
A Simple LC-MS/MS Method for the Quantification of PDA-66 in Human Plasma


 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Calibration Curve
2.2. Selectivity, Specificity and Carryover
2.3. Sensitivity, Accuracy and Precision
2.4. Matrix Effect and Recovery
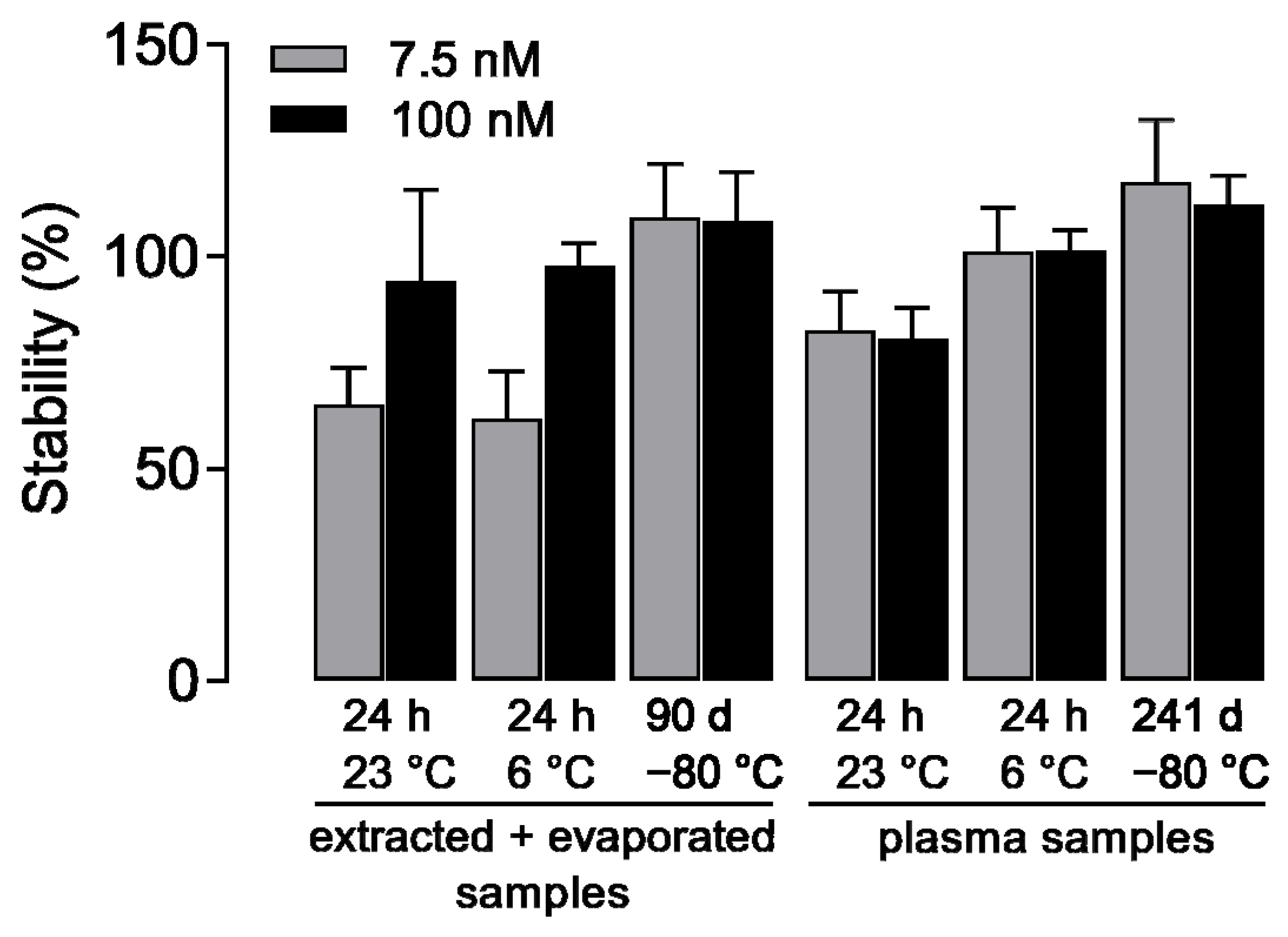
2.5. Stability
2.6. Quantification of PDA-66 in Mouse Plasma and Sera
3. Materials and Methods
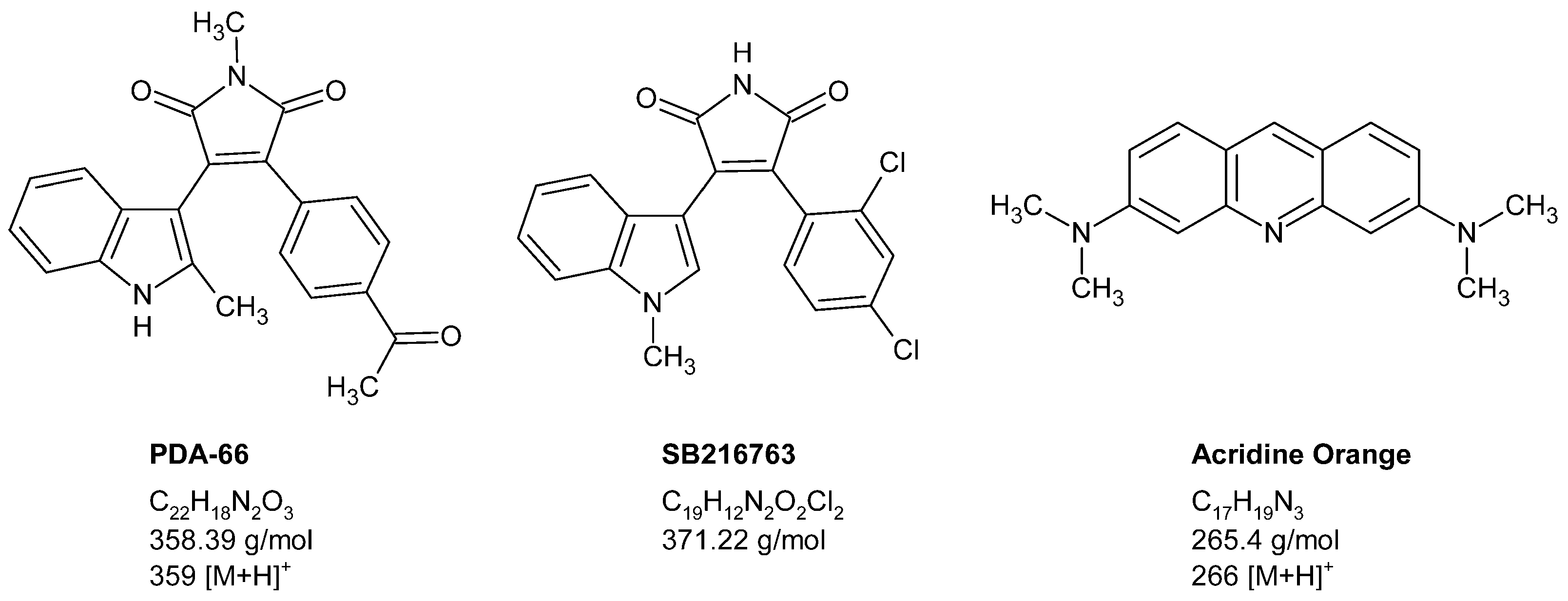
3.1. Chemical Reagents
3.2. Standard Preparation
3.3. LC-MS/MS Analysis
3.4. Validation
3.5. Preparation of Mouse Plasma and Sera
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Coghlan, M.P.; Culbert, A.A.; Cross, D.A.; Corcoran, S.L.; Yates, J.W.; Pearce, N.J.; Rausch, O.L.; Murphy, G.J.; Carter, P.S.; Roxbee Cox, L.; et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7, 793–803. [Google Scholar] [CrossRef]
- Pews-Davtyan, A.; Tillack, A.; Ortinau, S.; Rolfs, A.; Beller, M. Efficient palladium-catalyzed synthesis of 3-aryl-4-indolylmaleimides. Org. Biomol. Chem. 2008, 6, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Eisenlöffel, C.; Schmöle, A.C.; Pews-Davtyan, A.; Brennführer, A.; Kuznetsov, S.A.; Hübner, R.; Frech, S.; Schult, C.; Junghanss, C.; Beller, M.; et al. Interference of a novel indolylmaleimide with microtubules induces mitotic arrest and apoptosis in human progenitor and cancer cells. Biochem. Pharmacol. 2013, 85, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, C.; Roolf, C.; Langhammer, T.S.; Sekora, A.; Pews-Davtyan, A.; Beller, M.; Frech, M.J.; Eisenlöffel, C.; Rolfs, A.; Junghanss, C. The novel arylindolylmaleimide PDA-66 displays pronounced antiproliferative effects in acute lymphoblastic leukemia cells. BMC Cancer 2014, 14, 71. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, W.; Beck, J.; Schmidt, L.C.; Roolf, C.; Pews-Davtyan, A.; Rütgen, B.C.; Hammer, S.; Willenbrock, S.; Sekora, A.; Rolfs, A.; et al. Characterization of the novel indolylmaleimides PDA-66 and PDA-377 effect on canine lymphoma cells. Oncotarget 2016, 7, 35379–35389. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM). Bioanalytical Method Validation—Guidance for Industry. 2018. Available online: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf (accessed on 20 January 2022).
- Plant, N. Strategies for using in vitro screens in drug metabolism. Drug Discov. Today 2004, 9, 328–336. [Google Scholar] [CrossRef]
- Li, W.; Zhang, J.; Tse, F.L. Strategies in quantitative LC-MS/MS analysis of unstable small molecules in biological matrices. Biomed. Chromatogr. 2011, 25, 258–277. [Google Scholar] [CrossRef] [PubMed]
- Annesley, T.M. Ion suppression in mass spectrometry. Clin. Chem. 2003, 49, 1041–1044. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Used Concentration (nM) | Intra-Day (Day 1) | Intra-Day (Day 2) | Intra-Day (Day 3) | Inter-Day | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Measured Concentration (nM) | RE (%) | CV (%) | Measured Concentration (nM) | RE (%) | CV (%) | Measured Concentration (nM) | RE (%) | CV (%) | RE (%) | CV (%) | |
| 2.5 | 2.30 ± 0.2 | −8.04 ± 9.1 | 9.89 | 2.56 ± 0.2 | 2.33 ± 7.9 | 7.73 | 2.61 ± 0.2 | 4.45 ± 8.8 | 8.44 | −0.42 ± 9.8 | 9.82 |
| 7.5 | 6.78 ± 0.4 | −9.62 ± 4.7 | 5.20 | 7.26 ± 0.6 | −3.15 ± 8.3 | 8.54 | 7.68 ± 0.7 | 2.34 ± 8.9 | 8.66 | −3.48 ± 8.6 | 8.91 |
| 50 | 48.46 ± 4.5 | −3.08 ± 9.0 | 9.23 | 46.81 ± 2.3 | −6.38 ± 4.5 | 4.82 | 47.83 ± 2.4 | −4.35 ± 4.7 | 4.91 | −4.60 ± 6.1 | 6.37 |
| 100 | 96.64 ± 10.0 | −3.36 ± 10.0 | 10.32 | 109.62 ± 5.3 | 9.62 ± 5.3 | 4.79 | 102.68 ± 7.6 | 2.68 ± 7.6 | 7.37 | 2.98 ± 9.1 | 8.83 |
| PDA-66 | Acridine Orange | ||||
|---|---|---|---|---|---|
| Concentration (nM) | Recovery (%) | Matrix Effect (%) | Concentration (nM) | Recovery (%) | Matrix Effect (%) |
| 7.5 | 102.0 ± 6.8 | 31.0 ± 9.1 | 50 | 65.89 ± 5.3 | 21.4 ± 5.8 |
| 50 | 79.0 ± 7.4 | 28.0 ± 9.3 | |||
| 100 | 85.0 ± 6.7 | 28.3 ± 14.7 | |||
| Parameters for MS Instrument | |
|---|---|
| Capillary voltage | 3.5 kV |
| Source temperature | 120 °C |
| Desolvation temperature | 350 °C |
| Desolvation gas flow rate | 550 L h−1 |
| Cone gas flow rate | 50 L h−1 |
| Dwell times | 0.1 s |
| Delay times | 0.1 s |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarz, R.; Seiler, E.R.D.; Sender, S.; Pews-Davtyan, A.; Murua Escobar, H.; Zechner, D.; Beller, M.; Junghanß, C.; Hinz, B. A Simple LC-MS/MS Method for the Quantification of PDA-66 in Human Plasma. Molecules 2022, 27, 974. https://doi.org/10.3390/molecules27030974
Schwarz R, Seiler ERD, Sender S, Pews-Davtyan A, Murua Escobar H, Zechner D, Beller M, Junghanß C, Hinz B. A Simple LC-MS/MS Method for the Quantification of PDA-66 in Human Plasma. Molecules. 2022; 27(3):974. https://doi.org/10.3390/molecules27030974
Chicago/Turabian StyleSchwarz, Rico, Elisabeth R. D. Seiler, Sina Sender, Anahit Pews-Davtyan, Hugo Murua Escobar, Dietmar Zechner, Matthias Beller, Christian Junghanß, and Burkhard Hinz. 2022. "A Simple LC-MS/MS Method for the Quantification of PDA-66 in Human Plasma" Molecules 27, no. 3: 974. https://doi.org/10.3390/molecules27030974
APA StyleSchwarz, R., Seiler, E. R. D., Sender, S., Pews-Davtyan, A., Murua Escobar, H., Zechner, D., Beller, M., Junghanß, C., & Hinz, B. (2022). A Simple LC-MS/MS Method for the Quantification of PDA-66 in Human Plasma. Molecules, 27(3), 974. https://doi.org/10.3390/molecules27030974