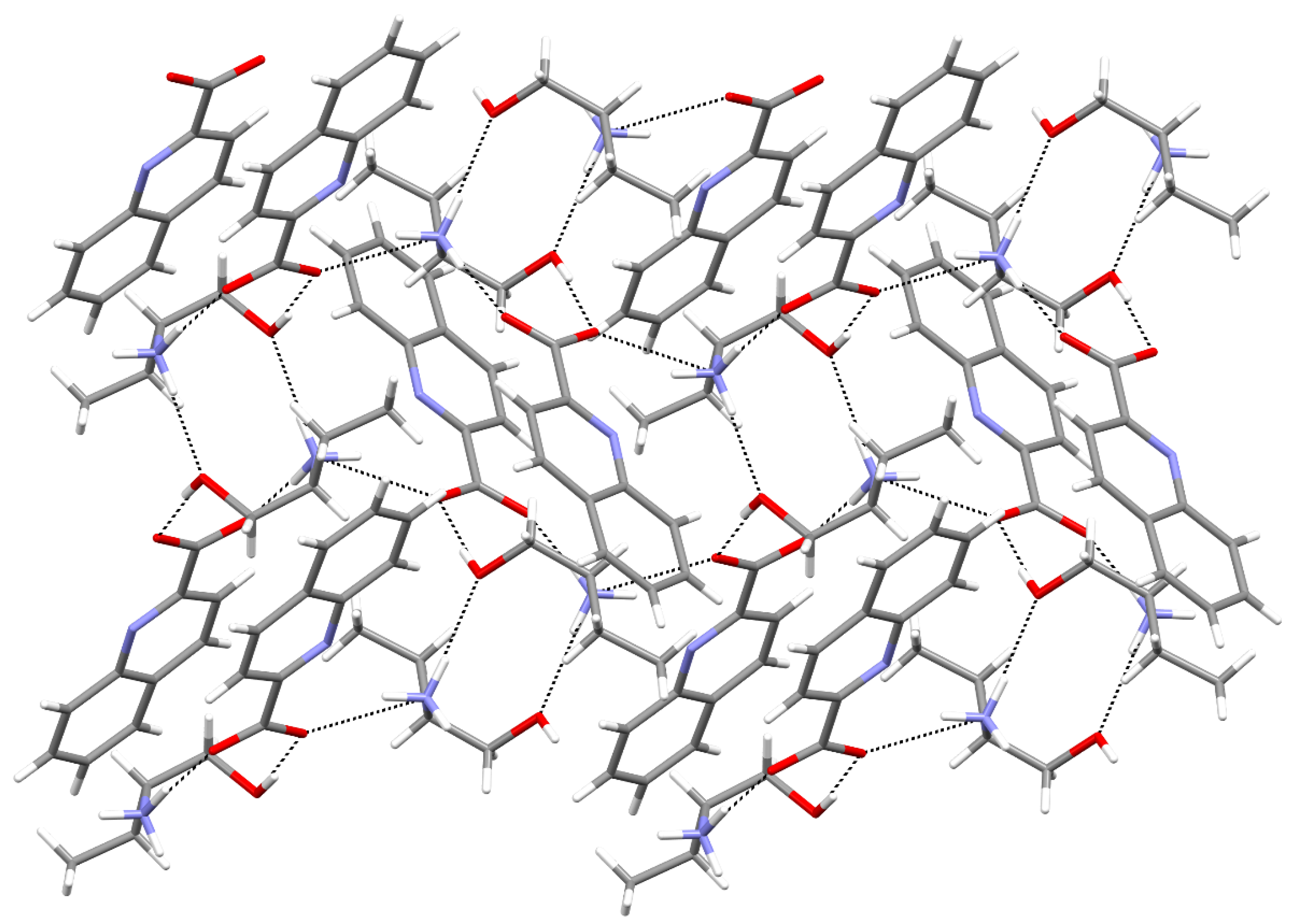
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Desiraju, G.R.; Vittal, J.J.; Ramanan, A. Crystal Engineering: A Textbook; World Scientific Publishing Co.: Singapore, 2011; pp. 1–3. [Google Scholar]
- Corpinot, M.K.; Bučar, D.-K. A practical guide to the design of molecular crystals. Cryst. Growth Des. 2019, 19, 1426–1453. [Google Scholar] [CrossRef] [Green Version]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular synthons in crystal engineering—A new organic synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Bučar, D.-K.; Lancaster, R.W.; Bernstein, J. Disappearing polymorphs revisited. Angew. Chem. Int. Ed. 2015, 54, 6972–6993. [Google Scholar] [CrossRef] [Green Version]
- Corpinot, M.K.; Stratford, S.A.; Arhangelskis, M.; Anka-Lufford, J.; Halasz, I.; Judaš, N.; Jones, W.; Bučar, D.-K. On the predictability of supramolecular interactions in molecular cocrystals—The view from the bench. CrysEngComm 2016, 18, 5434–5439. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2002; pp. 1–9. [Google Scholar]
- Gavezzotti, A.; Filippini, G. Polymorphic forms of organic crystals at room conditions: Thermodynamic and structural implications. J. Am. Chem. Soc. 1995, 117, 12299–12305. [Google Scholar] [CrossRef]
- McCrone, W.C. Physics and Chemistry of the Organic Solid State; Fox, D., Labes, M.M., Weissberger, A., Eds.; Interscience: New York, NY, USA, 1965; Volume 2, pp. 725–767. [Google Scholar]
- Morissette, S.L.; Almarsson, Ö.; Peterson, M.L.; Remenar, J.F.; Read, M.J.; Lemmo, A.V.; Ellis, S.; Cima, M.J.; Gardner, C.R. High-throughput crystallization: Polymorphs, salts, co-crystals and solvates of pharmaceutical solids. Adv. Drug Delivery Rev. 2004, 56, 275–300. [Google Scholar] [CrossRef] [PubMed]
- David, W.I.F.; Shankland, K.; Pulham, C.R.; Blagden, N.; Davey, R.J.; Song, M. Polymorphism in benzamide. Angew. Chem. Int. Ed. 2005, 44, 7032–7035. [Google Scholar] [CrossRef]
- Stahly, G.P. Diversity in single- and multiple-component crystals. The search for and prevalence of polymorphs and cocrystals. Cryst. Growth Des. 2007, 7, 1007–1026. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Cabeza, A.J.; Reutzel-Edens, S.M.; Bernstein, J. Facts and fictions about polymorphism. Chem. Soc. Rev. 2015, 44, 8619–8635. [Google Scholar] [CrossRef]
- Podjed, N.; Modec, B.; Clérac, R.; Rouzières, M.; Alcaide, M.M.; López-Serrano, J. Structural diversity and magnetic properties of copper(II) quinaldinate compounds with amino alcohols. Manuscript in Preparation. University of Ljubljana, Faculty of Chemistry and Chemical Technology: Ljubljana, Slovenia, 2022. [Google Scholar]
- Tudor, V.; Mocanu, T.; Tuna, F.; Madalan, A.M.; Maxim, C.; Shova, S.; Andruh, M. Mixed ligand binuclear alkoxo-bridged copper(II) complexes derived from aminoalcohols and nitrogen ligands. J. Mol. Struct. 2013, 1046, 164–170. [Google Scholar] [CrossRef]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J. Acid–base crystalline complexes and the pKa rule. CrystEngComm 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Childs, S.L.; Stahly, G.P.; Park, A. The salt−cocrystal continuum: The influence of crystal structure on ionization state. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef] [Green Version]
- Haynes, W.M.; Lide, D.R.; Bruno, T.J. CRC Handbook of Chemistry and Physics, 97th ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 5–91. [Google Scholar]
- Braga, D.; Maini, L. Solid-state versus solution preparation of two crystal forms of [HN(CH2CH2)3NH][OOC(CH2)COOH]2. Polymorphs or hydrogen bond isomers? Chem. Commun. 2004, 976–977. [Google Scholar] [CrossRef]
- Bernstein, J.; Davey, R.J.; Henck, J.-O. Concomitant polymorphs. Angew. Chem. Int. Ed. 1999, 38, 3440–3461. [Google Scholar] [CrossRef]
- Williams, D.B.G.; Lawton, M. Drying of organic solvents: Quantitative evaluation of the efficiency of several desiccants. J. Org. Chem. 2010, 75, 8351–8354. [Google Scholar] [CrossRef]
- Modec, B.; Podjed, N.; Lah, N. Beyond the simple copper(II) coordination chemistry with quinaldinate and secondary amines. Molecules 2020, 25, 1573. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Willcott, M.R. MestRe Nova. J. Am. Chem. Soc. 2009, 131, 13180. [Google Scholar] [CrossRef]
- Agilent. CrysAlis PRO; Agilent Technologies Ltd.: Yarnton, UK, 2014. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]

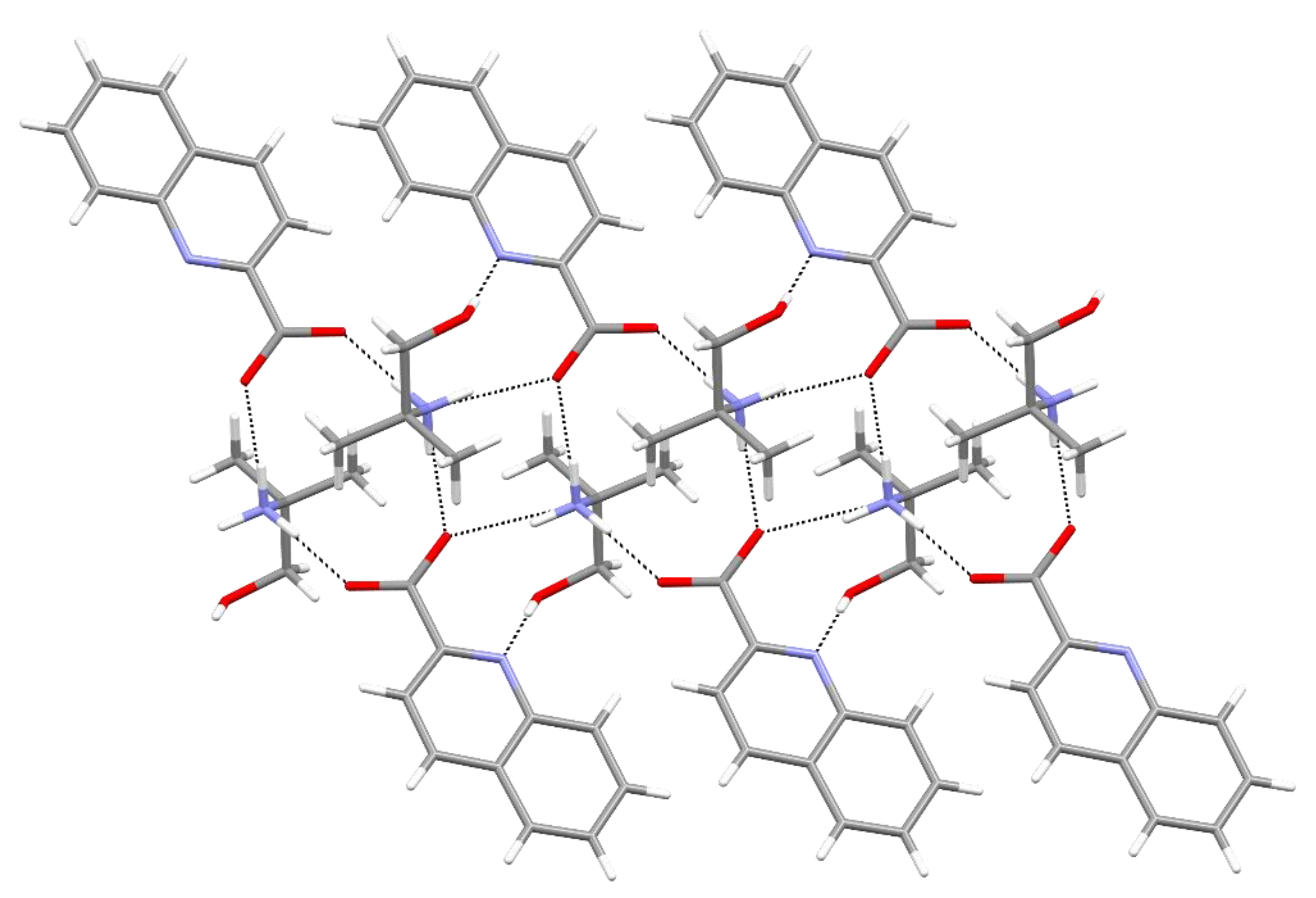
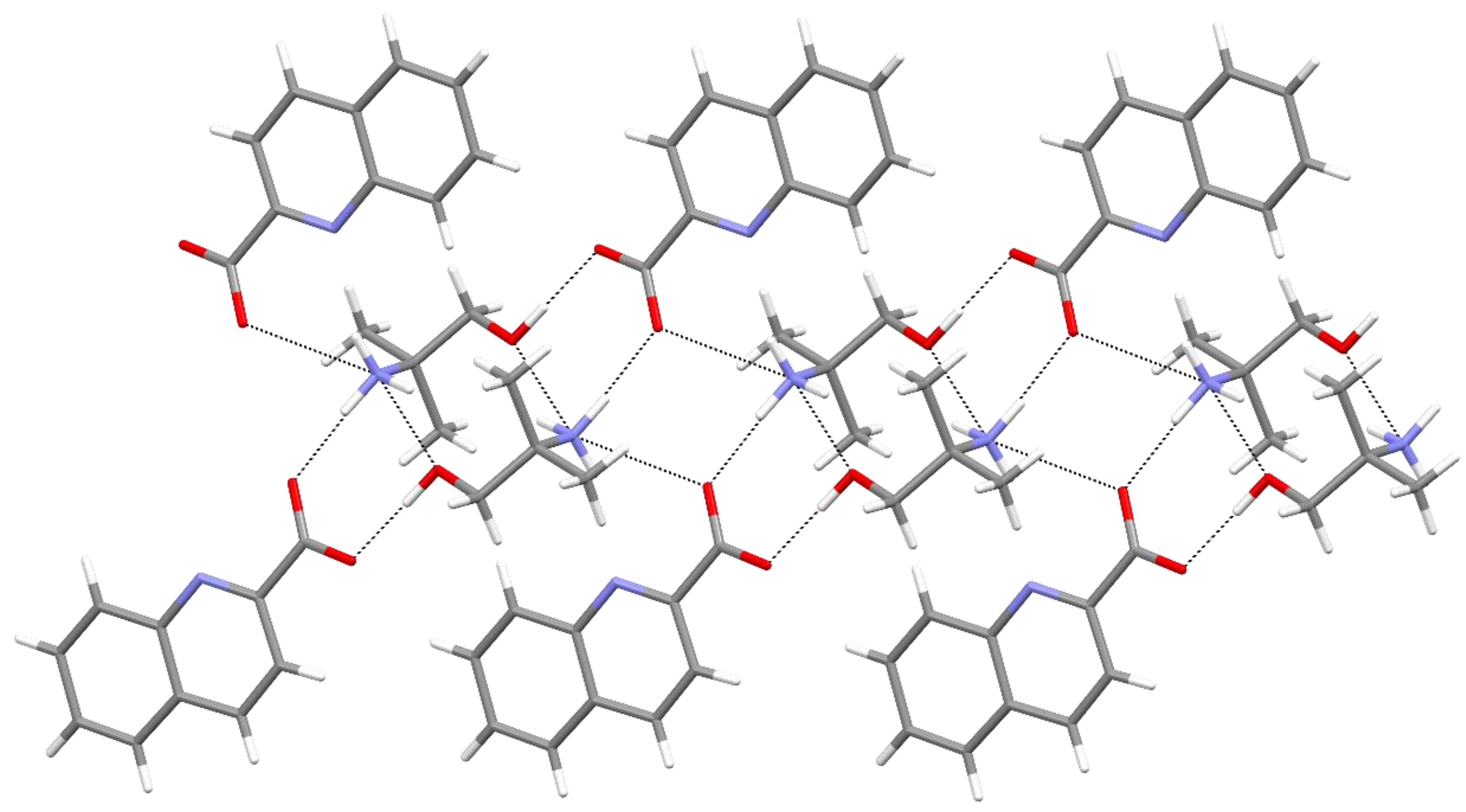

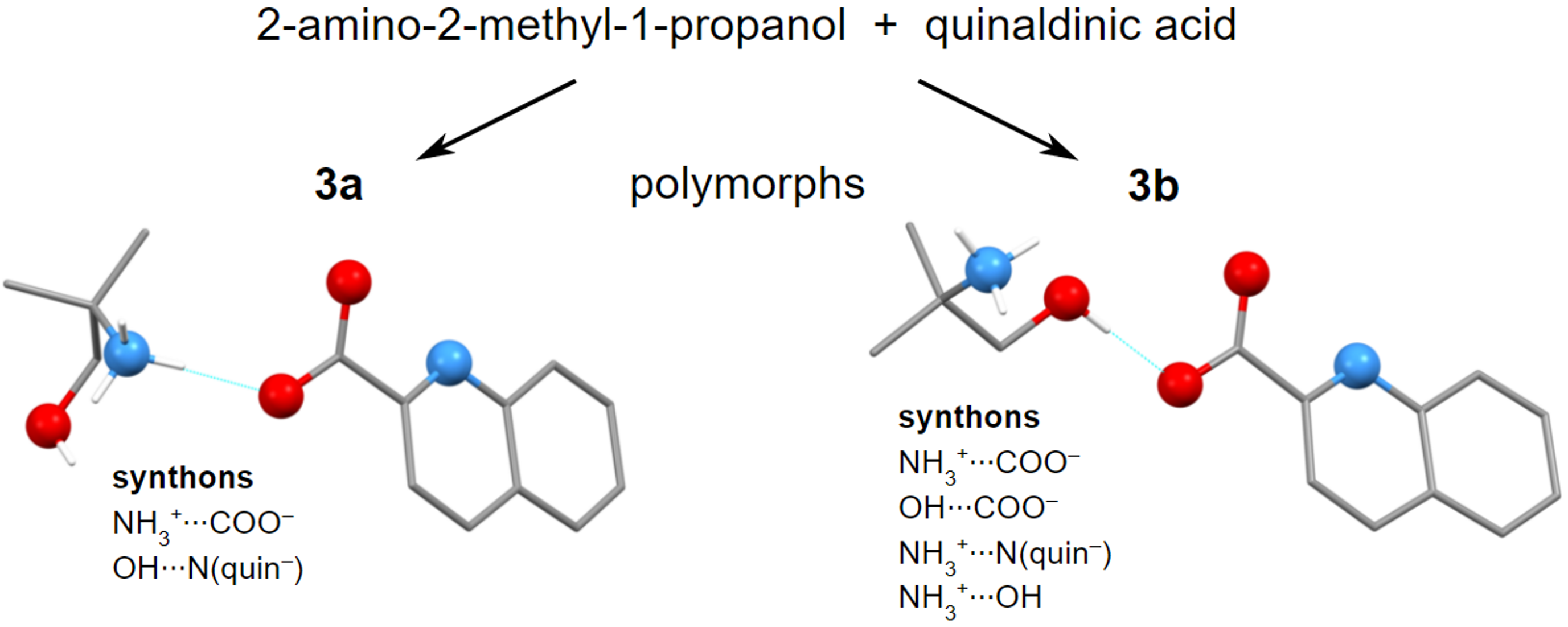{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 |
|---|
| π∙∙∙π Stacking Interactions Py∙∙∙Py [1−x, −y, 1−z], Cg∙∙∙Cg = 3.7402(15), dihedral angle = 0.02(11), shift distance = 1.552 Ph∙∙∙Py[1−x, −y, 1−z], Cg∙∙∙Cg = 3.6571(15), dihedral angle = 0.41(11), shift distance = 1.346 Ph∙∙∙Py[1−x, −1−y, 1−z], Cg∙∙∙Cg = 3.9681(16), dihedral angle = 0.41(11), shift distance = 1.656 Ph∙∙∙Ph[1−x, −1−y, 1−z], Cg∙∙∙Cg = 3.6899(16), dihedral angle = 0.00(11), shift distance = 0.821 |
| 2a |
| π∙∙∙π Stacking Interactions Py∙∙∙Py[−x, 1−y, 1−z], Cg∙∙∙Cg = 3.7080(10), dihedral angle = 0.02(6), shift distance = 1.635 Ph∙∙∙Py[−x, 1−y, 1−z], Cg∙∙∙Cg = 3.5163(9), dihedral angle = 0.39(7), shift distance = 1.123 |
| C–H∙∙∙π Interactions C–H∙∙∙Ph[1−x, 1−y, 1−z], H∙∙∙Cg = 3.00, C–H∙∙∙Cg = 149, C∙∙∙Cg = 3.8620(17) |
| 2b |
| C–H∙∙∙π Interactions C–H∙∙∙Ph[1+x, 1+y, z], H∙∙∙Cg = 2.79, C–H∙∙∙Cg = 131, C∙∙∙Cg = 3.495(3) |
| 3a |
| C–H∙∙∙π Interactions C–H∙∙∙Py[2.5−x, 0.5+y, 0.5−z], H∙∙∙Cg = 2.95, C–H∙∙∙Cg = 150, C∙∙∙Cg = 3.7898(19) C–H∙∙∙Ph[1.5−x, −0.5+y, 0.5−z], H∙∙∙Cg = 2.78, C–H∙∙∙Cg = 140, C∙∙∙Cg = 3.5438(18) |
| 3b |
| π∙∙∙π Stacking Interactions Ph∙∙∙Py[−x, −y, 1−z], Cg∙∙∙Cg = 3.8194(7), dihedral angle = 2.51(6), shift distance = 1.462 |
| C–H∙∙∙π Interactions C–H∙∙∙Py[1+x, 1+y, z], H∙∙∙Cg = 2.85, C–H∙∙∙Cg = 162, C∙∙∙Cg = 3.7689(15) C–H∙∙∙Ph[1+x, 1+y, z], H∙∙∙Cg = 2.86, C–H∙∙∙Cg = 167, C∙∙∙Cg = 3.8079(14) |
| Compound | Synthon | Details |
|---|---|---|
| 1 | NH3+∙∙∙−OOC | N∙∙∙O[2−x, 1−y, 2−z] = 2.740(3) |
| NH3+∙∙∙−OOC | N∙∙∙O[x, 1+y, z] = 2.817(3) | |
| OH∙∙∙−OOC | O∙∙∙O = 2.739(3) | |
| 2a | NH3+∙∙∙−OOC | N∙∙∙O = 2.8147(14) |
| NH3+∙∙∙−OOC | N∙∙∙O[0.5+x, 0.5−y, 0.5+z] = 2.8216(16) | |
| NH3+∙∙∙OH | N∙∙∙O[1−x, −y, 1−z] = 2.9409(14) | |
| OH∙∙∙−OOC | O∙∙∙O = 2.6178(12) | |
| 2b | NH3+∙∙∙−OOC | N∙∙∙O = 2.772(3) |
| NH3+∙∙∙−OOC | N∙∙∙O[1+x, y, z] = 2.948(2) | |
| NH3+∙∙∙−OOC | N∙∙∙O[1−x, 1−y, 1−z] = 3.008(2) | |
| NH3+∙∙∙N(quin−) | N∙∙∙N[1+x, y, z] = 3.080(3) | |
| OH∙∙∙−OOC | O∙∙∙O = 2.791(2) | |
| 3a | NH3+∙∙∙−OOC | N∙∙∙O = 2.7404(18) |
| NH3+∙∙∙−OOC | N∙∙∙O[−1+x, y, z] = 2.7768(18) | |
| NH3+∙∙∙−OOC | N∙∙∙O[1−x, 1−y, 1−z] = 2.7976(16) | |
| OH∙∙∙N(quin−) | O∙∙∙N[−1+x, y, z] = 2.8187(17) | |
| 3b | NH3+∙∙∙−OOC | N∙∙∙O[1−x, 2−y, 2−z] = 2.7307(13) |
| NH3+∙∙∙−OOC | N∙∙∙O[x, 1+y, z] = 2.8525(13) | |
| NH3+∙∙∙OH | N∙∙∙O[1−x, 2−y, 2−z] = 2.8148(12) | |
| OH∙∙∙−OOC | O∙∙∙O = 2.6222(12) |
| 1 | 2a | 2b | 3a | 3b | |
|---|---|---|---|---|---|
| NH3+∙∙∙−OOC | ✓ | ✓ | ✓ | ✓ | ✓ |
| OH∙∙∙−OOC | ✓ | ✓ | ✓ | ✓ | |
| NH3+∙∙∙N(quin−) | ✓ [a] | ✓ [a] | ✓ | ✓ [a] | |
| NH3+∙∙∙OH | ✓ | ✓ | |||
| OH∙∙∙N(quin−) | ✓ |
| 1 | 2a | 2b | 3a | 3b | |
|---|---|---|---|---|---|
| Empirical Formula | C13H16N2O3 | C14H18N2O3 | C14H18N2O3 | C14H18N2O3 | C14H18N2O3 |
| Formula Weight | 248.28 | 262.30 | 262.30 | 262.30 | 262.30 |
| Crystal System | triclinic | monoclinic | monoclinic | monoclinic | triclinic |
| Space Group | P−1 | P 21/n | P 21/n | P 21/n | P−1 |
| T (K) | 150.00(10) | 150.00(10) | 150.00(10) | 150.00(10) | 150.00(10) |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| a (Å) | 7.1378(16) | 12.1437(11) | 6.5579(4) | 6.5428(4) | 7.1342(4) |
| b (Å) | 7.5269(7) | 10.1451(5) | 10.2309(6) | 9.0723(4) | 8.4346(3) |
| c (Å) | 11.8314(14) | 12.2312(15) | 19.8329(13) | 23.1232(10) | 12.5059(7) |
| α (°) | 99.172(9) | 90 | 90 | 90 | 96.139(4) |
| β (°) | 95.916(14) | 119.527(14) | 97.837(6) | 93.835(5) | 105.187(5) |
| γ (°) | 90.647(13) | 90 | 90 | 90 | 104.829(4) |
| V (Å3) | 623.93(17) | 1311.2(3) | 1318.22(14) | 1369.48(12) | 689.74(6) |
| Z | 2 | 4 | 4 | 4 | 2 |
| Dcalc (g/cm3) | 1.322 | 1.329 | 1.322 | 1.272 | 1.263 |
| μ (mm−1) | 0.095 | 0.094 | 0.094 | 0.090 | 0.090 |
| Collected Reflections | 5349 | 11,880 | 6877 | 12,825 | 12,006 |
| Unique Reflections | 3186 | 3524 | 3413 | 3696 | 3689 |
| Observed Reflections | 1937 | 2780 | 2459 | 2528 | 2933 |
| Rint | 0.0587 | 0.0285 | 0.0233 | 0.0482 | 0.0224 |
| R1 (I > 2σ(I)) | 0.0878 | 0.0413 | 0.0671 | 0.0513 | 0.0429 |
| wR2 (all data) | 0.2559 | 0.1168 | 0.2000 | 0.1225 | 0.1274 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Podjed, N.; Modec, B. Hydrogen Bonding and Polymorphism of Amino Alcohol Salts with Quinaldinate: Structural Study. Molecules 2022, 27, 996. https://doi.org/10.3390/molecules27030996
Podjed N, Modec B. Hydrogen Bonding and Polymorphism of Amino Alcohol Salts with Quinaldinate: Structural Study. Molecules. 2022; 27(3):996. https://doi.org/10.3390/molecules27030996
Chicago/Turabian StylePodjed, Nina, and Barbara Modec. 2022. "Hydrogen Bonding and Polymorphism of Amino Alcohol Salts with Quinaldinate: Structural Study" Molecules 27, no. 3: 996. https://doi.org/10.3390/molecules27030996
APA StylePodjed, N., & Modec, B. (2022). Hydrogen Bonding and Polymorphism of Amino Alcohol Salts with Quinaldinate: Structural Study. Molecules, 27(3), 996. https://doi.org/10.3390/molecules27030996