
Disruption of Crystal Packing in Thieno[2,3-b]pyridines Improves Anti-Proliferative Activity




Abstract
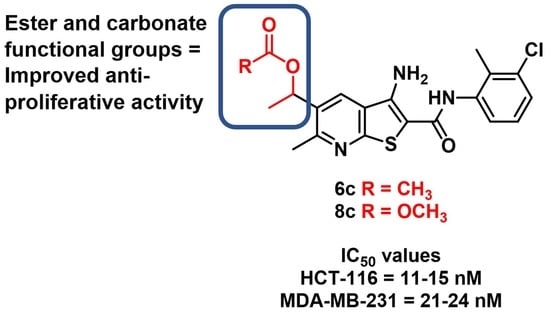
:
1. Introduction
2. Results and Discussion
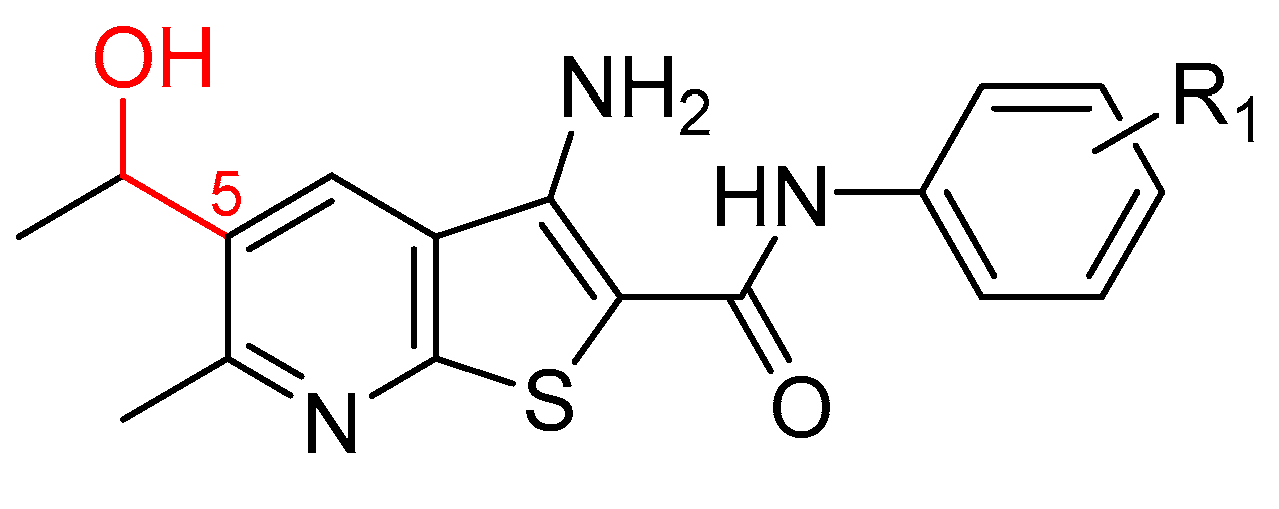
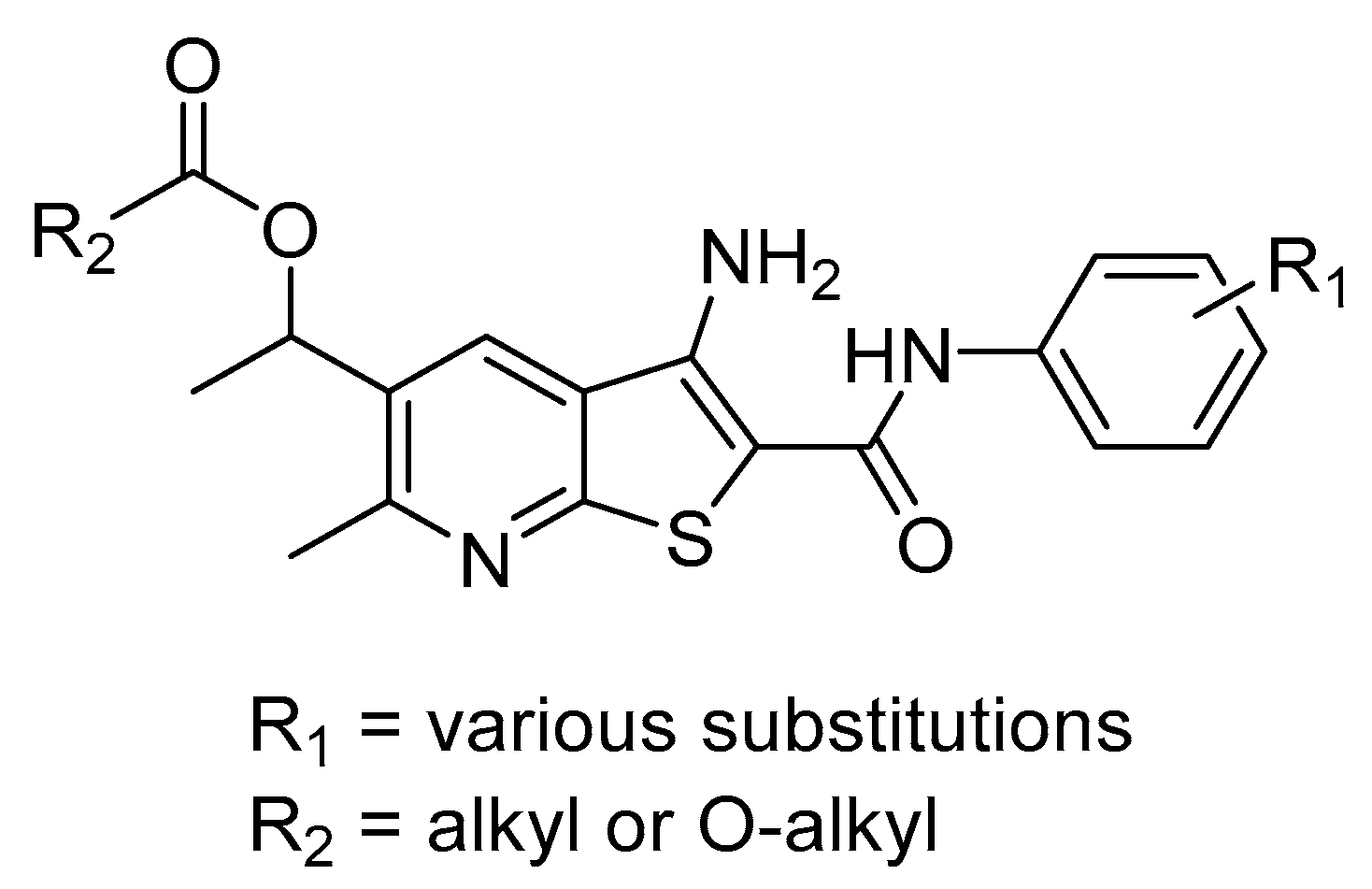
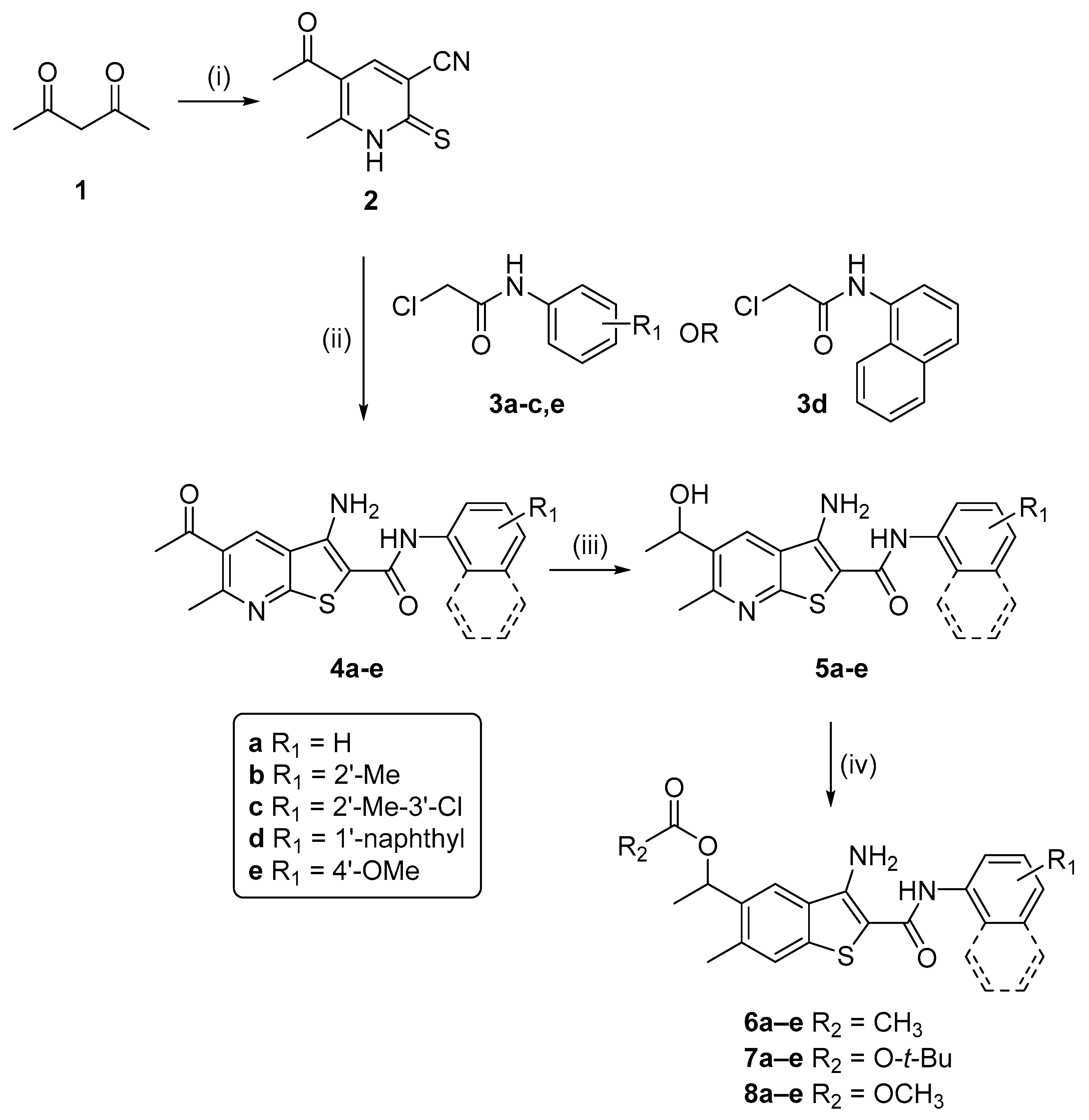
2.1. Synthesis of Targeted Compounds
2.2. Anti-Proliferative Activity
2.3. Molecular Modelling Study
2.4. Summary
3. Materials and Methods
3.1. Synthesis of the Compounds
3.2. Cell Proliferation Assay
3.3. Molecular Modelling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Haverkate, N.A.; van Rensburg, M.; Kumara, S.; Reynisson, J.; Leung, E.; Pilkington, L.I.; Barker, D. Improving the solubility of anti-proliferative thieno[2,3-b]quinoline-2-carboxamides. Bioorg. Med. Chem. 2021, 37, 116092. [Google Scholar] [CrossRef]
- Reynisson, J.; Court, W.; O’Neill, C.; Day, J.; Patterson, L.; McDonald, E.; Workman, P.; Katan, M.; Eccles, S.A. The identification of novel PLC-γ inhibitors using virtual high throughput screening. Bioorg. Med. Chem. 2009, 8, 3169–3176. [Google Scholar] [CrossRef]
- Leung, E.; Pilkington, L.I.; van Rensburg, M.; Jeon, C.Y.; Song, M.; Arabshahi, H.J.; De Zoysa, G.H.; Sarojini, V.; Denny, W.A.; Reynisson, J.; et al. Synthesis and cytotoxicity of thieno[2,3-b]quinoline-2-carboxamide and cycloalkyl[b]thieno[3,2-e]pyridine-2-carboxamide derivatives. Bioorg. Med. Chem. 2016, 5, 1142–1154. [Google Scholar] [CrossRef]
- Sala, G.; Dituri, F.; Raimondi, C.; Previdi, S.; Maffucci, T.; Mazzoletti, M.; Rossi, C.; Iezzi, M.; Lattanzio, R.; Piantelli, M.; et al. Phospholipase Cγ1 is required for metastasis development and progression. Cancer Res. 2008, 24, 10187–10196. [Google Scholar] [CrossRef] [Green Version]
- Reynisson, J.; Barker, D.; Jaiswal, J.K.; Denny, W.A.; Baguley, B.C.; D’Mello, S.A.N.; Leung, E.Y. Evidence that phospholipase C is involved in the antitumour action of NSC768313, a new thieno[2,3-b]pyridine derivative. Cancer Cell Int. 2016, 16, 18. [Google Scholar] [CrossRef] [Green Version]
- Leung, E.; Patel, J.; Hollywood, J.A.; Zafar, A.; Tomek, P.; Barker, D.; Pilkington, L.I.; van Rensburg, M.; Langley, R.J.; Helsby, N.A.; et al. Validating TDP1 as an Inhibition Target for the Development of Chemosensitizers for Camptothecin-Based Chemotherapy Drugs. Oncol. Ther. 2021, 2, 541–556. [Google Scholar] [CrossRef]
- Binsaleh, N.K.; Wigley, C.A.; Whitehead, K.A.; van Rensburg, M.; Reynisson, J.; Pilkington, L.I.; Barker, D.; Jones, S.; Dempsey-Hibbert, N.C. Thieno[2,3-b]pyridine derivatives are potent anti-platelet drugs, inhibiting platelet activation, aggregation and showing synergy with aspirin. Eur. J. Med. Chem. 2018, 143, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Zafar, A.; Sari, S.; Leung, E.; Pilkington, L.I.; Van Rensburg, M.; Barker, D.; Reynisson, J. GPCR modulation of thieno[2,3-b]pyridine anti-proliferative agents. Molecules 2017, 12, 2254. [Google Scholar] [CrossRef] [Green Version]
- Arabshahi, H.J.; van Rensburg, M.; Pilkington, L.I.; Jeon, C.Y.; Song, M.; Gridel, L.-M.; Leung, E.; Barker, D.; Vuica-Ross, M.; Volcho, K.P.; et al. A synthesis, in silico, in vitro and in vivo study of thieno[2,3-b]pyridine anticancer analogues. MedCheComm 2015, 11, 1987–1997. [Google Scholar] [CrossRef]
- Eurtivong, C.; Semenov, V.; Semenova, M.; Konyushkin, L.; Atamanenko, O.; Reynisson, J.; Kiselyov, A. 3-Amino-thieno[2,3-b]pyridines as microtubule-destabilising agents: Molecular modelling and biological evaluation in the sea urchin embryo and human cancer cells. Bioorg. Med. Chem. 2017, 2, 658–664. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Kimatrai Salvador, M.; Preti, D.; Aghazadeh Tabrizi, M.; Bassetto, M.; Brancale, A.; Hamel, E.; Castagliuolo, I.; Bortolozzi, R.; et al. Synthesis and biological evaluation of 2-(alkoxycarbonyl)-3-anilinobenzo[b]thiophenes and thieno[2,3-b]pyridines as new potent anticancer agents. J. Med. Chem. 2013, 6, 2606–2618. [Google Scholar] [CrossRef] [Green Version]
- Katritch, V.; Jaakola, V.-P.; Lane, J.R.; Lin, J.; Ijzerman, A.P.; Yeager, M.; Kufareva, I.; Stevens, R.C.; Abagyan, R. Structure-based discovery of novel chemotypes for adenosine A2A receptor antagonists. J. Med. Chem. 2010, 4, 1799–1809. [Google Scholar] [CrossRef] [Green Version]
- Markovic, M.; Ben-Shabat, S.; Dahan, A. Prodrugs for improved drug delivery: Lessons learned from recently developed and marketed products. Pharmaceutics 2020, 11, 1031. [Google Scholar] [CrossRef]
- Riccardi, C.; Musumeci, D.; Irace, C.; Paduano, L.; Montesarchio, D. Ru III Complexes for anticancer therapy: The importance of being nucleolipidic. Eur. J. Med. Chem. 2017, 2017, 1100–1119. [Google Scholar]
- Zafar, A.; Pilkington, L.I.; Haverkate, N.A.; van Rensburg, M.; Barker, D.; Reynisson, J.; Leung, E.; Kumara, S.; Denny, W.A.; Alsuraifi, A.; et al. Investigation into improving the aqueous solubility of the thieno[2,3-b]pyridine anti-proliferative agents. Molecules 2018, 1, 145. [Google Scholar] [CrossRef] [Green Version]
- Leung, E.; Hung, J.M.; Barker, D.; Reynisson, J. The effect of a thieno[2,3-b]pyridine PLC-γ inhibitor on the proliferation, morphology, migration and cell cycle of breast cancer cells. MedChemComm 2014, 1, 99–106. [Google Scholar] [CrossRef]
- Rautio, J.; Meanwell, N.A.; Di, L.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018, 8, 559–587. [Google Scholar] [CrossRef]
- Stella, V.J. Prodrugs: My initial exploration and where it led. J. Pharm. Sci. 2020, 12, 3514–3523. [Google Scholar] [CrossRef]
- van Rensburg, M.; Leung, E.; Haverkate, N.A.; Eurtivong, C.; Pilkington, L.I.; Reynisson, J.; Barker, D. Synthesis and antiproliferative activity of 2-chlorophenyl carboxamide thienopyridines. Bioorg. Med. Chem. Lett. 2017, 2, 135–138. [Google Scholar] [CrossRef]
- Haverkate, N.A.; Leung, E.; Pilkington, L.I.; Barker, D. Tethered aryl groups increase the activity of anti-proliferative thieno[2,3-b]pyridines by targeting a lipophilic region in the active site of PI-PLC. Pharmaceutics 2021, 12, 2020. [Google Scholar] [CrossRef]
- Haghnavaz, N.; Asghari, F.; Elieh Ali Komi, D.; Shanehbandi, D.; Baradaran, B.; Kazemi, T. HER2 positivity may confer resistance to therapy with paclitaxel in breast cancer cell lines. Artif. Cells Nanomed. Biotechnol. 2018, 3, 518–523. [Google Scholar] [CrossRef]
- Wen, S.-H.; Su, S.-C.; Liou, B.-H.; Lin, C.-H.; Lee, K.-R. Sulbactam-enhanced cytotoxicity of doxorubicin in breast cancer cells. Cancer Cell Int. 2018, 18, 128. [Google Scholar] [CrossRef] [Green Version]
- Arabshahi, H.J.; Leung, E.; Barker, D.; Reynisson, J. The development of thieno[2,3-b]pyridine analogues as anticancer agents applying in silico methods. MedChemComm 2014, 2, 186–191. [Google Scholar] [CrossRef]
- Hung, J.M.; Arabshahi, H.J.; Leung, E.; Reynisson, J.; Barker, D. Synthesis and cytotoxicity of thieno[2,3-b]pyridine and furo[2,3-b]pyridine derivatives. Eur. J. Med. Chem. 2014, 86, 420–437. [Google Scholar] [CrossRef]
- Haverkate, N.A. Synthesis and Investigation of Anti-Proliferative Thieno[2,3-b]pyridine Derivatives with Enhanced Solubility; University of Auckland: Auckland, New Zealand, 2017. [Google Scholar]
- Abu-Shanab, F.A.; Redhouse, A.D.; Thompson, J.R.; Wakefield, B.J. Synthesis of 2,3,5,6-tetrasubstituted pyridines from enamines derived from N,N-dimethylformamide dimethyl acetal. Synthesis 1995, 5, 557–560. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean Relative Growth of 1 μM in Cancer Cell Line (%) | IC50 (nM) | |||
|---|---|---|---|---|
| HCT-116 | MDA-MB-231 | HCT-116 | MDA-MB-231 | |
| 5a * | 95.3 | 107.4 | - | - |
| 5b * | 99.9 | 97.9 | - | - |
| 5c * | 2.2 | 11.7 | 72 | 76 |
| 5d * | 1.7 | 9.9 | 171 | 81 |
| 5e * | 97.5 | 114.4 | - | - |
| 6a | 0.8 | 8.0 | 224 | 274 |
| 6b | 1.7 | 11.7 | 387 | 448 |
| 6c | 0.2 | 4.6 | 11 | 24 |
| 6d | 3.1 | 7.1 | - | - |
| 6e | 70.1 | 77.0 | - | - |
| 7a | 4.0 | 24.6 | 667 | 768 |
| 7b | 7.8 | 33.0 | 776 | 791 |
| 7c | 0.7 | 5.7 | 61 | 90 |
| 7d | 5.4 | 7.3 | 473 | 449 |
| 7e | 94.8 | 96.4 | - | - |
| 8a | 7.0 | 15.2 | - | - |
| 8b | 11.1 | 16.7 | - | - |
| 8c | 0.2 | 3.1 | 15 | 21 |
| 8d | 0.2 | 3.1 | 24 | 32 |
| 8e | 101.1 | 107.0 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haverkate, N.A.; Leung, E.; Pilkington, L.I.; Barker, D. Disruption of Crystal Packing in Thieno[2,3-b]pyridines Improves Anti-Proliferative Activity. Molecules 2022, 27, 836. https://doi.org/10.3390/molecules27030836
Haverkate NA, Leung E, Pilkington LI, Barker D. Disruption of Crystal Packing in Thieno[2,3-b]pyridines Improves Anti-Proliferative Activity. Molecules. 2022; 27(3):836. https://doi.org/10.3390/molecules27030836
Chicago/Turabian StyleHaverkate, Natalie A., Euphemia Leung, Lisa I. Pilkington, and David Barker. 2022. "Disruption of Crystal Packing in Thieno[2,3-b]pyridines Improves Anti-Proliferative Activity" Molecules 27, no. 3: 836. https://doi.org/10.3390/molecules27030836
APA StyleHaverkate, N. A., Leung, E., Pilkington, L. I., & Barker, D. (2022). Disruption of Crystal Packing in Thieno[2,3-b]pyridines Improves Anti-Proliferative Activity. Molecules, 27(3), 836. https://doi.org/10.3390/molecules27030836