Abstract
Lung cancer has a high prevalence, with a growing number of new cases and mortality every year. Furthermore, the survival rate of patients with non-small-cell lung carcinoma (NSCLC) is still quite low in the majority of cases. Despite the use of conventional therapy such as tyrosine kinase inhibitor for Epidermal Growth Factor Receptor (EGFR), which is highly expressed in most NSCLC cases, there was still no substantial improvement in patient survival. This is due to the drug’s ineffectiveness and high rate of resistance among individuals with mutant EGFR. Therefore, the development of new inhibitors is urgently needed. Understanding the EGFR structure, including its kinase domain and other parts of the protein, and its activation mechanism can accelerate the discovery of novel compounds targeting this protein. This study described the structure of the extracellular, transmembrane, and intracellular domains of EGFR. This was carried out along with identifying the binding pose of commercially available inhibitors in the ATP-binding and allosteric sites, thereby clarifying the research gaps that can be filled. The binding mechanism of inhibitors that have been used clinically was also explained, thereby aiding the structure-based development of new drugs.
1. Introduction
Epidermal Growth Factor Receptor (EGFR) is one of the receptor tyrosine kinases (RTKs), members of the ErbB/HER family which consists of ErbB1 (EGFR or HER1), ErbB2 (HER2 or Neu), ErbB3 (HER3), and ErbB4 (HER4). They share similar basic structures such as an extracellular ligand binding, an α-helix transmembrane, a cytoplasmic tyrosine kinase (except ErbB3), and carboxy-terminal signaling domains [1]. The growth factors of the ErbB family are divided into three groups, namely the specific ligand of ErbB1 or EGFR (such as EGF, TGF-α, and amphiregulin), those that bind to ErbB1 and ErbB4 (such as heparin-binding EGF, betacellulin, and epiregulin), and ErbB3/ErbB4 (such as neuregulins and heregulins) [1]. However, unlike the others, ErbB2 has no specific ligand. It binds to any ligand similar to the one that can activate it and form a dimer [2].The ligand-binding process induces homodimerization and heterodimerization with other ErbB family members and activates the tyrosine kinase domain [1]. The activation of this intracellular domain causes autophosphorylation and enables it to interact with signaling components to downstream signaling pathways. Furthermore, this was performed through the RAS-RAF-MEK-MAPK, PI3KPTEN-AKT, and STAT pathways [3,4] to enhance cell proliferation and inhibit apoptosis (Figure 1) [5].
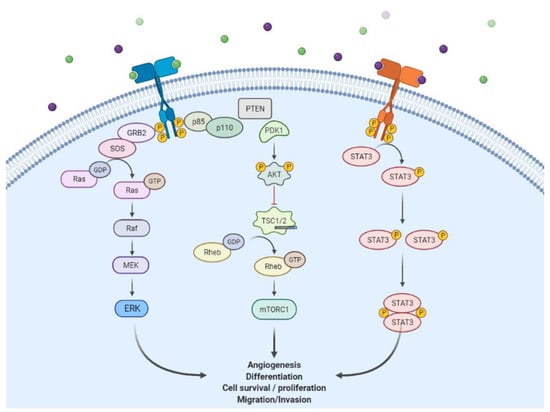
Figure 1.
HER/ErbB signaling (created by BioRender.com on 14 October 2021). Green and purple spheres indicate the EGFR-activating ligands.
EGFR signaling was observed in most lung cancer cases. Overexpression of this receptor has been identified in 40 to 89% of NSCLC cases, with the highest and lowest rates in squamous tumors and adenocarcinoma [6,7]. Besides its high rate in NSCLC, the level of EGFR activation is also related to bad prognosis and tumor regression rate [8]. The phosphorylation activity in the kinase domain of EGFR may also be irregulated by several mechanisms, including EGFR mutation and overexpression, which is commonly found in tumor cells [9]. This indicates that improper activation of tyrosine kinase promotes tumor progression and inhibits cell apoptosis [10]. Spontaneous oligomerization was also reported in several studies for mutated EGFR which lacked part of residues in the extracellular domain and induced autophosphorylation [11,12].
EGFR also interacts with the integrin pathway and triggers matrix metalloproteinases to alter and stimulate cell adhesion, motility and invasion, and promote metastases [13,14,15]. Furthermore, the overexpression or enhanced activation of EGFR mutations occurs in several NSCLC cases, leading to constitutive TK activity. This makes it a rational target for therapeutic intervention and also promotes the development of novel anticancer agents targeting EGFR.
2. Structure of EGFR
2.1. Extracellular Domain
The human EGFR encodes 1210 amino acids with a molecular mass of approximately 134 kDa [16]. It is located in the 7p12-14 region of chromosome 7, which consist of 28 exons (Figure 2a) [17]. The first 24 amino acids are the signal peptide of this protein that are often excluded in the structural numbering.
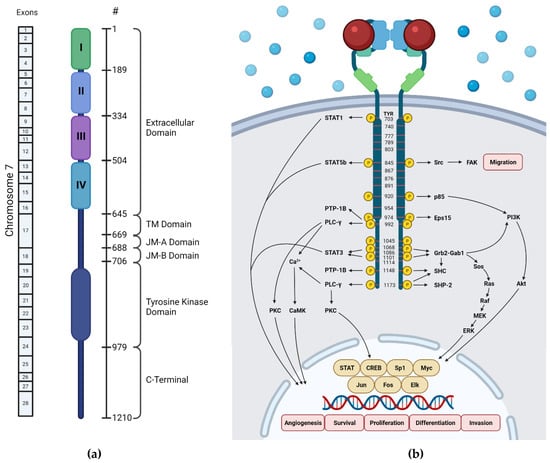
Figure 2.
Schematic diagrams of EGFR domains. (a) Domain structure of human EGFR and exons encoding it (created by BioRender.com on 14 October 2021), (b) EGFR phosphorylation sites [18]. Blue spheres indicate the molecules present outside the cell, and red spheres indicate the EGFR-activating ligand.
The extracellular ligand-binding domain of EGFR contains 620 amino acids (25-645) which are divided into four subdomains, namely I (L1), II (CR1), III (L2), and IV (CR2). L1 and L2 are both leucine-rich domains with a β-helix structure, and are responsible for growth-factor binding [19]. Both CR1 and CR2 are cysteine-rich regions with disulfide bonds. CR1 plays a role in the homo- or hetero-dimer formation of EGFR with other ErbB family members [19,20,21]. L1, CR1, and L2 form a C shape which accommodate EGF to bind between L1 and L2. Based on the interaction of EGFR in the binding site, three sites were defined in both subdomains (Figure 3). The B loop of EGF interacts with site 1 of L1 by hydrophobic interaction with Leu14, Tyr45, Leu69, and Leu98, and hydrogen bonds to residue 16 to 18 which forms a parallel β-sheet. The A loop of EGF hydrophobically interacts with Val350 and Phe357 on site 2 of L2, while Arg41 of EGF forms a salt bridge with Asp355 in L2. The C-terminal region of EGF forms a hydrophobic interaction with Leu382, Phe412, and Ile438 as third site of L2. The residues Gln43 and Arg45 are also able to form hydrogen bonds with the side chain of Gln384. Furthermore, in the dimeric EGF–EGFR complex, the two EGF ligands are located on the opposite side of the dimer which are 79 Å apart from each other [21].
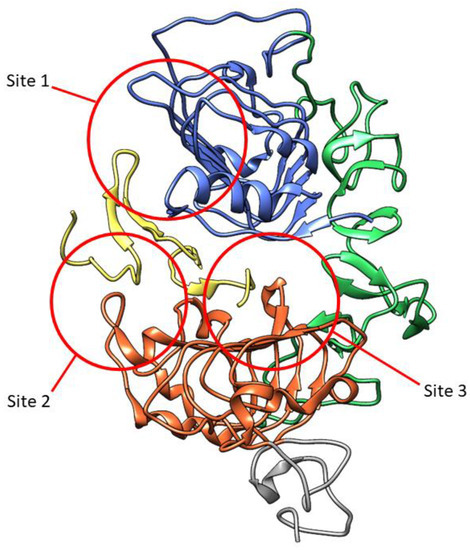
Figure 3.
Three-dimensional visualization of EGFR extracellular domain in complex with EGF (yellow) (PDB ID: 1IVO). Subdomains are marked in colors: L1, blue; CR1, green; L2, orange; part of CR2, grey. The three sites interacting with EGF are marked in red circles.
2.2. Transmembrane Domain
The transmembrane (TM) domain consists of ~22 amino acids (646–668) which connect the extracellular and intracellular domain of EGFR. Previous studies revealed the critical role of the TM domain in the allosteric modulation of EGFR by two activation pathways that involve pivoting and rotational motion of the TM helices [22,23]. The three-dimensional structure of the TM domain in the presence of the juxtamembrane domain shows interhelix contact at the N-terminal small-X3-small motif, which is experimentally stable [22,24]. Furthermore, the lipid environment of TM helix dimers influences the stability of certain conformations, as shown in the Coarse-Grained MetaDynamics (CG-MetaD) simulation conducted by Lelimousin et al. (2016). It was found that the thickness of the bilayer can determine the motions of the TM helix, thereby affecting the stability and receptor activation [22,25,26,27,28,29].
2.3. Juxtamembrane Domain
This links the C-terminus of the TM domain to the kinase domain of EGFR and plays important role in its dimerization and activation [30,31,32]. The juxtamembrane domain is a flexible region that is often absent from crystal structures. Furthermore, interdisciplinary approaches using X-ray crystallography and NMR were successful in determining high-resolution JM domain structures which were also useful in silico study [33,34]. Even though it is a short region in EGFR, containing only ~37 residues, it consists of lysosomal and basolateral sorting motifs [35,36], a nuclear localization sequence [37], calmodulin-binding site [38], protein kinase C [39,40], and MAPK phosphorylation sites [32,41]. A study revealed that mutation of Thr654 within the JM domain caused an increase in kinase phosphorylation. It was also found that the phosphorylation of Thr-654 diminishes the ability of this region to modulate the receptor activation [32].
2.4. Kinase Domain
The EGFR tyrosine kinase (TK) domain consists of an NH2-terminal lobe (N-lobe) which comprises five β-sheet strands (β1-5) and one αC-helix spanning from residue 729 to 744, and a larger COOH-terminal lobe (C-lobe) comprising five α helices (αE, αF, αG, αH, and αI). The ATP-binding site is located in a cleft between the two lobes, beneath a highly conserved glycine-rich phosphate-binding loop that links β1 and β2 in the N-lobe [42]. The glycine-rich loop coordinates closely with the phosphates of ATP via backbone interactions [43].
In the active state, the conserved glutamate Glu738 in the αC helix forms an ion pair with Lys721 in β3 that interacts with the phosphate groups of ATP. The C-lobe surrounds the ATP-binding cleft from below and contributes to a highly conserved catalytic loop (Asp812-Asn818). Furthermore, the Asp812 interacts with the attacking hydroxyl side chain of the tyrosine substrate, while Asn818 forms hydrogen bond interactions that orient Asp812. The C-lobe also contributes to the regulatory activation loop Asp831-Val852 which has a conserved Asp-Phe-Gly (DFG) motif as its base [44].
2.5. C-Terminal
According to kinetic studies, the C-terminal domain of EGFR consists of 229 amino acids (residue 982–1210) and plays an important role in regulating receptor activation by suppressing kinase activity in the absence of autophosphorylation [45,46]. This domain is a proline-rich residue that has phosphorylation sites [1]. Due to its flexible structure, it is often disordered in the published crystal structures of EGFR, especially the polypeptide chain (residue 990–1005) which is highly fluctuated in MD simulation [47]. It has a proximal tail that is important in the autoinhibition activity of the receptor and has been studied structurally [48,49]. Residue 997–1001 forms an α-helix structure called AP-2 helix because it interacts with clathrin-associated protein complex AP-2 [50]. By interacting with the N-lobe of the second kinase in the dimer, this helix maintains two kinase domains in an inactive dimer [51,52]. The AP-2 helix is followed by a hook spanning from residue 1003 to 1022, which are acidic and interact with the hinge region of the kinase. This hook mediates an inhibitory dimerization of the receptor by electrostatic interaction, thus it is called an electrostatic hook. However, these interactions are destabilized by phosphorylation [49]. The last part of this hook forms a β-strand that prevents the formation of the JM latch [48]. The deletion of the distal residue Tyr1210, which is also part of the NPXY motif, significantly reduces the phosphorylation of Tyr869 in the kinase domain [48]. Therefore, these factors conclude that Tyr1210 is important for kinase activation.
3. Active–Inactive Conformation
Kinase is an enzyme that catalyzes phosphorylation processes. It facilitates the transfer of phosphate groups from a phosphate donor such as ATP to a specific substrate. There are more than 500 protein kinases encoded by the human genome, which have a highly conserved catalytic domain formed by α-helix C-terminal and β-strand N-terminal lobes as ATP-binding sites [53]. The activation loop within those kinases contains tyrosine, threonine, or serine that is phosphorylated and regulates kinase activity [54]. Two regions are commonly used to indicate the confirmation of active and inactive kinase, they are the αC helix and DFG motif in the activation segment. Furthermore, the kinase and αC helix located at residue 753–767 in EGFR are twisted inward against the N-lobe and towards the active site. This conformation shortens the distance between Glu762 of αC helix and Lys745 of β3 strands, allowing a salt-bridge formation and further interaction with α- and β-phosphate groups of ATP [55].
The activation segment of EGFR is located at residue 855-884. The Asp-Phe-Gly (DFG) motif at the beginning of the activation loop plays an important role in protein catalysis [56]. This DFG motif shows conformation in the active kinases where the aspartate is pointing to the ATP-binding site, allowing the magnesium ion to bind to the β- and γ- phosphate groups of ATP, which are known as DFG-in conformation [57]. However, this DFG motif flips outward in the inactive kinases, causing the aspartate to no longer coordinate the magnesium ion at the catalytic site, which is further known as DFG-out conformation [58]. An alteration of aspartate and phenylalanine orientation is found in this out form, where phenylalanine occupies the in-position of aspartate [55]. Based on the mutagenesis study, a residue was proposed to influence the sensitivity of kinases towards inhibitors that stabilized DFG-out conformation. It acts as “gatekeeper” since it has a bulky size and occupies the position that prevents ATP or other molecules from accessing the inner hydrophobic pocket [59]. Moreover, the “gatekeeper” residue Tyr790 mutation to methionine has been known to cause resistance to inhibitors due to the enhanced affinity for ATP [60].
In a study by Zhao et al., (2019), EGFR kinase conformations are classified into six classes which consist of two classes of active conformation and four classes of inactive conformation (Figure 4). In all classes, the side chain of aspartate has three different positions, namely DFG-in, DFG-out, and DFG-½in. Unlike the others, the side chain of Asp points to the upside [61]. The αC helix also shows three states, namely in, out, and in between or ½out. The first two classes of active kinase have DFG-in conformation with the αC helix in the in and ½out states. In the inactive EGFR, the DFG motifs have three different positions, while all the αC helices are in the out positions [62]. These various conformations of the EGFR kinase binding site provide essential information for all possible sites outside the ATP-binding site in order to be a benchmark for the discovery of novel inhibitors. The binding pocket sizes of Class 1-5 were not significantly different and ranged from 950 to 1119 Å3. However, the volume was significantly increased in Class-6 conformation to 1913 Å3, allowing it to bind with various ligands [62].
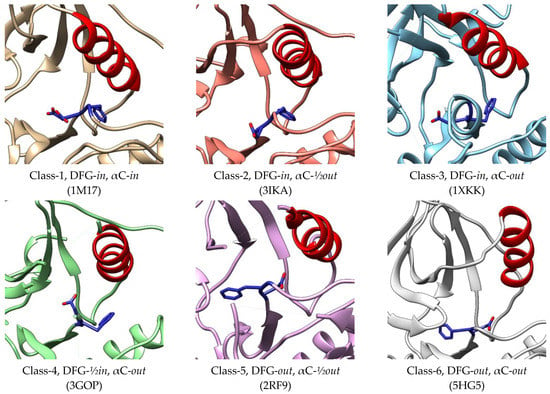
Figure 4.
Conformations of active and inactive EGFR classified by Zhao et al. (2019) [62]. The crystal structures 1M17 [42] and 3IKA [63] represent class-1 and class-2 of the active conformations, while 1XKK [64], 3GOP [30], 2RF9 [65], and 5HG5 [66] represent class-3, class-4, class-5, and class-6 of the inactive conformations, respectively.
There are other common, major regions in the kinase receptors beside the αC helix and activation loop, namely the N-lobe, ATP binding and allosteric sites, and the hinge region. The N-lobe consists of one α-helix and five antiparallel β-sheets, and it is connected to the C-lobe via a hinge region in the ATP-binding site [67]. The ligand binding causes conformational changes in this protein structure and enables autophosphorylation. In the structural view, the C-lobe also shrinks and moves closer to the N-lobe during phosphorylation and activates the kinase receptor [68].
4. Mutation of EGFR and Resistance Mechanism
Somatic mutations of EGFR tyrosine kinase domain in NSCLC patients were first reported in 2004, in which there was a deletion in exon 19 and point mutations in exon 21 [6,69]. According to the type of nucleotide changes, the EGFR mutations are grouped into three classes. Class I mutations involve short deletions that result in the loss of four to six residues between E746 to S752 encoded by exon 19. Class II mutations involve substitution of a single residue which occurs between exon 18 to 21. Class III mutations include duplications or insertions that mostly occur in exon 20 [70,71]. Most tyrosine kinase domain mutations (85–90%) had a deletion in exon 19 and substitution of L858R in exon 21. Recent studies have also reported a rare exon 22 mutation (E884K) that may reduce sensitivity to different EGFR inhibitors [72]. Several mutations at the cytoplasmic region cause destabilization of the conformation, upregulation of kinase activity, and irregularly promote downstream signaling pathways by avoiding cell apoptosis [73,74,75].
The mutated EGFR shows a resistance mechanism to the inhibitor used clinically and limited drug efficacy. This was found in lung cancer patients with substitution of threonine in the position of 790 to methionine (T790M) [76,77,78]. Half of all cases of resistance to the first-generation EGFR kinase inhibitor were caused by the T790M mutation [79,80]. Thr790 is also referred to as a “gatekeeper” residue because it is located in the entrance of the hydrophobic pocket of the ATP-binding pocket which determines the specificity of the inhibitor in the kinases. Substitution of Thr790 to a bulkier residue such as methionine causes steric hindrance for inhibitor binding [76,77,78].
Though this mutation had resistance to gefitinib and erlotinib, it is still sensitive to irreversible inhibitors such as EKB-569 and HKI-272 [77,78,81,82]. Despite the fact they share the same quinazoline core and aniline group as the first-generation inhibitors, they also contain a crotonamide as a Michael-acceptor group that is able to form a covalent bond with Cys797 at the ATP-binding site [60]. This prompted the investigation into why the irreversible inhibitors are able to interact with the binding pocket even though it has an aniline group that caused steric hindrance for gefitinib. However, this has been discovered by a study that revealed the resistance mechanism of this secondary mutation. The T790M substitution increases the ATP affinity to the binding pocket and allows it to compete with the inhibitors [BioRender object] resulting in a resistance mechanism [60].
The second mutation occurred in about half of the patients that were treated with EGFR-TKIs, despite the fact that occurrence before therapy was uncommon. Furthermore, the T790M mutation is also found in combination with the others that mostly coexist with an L858R point mutation [83,84]. The double mutation L858R/T790M activates EGFR and reduces ATP’s affinity to the binding site at the same time. The L858R/T790M mutation is more resistant to EGFR TKIs compared to the single L858R mutation or exon-19 deletion in the NSCLC cell line [76,85,86].
The other non-T790M mutation that was first discovered is D761Y, which commonly coexists with an L858R mutation. The L858R/D761Y mutation was more resistant to gefitinib compared to the cell with L858R alone, although it was still less resistant than L858R/T790M. This mutation was found to be less sensitive to the irreversible inhibitor, HKI-272, compared to the gefitinib response, which suggests that different mutations of EGFR will cause different responses to the reversible and irreversible inhibitors [80]. Though the resistance mechanism of this mutation is still unclear, it induces a conformational change between active and inactive states of EGFR and affects the binding ability of the inhibitor [80,87]. Recently, an L858R/T854A mutation was also discovered in a NSCLC patient that received long-term treatment of first-generation TKIs [88]. This mutation was resistant to erlotinib, showing a 3-fold increase in erlotinib concentration compared to the L858R single mutation. Furthermore, L858R/T854A was still more sensitive to erlotinib than L858R/T790M by more than 300-fold, and had more resistance than L858R [89]. Recently, a list of rare mutations of EGFR exons 18–21 in NSCLC patients has been reported [90]. Some that were discovered were not listed in the COSMIC database and not catalogued in cobas or IdyllaTM kit, such as L747_A750delinsNRQG, A763_Y764insLQEA, N771delinsHH, D770_N771insGV [91], W817X [92,93], K823E [94], and G857Efs*40 [90]. Furthermore, one of the 1228 patients tested positive for the K823E mutation. It was found that this mutation was able to significantly reduce EGFR activity by decreasing phosphorylation [90,94].
EGFR and its signaling elements are used as targets for the development of new anticancer molecules, such as monoclonal antibodies panitumumab and cetuximab, and tyrosine kinase inhibitors (TKIs), namely gefitinib, erlotinib, afatinib, and osimertinib [95,96,97,98,99]. However, the effectiveness of TKIs has been limited due to the resistance caused by EGFR mutations, and they need further development [98].
5. ATP-Binding Site
The ATP-binding site is highly conserved in kinase receptors. Therefore, to develop a more potent inhibitor of EGFR, it is necessary to understand the kinase–inhibitor interaction in the molecular level [99]. Based on structural analysis using several published EGFR tyrosine kinase structures in relation to ligands located at the ATP-binding pocket, 39 residues were found to be close to the binding site and were located in β-sheets, hinge regions, and α-helix. Residues Leu718, Val726, Ala743, Met793, and Leu844 have the most frequent contact in the crystal structures, which are located at β1-3, β-6, and the hinge region. These residues form a core binding pocket that is highly hydrophobic and conserved [62].
Zhao et al. (2019) has grouped the binding modes of all cocrystallized ligands into six clusters by hierarchical cluster analysis. Most of the ligands fall into cluster-1, which showed that the ligands are located at the ATP-binding site and form hydrogen bonds with the amino acids located in the hinge region. The ligands have no interaction with αC-helix. However, the inactive state of EGFR is observed in this cluster, which belongs to the class-6 conformation as described in Section 3. In addition to having a comparable ATP-competitive binding mechanism to cluster-1, the ligand in cluster-3 features a hydrophobic group that penetrates deeply into the hydrophobic pocket at the back of the ATP-binding site, which is often targeted to increase inhibitor selectivity [62]. However, the mutation of the “gatekeeper” residues that is commonly observed in NSCLC patients has a direct effect on the effectiveness of this cluster-3 inhibitor (see Section 4) [60]. The ligands in cluster-4 bind to the hydrophobic and allosteric sites instead of the ATP-binding site, in order not to have a non-ATP-competitive property. Crystal structures of this cluster showed the out conformation for the αC-helix, and opened the allosteric site in order for the ligands to interact with the DFG motif in activation loop as well as the αC-helix, and to provide allosteric modulation that has high selectivity to the mutated “gatekeeper” EGFR [100].
The phenyl groups of ligands in cluster-5 interact with the hydrophobic pocket, while the tails interact with aspartate in the DFG-in motif, and Asp842 at β-6. This is similar to other ligands positioned in cluster-2. However, their tail interacts with phenylalanine of the DFG-out motif and the G-rich loop of the protein. The furan group of ligands in cluster-6 has different characteristic by forming hydrophobic interactions with the pocket as well as hydrogen bonds with Lys745, Gly762, and the DFG motif [62].
6. Allosteric Site
There have been recent developments in compounds targeting the allosteric site in order to produce an alternative medicine that might solve the resistance issue to drugs used in current therapy [101]. Furthermore, the ligands binding to this site, which are also called allosteric modulators of EGFR, lead to conformational changes that may enhance the protein activity and orthosteric ligand binding, or vice versa [102,103,104]. They are divided into three types based on their properties, namely positive (PAM), neutral, and negative allosteric modulators (NAM) [103,105]. These modulators are able to tune the protein kinase activity by interfering with the dynamic interconversion between active and inactive states. In EGFR, the allosteric site of the tyrosine kinase domain is located in the inner pocket of the ATP-binding site. A crystal structure of the EGFR tyrosine kinase domain with an allosteric inhibitor called EAI001 is released into the Protein Data Bank along with the PDB ID of 5D41. Figure 5 shows that EAI001 binds to the allosteric pocket in close proximity to the αC-helix, and is able to inhibit EGFR activation without blocking ATP binding [100,106]. The binding mode of allosteric modulators was also shown along with ligands in cluster-3 and cluster-4, which were mentioned in the previous section.
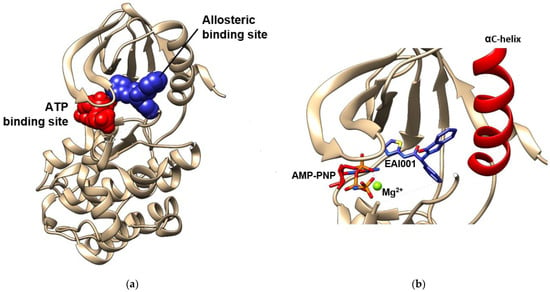
Figure 5.
ATP and allosteric binding site of EGFR TK domain. (a) Allosteric binding site is marked by blue spheres, while ATP-binding site is marked by red spheres. (b) The close visualization of the allosteric inhibitor EAI001 and AMP-PNP in the binding pocket. EAI001 binds to the allosteric site close to αC-helix. The visualizations are made using the crystal structure with PDB code 5D41 [100] by Chimera 1.15 (accessed on 13 August 2021).
This binding aims to prevent autophosphorylation and conformational changes of the kinase domain and achieve equilibrium [107]. Therefore, an allosteric modulator can be used to maintain the stability of the interaction between the inhibitor and the kinase ATP-binding pocket. This approach can be applied to solve the drug resistance issue that is often encountered in current treatment because of the inability of TKI inhibitors to bind to mutated EGFR.
7. EGFR Tyrosine Kinase Inhibitors
Several advances have been made in recent decades since the identification of activating mutations in the EGFR tyrosine kinase domain in NSCLC patients responding to the first-generation tyrosine kinase inhibitor (TKI) gefitinib, and first-line treatment with EGFR TKIs is now a well-established option in advanced EGFR-mutated NSCLC patients. Various EGFR TKIs have been developed, and some agents have been authorized in these selected patients (Figure 6). Osimertinib, the third-generation EGFR TKI used as the current first-line treatment for NSCLC patients who are EGFR-positive, showed disease-free survival (DFS) improvement in the overall population compared to first-generation EGFR TKIs. Osimertinib underwent phase I trial in 2013 and was approved by the FDA two years later to be used for EGFR T790M-positive NSCLC patients. In 18 April 2018, AstraZeneca announced that osimertinib was approved as the first-line treatment for patients with mutated EGFR (exon 19 deletion or exon 21 L858R substitution) [108]. In contrast, olmutinib, which was developed before Osimertinib, was withdrawn several months after its approval in South Korea due to the occurrence of Stevens–Johnson syndrome and consequent patient death [109].
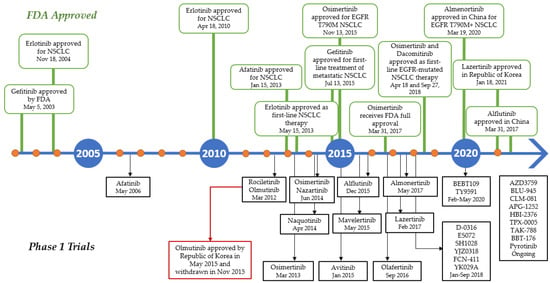
Figure 6.
Timeline in the development of EGFR TKIs during the last two decades. Green boxes above the line indicates the drugs approved by FDA, while the black boxes below the lines indicates the phase I clinical trial of the TKIs.
7.1. First-Generation Inhibitors
EGFR was overexpressed in the majority of NSCLC cases. Therefore, treatment was directed towards using inhibitors of this protein kinase. Gefitinib (IressaTM, ZD-1839) was the first EGFR tyrosine kinase inhibitor (TKI) that was approved by the FDA, in 2003, to be used as the first line of treatment for lung cancer with mutated EGFR (Figure 7) [110,111]. This anilinoquinazoline compound selectively inhibits EGFR by reversibly binding to the ATP-binding site and blocking the signal transduction pathways, thus inhibiting cell growth and inducing cell apoptosis [112,113]. Crystal structures of gefitinib and wild-type and mutated EGFR TK have been deposited in RCSB protein databank. These show that gefitinib occupies the ATP-binding site by interacting with several residues in the pocket. The N1 of the quinazoline ring forms a hydrogen bond with Met793 in the hinge region. It also hydrophobically interacts with Leu718, Val726, Lys745, Met766, Leu788, Thr790, and Leu844. Furthermore, gefitinib is classified as a class I inhibitor since it interacts with the active conformation of EGFR with αC-in and DFG-in [114]. The two randomized phase II trials of gefitinib, IDEAL I and IDEAL II, were conducted as monotherapy in advanced or metastatic NSCLC patients that failed prior chemotherapy treatments. It was found that neither of the trials demonstrated any additional advantage in the survival, progression, or response rates compared to conventional chemotherapy, although they had a more favorable safety profile [113,115,116,117,118]. In the phase III trial (ISEL), gefitinib was able to increase progression-free survival rate, but generally not in advanced-NSCLC patients that had previously undergone chemotherapy [119,120,121,122].
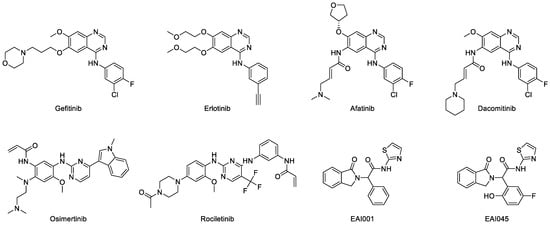
Figure 7.
Chemical structure of EGFR TKI (first to fourth generations).
Erlotinib (TarcevaTM, CP-358774) was approved by the FDA in 2004 as the first-line treatment of metastatic NSCLC with mutated EGFR, such as deletion in exon 19 and L858R substitution [110]. It is also used as the first line of treatment for NSCLC patients that show progression following at least one chemotherapy regimen. It has the same anilinoquinazoline scaffold as gefitinib but with a symmetrical 2-methoxyethoxy hand in the quinazoline core and an ethyne in the meta position of phenyl group. Furthermore, erlotinib is a type I inhibitor that competes with ATP binding and has similar interaction pattern as gefitinib. It is also a type I½B inhibitor that binds to the EGFR with inactive DFG-in and αC-out conformation [42,114,123]. A randomized phase II clinical trial using erlotinib alone showed a modest improvement in progression-free survival of none- or light-smoker patients, while a higher rate of adverse events was observed in the use of erlotinib combined with conventional chemotherapy [124]. A placebo-controlled phase III clinical trial in NSCLC patients that had progression after platinum-based therapy demonstrated a longer progression-free and overall survival rate with the use of erlotinib combined with bevacizumab [125,126]. However, a phase II randomized clinical trial showed that there was no superior efficacy of both combinations compared to when using only erlotinib [127].
Since both drugs interact with the gatekeeper residue Thr790, its mutation results in the resistance of gefitinib and erlotinib due to steric clash with aromatic ring [128]. Furthermore, the cell line with L858R/T790M mutation showed a 100-fold lower sensitivity to gefitinib and erlotinib, indicating the resistance of the drugs [76]. The use of gefitinib was restricted in 2005 and withdrawn in 2012 by the US-FDA due to the unfavorable phase III trial results. Therefore, the development of the first-generation TKI is required to increase the effectiveness of NSCLC treatment.
7.2. Second-Generation Inhibitors
Anilinoquinazoline derivatives such as afatinib and dacomitinib (Figure 7) were developed to overcome the limitation of the first generation of TKI. Afatinib (GiotrifTM, BIBW-2992) is an irreversible inhibitor of EGFR with exon-19 deletion and L858R substitution, which causes resistance to gefitinib and erlotinib [129,130]. The two crystal structures of afatinib in complex with wild-type and T790M EGFR were deposited in the protein databank with ID 4G5J and 4G5P, respectively [131]. Similar to gefitinib, afatinib forms hydrogen bonds with the backbone of Met793 in the hinge region. It also interacts with the hydrophobic region in the same way as gefitinib. The furanyl group was exposed to the solvent and the 3-chloro-4-fluorophenyl group was located close to the gatekeeper residue. It was classified as a type VI inhibitor because its acrylamide group binds covalently to the Cys797 of the active conformation of EGFR [114]. Furthermore, two randomized phase II clinical trials were conducted to compare afatinib and platinum-doublet chemotherapy in patients with advanced lung carcinoma harboring mutated EGFR. Afatinib showed longer progression-free survival compared to the chemotherapy, although there was no significant difference in terms of overall survival [132,133,134]. Based on the pooled analysis of the overall survival data for patients with exon 19 deletion of EGFR, there was a significant increase in the median overall survival by 31.7 months compared to chemotherapy. However, there was no benefit in the analysis for patients with L858R EGFR. These data indicate that afatinib is the first EGFR TKI to show overall survival benefit compared to chemotherapy in patients with EGFR exon 19 deletion [135].
Dacomitinib (VizimproTM, PF-299804) is also an irreversible EGFR TKI that has been approved by the FDA to be used for patients with metastatic NSCLC with exon 19 deletion and exon 21 substitution [136,137,138,139]. It shares similar binding properties with afatinib and forms hydrogen bonds with the hinge residue and hydrophobic interactions with those in the binding pocket. It is also a type VI inhibitor because the acrylamide group covalently interacts with Cys797 [114]. This irreversibly covalent interaction is the characteristic of second-generation TKIs. Resistance to dacomitinib was found in EGFR with C797S substitution [140,141]. Furthermore, a randomized phase III clinical trial (ARCHER 1050) of dacomitinib versus gefitinib was carried out on NSCLC patients with mutated EGFR. It was found that dacomitinib has more promising results in terms of progression-free survival compared to gefitinib. However, more serious adverse effects were observed in patients that were given dacomitinib [142].
7.3. Third-Generation Inhibitors
The third-generation TKIs, namely osimertinib and rociletinib (Figure 7) were developed to overcome the resistance of EGFR secondary mutation and to reduce the cases of serious adverse effects. Osimertinib (TagrissoTM, AZD-9291) is the first TKI with a non-quinazoline core approved by the FDA to be used as the adjuvant therapy for NSCLC patients that have undergone resection and have EGFR with mutations in exon 19 and 21 [143]. It is an irreversible TKI that is used for the EGFR T790M mutation, but has less activity against wild-type EGFR [143,144,145]. The deposition of the crystal structures of osimertinib along with wild-type and mutated EGFR in the databank in good resolution [146,147] have made it possible to carry out interaction studies on osimertinib and he EGFR TK domain. Furthermore, due to the crystalline structures, the nitrogen in the pyrimidine ring is able to establish a hydrogen bond with the N-H backbone of Met793. This indicates that the pyrimidine is able to mimic the interaction of the quinazoline core of other TKIs in the binding site. The acrylamide group covalently interacts with Cys797, thus it is classified as type VI inhibitor [114]. The hydrophobic interactions of osimertinib in the binding pocket were similar to those of other TKIs. However, the acquired EGFR mutation (C797S substitution) causes resistance to this drug [141]. A double-blind phase III clinical trial (FLAURA) showed that osimertinib gave a significantly longer progression-free survival than other TKIs in advanced-NSCLC patients with previously untreated mutated EGFR [148]. It also has similar safety profile to other TKIs but with lower rates of grade 3 or higher adverse events, which suggests its superiority to the other TKIs [133,148,149,150,151].
Rociletinib (XegafriTM, CO-1686) is an irreversible inhibitor of mutated EGFR. According to preclinical and phase I/II clinical studies, this drug has minimal efficacy against wild-type and exon-20-insertion EGF [152,153]. Furthermore, the crystal structures of rociletinib in complex with EGFR T790M and L858R were also studied [154]. It was found that in the T790M EGFR, the anilinopyrimidine group in rociletinib forms two hydrogen bonds with Met793 amide and carbonyl backbone. Rociletinib was also able to form two hydrogen bonds in EGFR L858R. These include one between nitrogens in the pyrimidine group, and another between the fluoromethyl and Thr790, which became a hydrophobic interaction in the T790M structure. The acrylamide group in rociletinib covalently binds to Cys797 in both active (DFG-in/αC-in) conformation structures [154]. In addition, a randomized phase III trial (TIGER-3) showed that rociletinib has longer progression-free survival but higher rates of hyperglycemia compared to chemotherapy in advanced-NSCLC patients with mutated EGFR (excluding exon 20 insertion) [155].
7.4. Fourth-Generation Inhibitors
It was reported that EGFR mutation C797S causes resistance to irreversible second and third TKIs [140,141,156,157]. In order to solve this issue, a large library of compounds targeting EGFR L858R/T790M was screened, and two compounds were obtained as the next-generation TKIs, namely EAI001 and EAI045 (Figure 7) [100,158]. These compounds have high selectivity towards EGFR L858R/T790M compared to the wildtype. Furthermore, the high resolution of the EGFR T790M/V948R crystal structure in complex with EAI001 that was deposited in the databank had an allosteric binding property with the ligands. This indicates that EAI001 is a selective allosteric inhibitor of mutant EGFR [100]. Instead of binding near the hinge residues, EAI001 binds to the deeper pocket and forms a hydrogen bond with Asp855 in the allosteric site of EGFR (Figure 5). The mutation of Thr790 into methionine enables the thiazole group of EAI001 to form a pi-sulfur interaction with the mutated gatekeeper residue. The aromatic rings have hydrophobic interactions with Met766, Leu777, Leu788, and Phe856, that may have a key role in the allosteric activity. Furthermore, the binding of the allosteric inhibitor prevents autophosphorylation of kinase and stabilizes protein conformation [107].
The optimization of EAI001 resulted in the formation of EAI045, which is a highly selective allosteric inhibitor of EGFR L858R/T790M. The binding mechanism of EGFR T790M/C797S/V948R with EAI045 was disclosed in the 2.90 crystal structure of this ligand in the presence of the C797S substitution, which causes resistance to type 1 TKIs [158]. Furthermore, EAI045 is able to form more polar interactions than the former compound EAI001. It forms three hydrogen bonds, namely carbonyl oxygen of the isoindolin-1-one to Lys745, amide nitrogen to Asp855, and para-fluorophenol to Phe856 in the allosteric binding pocket, and hydrophobic interactions with Met766, Leu777, Leu788, and Phe856 side chain. These interactions lead to better affinity and solubility of the compound. However, the EAI045 interaction with Lys745 appears to be impossible in the actual condition. In the EGFR–EAI001 complex, Lys745 interacts with the negatively charged AMP-PNP rather than the ligand. This might be due to the strong polar interaction that “pulled” the side chain of Lys745 away from EAI001, causing it to preferably interact with the AMP-PNP rather than the allosteric ligand. Furthermore, due to the fact that the ATP molecule is missing from the crystal structure of EGFR T790M/C797S/V948R-EAI045, the binding affinity of these two complexes cannot be compared equally [158].
The in vitro assay showed that the quantity of EAI045 reduced but did not fully eliminate EGFR autophosphorylation or kinase function in NSCLC and an H1975 cell line with EGFR L858R/T790M. The efficacy of EAI045 was also evaluated and then was used in combination with cetuximab in an in vivo study using a mouse model with L858R/T790M mutant-driven lung cancer. The mice treated with EAI045 and cetuximab showed remarkable tumor regression, while the one that was only given EAI045 showed no response. The same result was also found in L858R/T790M/C797S-engineered Ba/F3 cells as well as mice harboring L858R/T790M/C797S tumor xenografts, which demonstrated that EAI045 is superior to other TKIs. It was also able to overcome acquired resistance to T790M and C797S mutations [159]. Therefore, clinical trials are required to confirm its effectiveness in advanced-lung-cancer patients.
7.5. Other Allosteric Inhibitors
A high-throughput docking was performed using the mutated EGFR–EAI045 complex, a L858R/T790M mutant EGFR, and a homology model of EGFR, which produced 92 small molecules that were predicted to have allosteric behavior with the kinase protein. These compounds were further screened using in vitro assay, and compound N-((2-methyl-1H-indol-3-yl)(pyridin-2-yl)methyl)-3-nitroaniline (refer to compound 8 in the publication) was obtained as the most promising allosteric inhibitor that is able to interact with wild-type, T790M, and L858R/T790M EGFR [160]. Furthermore, in order to determine the effect of the stereochemistry, two enantiomers of compound 8 were docked to the protein. The nitrophenylalanine of (S)-enantiomer formed a hydrogen bond with the carboxyl group of Asp855 in the allosteric binding site. It also had hydrophobic interaction with several residues in the pocket. In addition, the pyridine and indole group were flipped in the (R)-enantiomer to enable it form three hydrogen bonds, including a polar contact with Asp855. The pyridine ring of (R)-enantiomer formed a hydrogen bond with Lys745, while the indole group interacted with Phe856. This latter enantiomer interacts hydrophobically with residues that are similar to those found in the (S)-enantiomer. Therefore, this indicates that both enantiomers are capable of interacting favorably inside the allosteric region. The compound was able to inhibit the activity of wild-type and L858R/T790M double-mutation EGFR in 4.7 μM and 6.2 μM, respectively. The dose–response curve did not change when the concentration of ATP varied. This indicates that the compound inhibits the double-mutant EGFR via an allosteric mechanism. In addition, according to a ligand-based similarity analysis, compound 8 differs structurally from the allosteric inhibitors EAI001 and EAI045 [160].
7.6. Small Molecules in the Clinical Trials
Several novel TKIs have been developed to address resistance issues caused by both common and rare EGFR mutationss. Table 1 lists the drugs which are now undergoing clinical trials, followed by the condition being studied.

Table 1.
Investigational TKIs in the pipeline.
8. Future Direction
The current generation of EGFR TK inhibitors has experienced drawbacks, and most notable is the issue of mutant EGFR resistance. A number of common and rare mutations discovered in NSCLC patients diminish the sensitivity to TKIs given as therapy, and lower overall survival rate. Furthermore, drug safety must be considered. Olmutinib, which had been previously approved by the Republic of Korea in May 2015, had to be withdrawn several months later due to Stevens–Johnson syndrome as its adverse reaction [109].
Therefore, more powerful and efficient inhibitors are required. The availability of crystal structures for both native and mutant EGFR has promoted the development of NSCLC medicines targeting EGFR. EAI045 was designed based on the optimization of current inhibitors used in therapy, although the in vivo finding revealed that EAI045 was only beneficial when coupled with cetuximab [161]. This compound needs to be further developed in the future to solve the problem of resistance, while maintaining the inhibitory activity of the molecule. One of the strategies is to retain the structure or group capable of forming interactions with the key amino acids in the kinase binding site. The other possible strategy is combining the TKI with other agents that can overcome the various resistant tumor clone.
Apart from the development of TK domain inhibitors, the extracellular region of the EGFR, which is known as the binding site for native ligands EGF [21] and the monoclonal antibodies panitumumab and cetuximab [161], may also be targeted in the development of NSCLC treatments. Understanding the important interactions as well as the appropriate peptide length for extracellular EGFR can also help in the development of new inhibitory compounds in this region [90]. The development of novel compounds capable of binding to the rare mutant EGFR would be an intriguing research area. Therefore, a greater understanding of EGFR activation and dimerization mechanisms may aid in the development of new TK inhibitors in the future.
Author Contributions
T.A. contributed to designing the review content, collecting and screening the references, visualization of the figures, and drafting the manuscript. D.H.T. conceptualized the review and corrected the manuscript, while R.E.K. and T.O. contributed to the supervision and correcting of the manuscript. D.H.T. also contributed with resources, funding acquisition, and supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported in part by P3MI 2020, Bandung Institute of Technology (No. 014/SK/I1.C03/KP/2020).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not available.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB Signalling Network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Zhang, G.; Haura, E.B. Targeting Epidermal Growth Factor Receptor: Central Signaling Kinase in Lung Cancer. Biochem. Pharmacol. 2010, 80, 613–623. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Carotenuto, A.; Rachiglio, A.; Gallo, M.; Maiello, M.R.; Aldinucci, D.; Pinto, A.; Normanno, N. The Role of the EGFR Signaling in Tumor Microenvironment. J. Cell. Physiol. 2008, 214, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Hidalgo, M. Pharmacogenomics of Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors. Biochim. Biophys. Acta (BBA) Rev. Cancer 2006, 1766, 217–229. [Google Scholar] [CrossRef]
- Mosesson, Y.; Yarden, Y. Oncogenic Growth Factor Receptors: Implications for Signal Transduction Therapy. Semin. Cancer Biol. 2004, 14, 262–270. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Al Olayan, A.; al Hussaini, H.; Rahman Jazieh, A. The Roles of Epidermal Growth Factor Receptor (EGFR) Inhibitors in the Management of Lung Cancer. J. Infect. Public Health 2012, 5, S50–S60. [Google Scholar] [CrossRef] [Green Version]
- Jänne, P.A.; Engelman, J.A.; Johnson, B.E. Epidermal Growth Factor Receptor Mutations in Non–Small-Cell Lung Cancer: Implications for Treatment and Tumor Biology. J. Clin. Oncol. 2005, 23, 3227–3234. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. EGFR Antagonists in Cancer Treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [Green Version]
- Woodburn, J.R. The Epidermal Growth Factor Receptor and Its Inhibition in Cancer Therapy. Pharmacol. Ther. 1999, 82, 241–250. [Google Scholar] [CrossRef]
- Huang, H.-J.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.-D.; Huang, C.-M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The Enhanced Tumorigenic Activity of a Mutant Epidermal Growth Factor Receptor Common in Human Cancers Is Mediated by Threshold Levels of Constitutive Tyrosine Phosphorylation and Unattenuated Signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, J.; Hsuan, J.; Waterfield, M. Analysis of Mammalian Fibroblast Transformation by Normal and Mutated Human EGF Receptors. Oncogene 1989, 4, 273–283. [Google Scholar]
- Hazan, R.B.; Norton, L. The Epidermal Growth Factor Receptor Modulates the Interaction of E-Cadherin with the Actin Cytoskeleton. J. Biol. Chem. 1998, 273, 9078–9084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Bunn, P.A. Targeting the Epidermal Growth Factor Receptor in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2003, 9, 5813–5824. [Google Scholar] [PubMed]
- Ellerbroek, S.M.; Halbleib, J.M.; Benavidez, M.; Warmka, J.K.; Wattenberg, E.V.; Stack, M.S.; Hudson, L.G. Phosphatidylinositol 3-Kinase Activity in Epidermal Growth Factor-Stimulated Matrix Metalloproteinase-9 Production and Cell Surface Association. Cancer Res. 2001, 61, 1855–1861. [Google Scholar] [PubMed]
- Roskoski, R. Small Molecule Inhibitors Targeting the EGFR/ErbB Family of Protein-Tyrosine Kinases in Human Cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Fu, L. Mechanisms of Resistance to EGFR Tyrosine Kinase Inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Chang, Y. Epidermal Growth Factor Receptor (EGFR) Phosphorylation, Signaling and Trafficking in Prostate Cancer. In Prostate Cancer—From Bench to Bedside; InTech: London, UK, 2011. [Google Scholar]
- Ullrich, A.; Schlessinger, J. Signal Transduction by Receptors with Tyrosine Kinase Activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J.; et al. Human Epidermal Growth Factor Receptor CDNA Sequence and Aberrant Expression of the Amplified Gene in A431 Epidermoid Carcinoma Cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.-H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal Structure of the Complex of Human Epidermal Growth Factor and Receptor Extracellular Domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Lelimousin, M.; Limongelli, V.; Sansom, M.S.P. Conformational Changes in the Epidermal Growth Factor Receptor: Role of the Transmembrane Domain Investigated by Coarse-Grained MetaDynamics Free Energy Calculations. J. Am. Chem. Soc. 2016, 138, 10611–10622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocharov, E.V.; Bragin, P.E.; Pavlov, K.V.; Bocharova, O.V.; Mineev, K.S.; Polyansky, A.A.; Volynsky, P.E.; Efremov, R.G.; Arseniev, A.S. The Conformation of the Epidermal Growth Factor Receptor Transmembrane Domain Dimer Dynamically Adapts to the Local Membrane Environment. Biochemistry 2017, 56, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Endres, N.F.; Das, R.; Smith, A.W.; Arkhipov, A.; Kovacs, E.; Huang, Y.; Pelton, J.G.; Shan, Y.; Shaw, D.E.; Wemmer, D.E.; et al. Conformational Coupling across the Plasma Membrane in Activation of the EGF Receptor. Cell 2013, 152, 543–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janosi, L.; Prakash, A.; Doxastakis, M. Lipid-Modulated Sequence-Specific Association of Glycophorin A in Membranes. Biophys. J. 2010, 99, 284–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, D.; Marrink, S.J. Lipid-Mediated Interactions Tune the Association of Glycophorin A Helix and Its Disruptive Mutants in Membranes. Phys. Chem. Chem. Phys. 2010, 12, 12987–12996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, A.S.; Polyansky, A.A.; Fleck, M.; Volynsky, P.E.; Efremov, R.G. Adaptable Lipid Matrix Promotes Protein–Protein Association in Membranes. J. Chem. Theory Comput. 2015, 11, 4415–4426. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, E.V.; Mayzel, M.L.; Volynsky, P.E.; Mineev, K.S.; Tkach, E.N.; Ermolyuk, Y.S.; Schulga, A.A.; Efremov, R.G.; Arseniev, A.S. Left-Handed Dimer of EphA2 Transmembrane Domain: Helix Packing Diversity among Receptor Tyrosine Kinases. Biophys. J. 2010, 98, 881–889. [Google Scholar] [CrossRef] [Green Version]
- Muhle-Goll, C.; Hoffmann, S.; Afonin, S.; Grage, S.L.; Polyansky, A.A.; Windisch, D.; Zeitler, M.; Bürck, J.; Ulrich, A.S. Hydrophobic Matching Controls the Tilt and Stability of the Dimeric Platelet-Derived Growth Factor Receptor (PDGFR) β Transmembrane Segment. J. Biol. Chem. 2012, 287, 26178–26186. [Google Scholar] [CrossRef] [Green Version]
- Brewer, M.R.; Choi, S.H.; Alvarado, D.; Moravcevic, K.; Pozzi, A.; Lemmon, M.A.; Carpenter, G. The Juxtamembrane Region of the EGF Receptor Functions as an Activation Domain. Mol. Cell 2009, 34, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Aifa, S.; Aydin, J.; Nordvall, G.; Lundström, I.; Svensson, S.P.S.; Hermanson, O. A Basic Peptide within the Juxtamembrane Region Is Required for EGF Receptor Dimerization. Exp. Cell Res. 2005, 302, 108–114. [Google Scholar] [CrossRef]
- Thiel, K.W.; Carpenter, G. Epidermal Growth Factor Receptor Juxtamembrane Region Regulates Allosteric Tyrosine Kinase Activation. Proc. Natl. Acad. Sci. USA 2007, 104, 19238–19243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arkhipov, A.; Shan, Y.; Kim, E.T.; Shaw, D.E. Membrane Interaction of Bound Ligands Contributes to the Negative Binding Cooperativity of the EGF Receptor. PLoS Comput. Biol. 2014, 10, e1003742. [Google Scholar] [CrossRef] [Green Version]
- Hedger, G.; Sansom, M.S.P.; Koldsø, H. The Juxtamembrane Regions of Human Receptor Tyrosine Kinases Exhibit Conserved Interaction Sites with Anionic Lipids. Sci. Rep. 2015, 5, 9198. [Google Scholar] [CrossRef] [Green Version]
- Kil, S.J.; Carlin, C. EGF Receptor Residues Leu679, Leu680 Mediate Selective Sorting of Ligand-Receptor Complexes in Early Endosomal Compartments. J. Cell. Physiol. 2000, 185, 47–60. [Google Scholar] [CrossRef]
- He, C.; Hobert, M.; Friend, L.; Carlin, C. The Epidermal Growth Factor Receptor Juxtamembrane Domain Has Multiple Basolateral Plasma Membrane Localization Determinants, Including a Dominant Signal with a Polyproline Core. J. Biol. Chem. 2002, 277, 38284–38293. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-Y.; Makino, K.; Xia, W.; Matin, A.; Wen, Y.; Kwong, K.Y.; Bourguignon, L.; Hung, M.-C. Nuclear Localization of EGF Receptor and Its Potential New Role as a Transcription Factor. Nat. Cell Biol. 2001, 3, 802–808. [Google Scholar] [CrossRef]
- Martín-Nieto, J.; Villalobo, A. The Human Epidermal Growth Factor Receptor Contains a Juxtamembrane Calmodulin-Binding Site. Biochemistry 1998, 37, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T.; Ling, N.; Cooper, J.A. Protein Kinase C Phosphorylation of the EGF Receptor at a Threonine Residue Close to the Cytoplasmic Face of the Plasma Membrane. Nature 1984, 311, 480–483. [Google Scholar] [CrossRef]
- Davis, R.J.; Czech, M.P. Tumor-Promoting Phorbol Diesters Cause the Phosphorylation of Epidermal Growth Factor Receptors in Normal Human Fibroblasts at Threonine-654. Proc. Natl. Acad. Sci. USA 1985, 82, 1974–1978. [Google Scholar] [CrossRef] [Green Version]
- Takishima, K.; Griswold-Prenner, I.; Ingebritsen, T.; Rosner, M.R. Epidermal Growth Factor (EGF) Receptor T669 Peptide Kinase from 3T3-L1 Cells Is an EGF-Stimulated “MAP” Kinase. Proc. Natl. Acad. Sci. USA 1991, 88, 2520–2524. [Google Scholar] [CrossRef] [Green Version]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jura, N.; Zhang, X.; Endres, N.F.; Seeliger, M.A.; Schindler, T.; Kuriyan, J. Catalytic Control in the EGF Receptor and Its Connection to General Kinase Regulatory Mechanisms. Mol. Cell 2011, 42, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Fernandez, M.L.; Clarke, D.T.; Roberts, S.K.; Zanetti-Domingues, L.C.; Gervasio, F.L. Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and Its Relevance to Non-Small Cell Lung Cancer. Cells 2019, 8, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertics, P.J.; Gill, G.N. Self-Phosphorylation Enhances the Protein-Tyrosine Kinase Activity of the Epidermal Growth Factor Receptor. J. Biol. Chem. 1985, 260, 14642–14647. [Google Scholar] [CrossRef]
- Bertics, P.J.; Chen, W.S.; Hubler, L.; Lazar, C.S.; Rosenfeld, M.G.; Gill, G.N. Alteration of Epidermal Growth Factor Receptor Activity by Mutation of Its Primary Carboxyl-Terminal Site of Tyrosine Self-Phosphorylation. J. Biol. Chem. 1988, 263, 3610–3617. [Google Scholar] [CrossRef]
- Mustafa, M.; Mirza, A.; Kannan, N. Conformational Regulation of the EGFR Kinase Core by the Juxtamembrane and C-Terminal Tail: A Molecular Dynamics Study. Proteins Struct. Funct. Bioinform. 2011, 79, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, E.; Das, R.; Wang, Q.; Collier, T.S.; Cantor, A.; Huang, Y.; Wong, K.; Mirza, A.; Barros, T.; Grob, P.; et al. Analysis of the Role of the C-Terminal Tail in the Regulation of the Epidermal Growth Factor Receptor. Mol. Cell. Biol. 2015, 35, 3083–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landau, M.; Fleishman, S.J.; Ben-Tal, N. A Putative Mechanism for Downregulation of the Catalytic Activity of the EGF Receptor via Direct Contact between Its Kinase and C-Terminal Domains. Structure 2004, 12, 2265–2275. [Google Scholar] [CrossRef] [Green Version]
- Sorkin, A.; Mazzotti, M.; Sorkina, T.; Scotto, L.; Beguinot, L. Epidermal Growth Factor Receptor Interaction with Clathrin Adaptors Is Mediated by the Tyr974-Containing Internalization Motif. J. Biol. Chem. 1996, 271, 13377–13384. [Google Scholar] [CrossRef] [Green Version]
- Bublil, E.M.; Pines, G.; Patel, G.; Fruhwirth, G.; Ng, T.; Yarden, Y. Kinase-Mediated Quasi-Dimers of EGFR. FASEB J. 2010, 24, 4744–4755. [Google Scholar] [CrossRef]
- Jura, N.; Endres, N.F.; Engel, K.; Deindl, S.; Das, R.; Lamers, M.H.; Wemmer, D.E.; Zhang, X.; Kuriyan, J. Mechanism for Activation of the EGF Receptor Catalytic Domain by the Juxtamembrane Segment. Cell 2009, 137, 1293–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canagarajah, B.J.; Khokhlatchev, A.; Cobb, M.H.; Goldsmith, E.J. Activation Mechanism of the MAP Kinase ERK2 by Dual Phosphorylation. Cell 1997, 90, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Hasenahuer, M.A.; Barletta, G.P.; Fernandez-Alberti, S.; Parisi, G.; Fornasari, M.S. Pockets as Structural Descriptors of EGFR Kinase Conformations. PLoS ONE 2017, 12, e0189147. [Google Scholar] [CrossRef] [Green Version]
- Nagar, B. C-Abl Tyrosine Kinase and Inhibition by the Cancer Drug Imatinib (Gleevec/STI-571). J. Nutr. 2007, 137, 1518S–1523S. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, S.R. Crystal Structure of the Activated Insulin Receptor Tyrosine Kinase in Complex with Peptide Substrate and ATP Analog. EMBO J. 1997, 16, 5572–5581. [Google Scholar] [CrossRef]
- Hubbard, S.R.; Wei, L.; Hendrickson, W.A. Crystal Structure of the Tyrosine Kinase Domain of the Human Insulin Receptor. Nature 1994, 372, 746–754. [Google Scholar] [CrossRef]
- Hari, S.B.; Merritt, E.A.; Maly, D.J. Sequence Determinants of a Specific Inactive Protein Kinase Conformation. Chem. Biol. 2013, 20, 806–815. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M Mutation in EGFR Kinase Causes Drug Resistance by Increasing the Affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013, 20, 146–159. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Xie, L.; Bourne, P.E. Structural Insights into Characterizing Binding Sites in Epidermal Growth Factor Receptor Kinase Mutants. J. Chem. Inf. Model. 2019, 59, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.-H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel Mutant-Selective EGFR Kinase Inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A Unique Structure for Epidermal Growth Factor Receptor Bound to GW572016 (Lapatinib). Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Pickin, K.A.; Bose, R.; Jura, N.; Cole, P.A.; Kuriyan, J. Inhibition of the EGF Receptor by Binding of MIG6 to an Activating Kinase Domain Interface. Nature 2007, 450, 741–744. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Nair, S.K.; Murray, B.W.; Almaden, C.; Bailey, S.; Baxi, S.; Behenna, D.; Cho-Schultz, S.; Dalvie, D.; Dinh, D.M.; et al. Discovery of 1-{(3 R,4 R)-3-[({5-Chloro-2-[(1-Methyl-1 H -Pyrazol-4-Yl)Amino]-7 H -Pyrrolo[2,3-d]Pyrimidin-4-Yl}oxy)Methyl]-4-Methoxypyrrolidin-1-Yl}prop-2-En-1-One (PF-06459988), a Potent, WT Sparing, Irreversible Inhibitor of T790M-Containing EGFR Mutants. J. Med. Chem. 2016, 59, 2005–2024. [Google Scholar] [CrossRef]
- Makinoshima, H.; Takita, M.; Saruwatari, K.; Umemura, S.; Obata, Y.; Ishii, G.; Matsumoto, S.; Sugiyama, E.; Ochiai, A.; Abe, R.; et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (MTOR) Axis Is Responsible for Aerobic Glycolysis Mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-Mutated Lung Adenocarcinoma. J. Biol. Chem. 2015, 290, 17495–17504. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, R.A.; Luwor, R.B.; Burgess, A.W. Epidermal Growth Factor Receptor: Structure-Function Informing the Design of Anticancer Therapeutics. Exp. Cell Res. 2018, 371, 1–9. [Google Scholar] [CrossRef]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Shigematsu, H.; Gazdar, A.F. Somatic Mutations of Epidermal Growth Factor Receptor Signaling Pathway in Lung Cancers. Int. J. Cancer 2006, 118, 257–262. [Google Scholar] [CrossRef]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and Biological Features Associated with Epidermal Growth Factor Receptor Gene Mutations in Lung Cancers. J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Choong, N.W.; Dietrich, S.; Seiwert, T.Y.; Tretiakova, M.S.; Nallasura, V.; Davies, G.C.; Lipkowitz, S.; Husain, A.N.; Salgia, R.; Ma, P.C. Gefitinib Response of Erlotinib-Refractory Lung Cancer Involving Meninges—Role of EGFR Mutation. Nat. Clin. Pract. Oncol. 2006, 3, 50–57. [Google Scholar] [CrossRef]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Petri, E.T.; Halmos, B.; Boggon, T.J. Structure and Clinical Relevance of the Epidermal Growth Factor Receptor in Human Cancer. J. Clin. Oncol. 2008, 26, 1742–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible Inhibitors of the EGF Receptor May Circumvent Acquired Resistance to Gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Yoshida, K.; Hida, T.; Tsuboi, M.; Tada, H.; Kuwano, H.; Mitsudomi, T. Analysis of Epidermal Growth Factor Receptor Gene Mutation in Patients with Non–Small Cell Lung Cancer and Acquired Resistance to Gefitinib. Clin. Cancer Res. 2006, 12, 5764–5769. [Google Scholar] [CrossRef] [Green Version]
- Balak, M.N.; Gong, Y.; Riely, G.J.; Somwar, R.; Li, A.R.; Zakowski, M.F.; Chiang, A.; Yang, G.; Ouerfelli, O.; Kris, M.G.; et al. Novel D761Y and Common Secondary T790M Mutations in Epidermal Growth Factor Receptor–Mutant Lung Adenocarcinomas with Acquired Resistance to Kinase Inhibitors. Clin. Cancer Res. 2006, 12, 6494–6501. [Google Scholar] [CrossRef] [Green Version]
- Carter, T.A.; Wodicka, L.M.; Shah, N.P.; Velasco, A.M.; Fabian, M.A.; Treiber, D.K.; Milanov, Z.V.; Atteridge, C.E.; Biggs, W.H.; Edeen, P.T.; et al. Inhibition of Drug-Resistant Mutants of ABL, KIT, and EGF Receptor Kinases. Proc. Natl. Acad. Sci. USA 2005, 102, 11011–11016. [Google Scholar] [CrossRef] [Green Version]
- Greulich, H.; Chen, T.-H.; Feng, W.; Jänne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R.; et al. Oncogenic Transformation by Inhibitor-Sensitive and -Resistant EGFR Mutants. PLoS Med. 2005, 2, e313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, Y.; Suda, K.; Kimura, H.; Matsumoto, K.; Arao, T.; Nagai, T.; Saijo, N.; Yatabe, Y.; Mitsudomi, T.; Nishio, K. Highly Sensitive Detection of EGFR T790M Mutation Using Colony Hybridization Predicts Favorable Prognosis of Patients with Lung Cancer Harboring Activating EGFR Mutation. J. Thorac. Oncol. 2012, 7, 1640–1644. [Google Scholar] [CrossRef] [Green Version]
- Su, K.-Y.; Chen, H.-Y.; Li, K.-C.; Kuo, M.-L.; Yang, J.C.-H.; Chan, W.-K.; Ho, B.-C.; Chang, G.-C.; Shih, J.-Y.; Yu, S.-L.; et al. Pretreatment Epidermal Growth Factor Receptor (EGFR) T790M Mutation Predicts Shorter EGFR Tyrosine Kinase Inhibitor Response Duration in Patients with Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2012, 30, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Ji, H.; Yuza, Y.; Meyerson, M.; Wong, K.-K.; Tenen, D.G.; Halmos, B. An Alternative Inhibitor Overcomes Resistance Caused by a Mutation of the Epidermal Growth Factor Receptor. Cancer Res. 2005, 65, 7096–7101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Mukohara, T.; Zejnullahu, K.; Lifshits, E.; Borras, A.M.; Gale, C.-M.; Naumov, G.N.; Yeap, B.Y.; Jarrell, E.; Sun, J.; et al. Allelic Dilution Obscures Detection of a Biologically Significant Resistance Mutation in EGFR-Amplified Lung Cancer. J. Clin. Investig. 2006, 116, 2695–2706. [Google Scholar] [CrossRef]
- Costa, D.B.; Halmos, B.; Kumar, A.; Schumer, S.T.; Huberman, M.S.; Boggon, T.J.; Tenen, D.G.; Kobayashi, S. BIM Mediates EGFR Tyrosine Kinase Inhibitor-Induced Apoptosis in Lung Cancers with Oncogenic EGFR Mutations. PLoS Med. 2007, 4, e315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bean, J.; Riely, G.J.; Balak, M.; Marks, J.L.; Ladanyi, M.; Miller, V.A.; Pao, W. Acquired Resistance to Epidermal Growth Factor Receptor Kinase Inhibitors Associated with a Novel T854A Mutation in a Patient with EGFR -Mutant Lung Adenocarcinoma. Clin. Cancer Res. 2008, 14, 7519–7525. [Google Scholar] [CrossRef] [Green Version]
- Avizienyte, E.; Ward, R.A.; Garner, A.P. Comparison of the EGFR Resistance Mutation Profiles Generated by EGFR-Targeted Tyrosine Kinase Inhibitors and the Impact of Drug Combinations. Biochem. J. 2008, 415, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Sousa, A.C.; Silveira, C.; Janeiro, A.; Malveiro, S.; Oliveira, A.R.; Felizardo, M.; Nogueira, F.; Teixeira, E.; Martins, J.; Carmo-Fonseca, M. Detection of Rare and Novel EGFR Mutations in NSCLC Patients: Implications for Treatment-Decision. Lung Cancer 2020, 139, 35–40. [Google Scholar] [CrossRef]
- Naidoo, J.; Sima, C.S.; Rodriguez, K.; Busby, N.; Nafa, K.; Ladanyi, M.; Riely, G.J.; Kris, M.G.; Arcila, M.E.; Yu, H.A. Epidermal Growth Factor Receptor Exon 20 Insertions in Advanced Lung Adenocarcinomas: Clinical Outcomes and Response to Erlotinib. Cancer 2015, 121, 3212–3220. [Google Scholar] [CrossRef] [Green Version]
- Weber, F.; Fukino, K.; Sawada, T.; Williams, N.; Sweet, K.; Brena, R.M.; Plass, C.; Caldes, T.; Mutter, G.L.; Villalona-Calero, M.A.; et al. Variability in Organ-Specific EGFR Mutational Spectra in Tumour Epithelium and Stroma May Be the Biological Basis for Differential Responses to Tyrosine Kinase Inhibitors. Br. J. Cancer 2005, 92, 1922–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preusser, M.; Berghoff, A.S.; Koller, R.; Zielinski, C.C.; Hainfellner, J.A.; Liebmann-Reindl, S.; Popitsch, N.; Geier, C.B.; Streubel, B.; Birner, P. Spectrum of Gene Mutations Detected by next Generation Exome Sequencing in Brain Metastases of Lung Adenocarcinoma. Eur. J. Cancer 2015, 51, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Gajiwala, K.S. EGFR: Tale of the C-Terminal Tail. Protein Sci. 2013, 22, 995–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Deng, G.; Liu, X.; Cho, W.C. Monoclonal Antibodies in Lung Cancer. Expert Opin. Biol. Ther. 2013, 13, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Riely, G.J.; Lovly, C.M. Therapeutic Strategies Utilized in the Setting of Acquired Resistance to EGFR Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2014, 20, 5898–5907. [Google Scholar] [CrossRef] [Green Version]
- Remon, J.; Morán, T.; Majem, M.; Reguart, N.; Dalmau, E.; Márquez-Medina, D.; Lianes, P. Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer: A New Era Begins. Cancer Treat. Rev. 2014, 40, 93–101. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, X.; Jin, H. EGFR-TKI Resistance in NSCLC Patients: Mechanisms and Strategies. Am. J. Cancer Res. 2014, 4, 411–435. [Google Scholar]
- Zhao, Z.; Xie, L.; Xie, L.; Bourne, P.E. Delineation of Polypharmacology across the Human Structural Kinome Using a Functional Site Interaction Fingerprint Approach. J. Med. Chem. 2016, 59, 4326–4341. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) Resistance with Mutant-Selective Allosteric Inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, P.A.; Moody, C.L.; Murali, R. Allosteric Modulation of Ras and the PI3K/AKT/MTOR Pathway: Emerging Therapeutic Opportunities. Front. Physiol. 2014, 5, 478. [Google Scholar] [CrossRef] [Green Version]
- Ling, Y.; Jing, M.; Wang, X. Allosteric Therapies for Lung Cancer. Cancer Metastasis Rev. 2015, 34, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.-J.; Jang, H. Dynamic Protein Allosteric Regulation and Disease. Adv. Exp. Med. Biol. 2019, 1163, 25–43. [Google Scholar] [PubMed]
- Tsai, C.-J.; Nussinov, R. Emerging Allosteric Mechanism of EGFR Activation in Physiological and Pathological Contexts. Biophys. J. 2019, 117, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussinov, R.; Tsai, C.-J. Allostery in Disease and in Drug Discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, L.; Rastelli, G. AC Helix Displacement as a General Approach for Allosteric Modulation of Protein Kinases. Drug Discov. Today 2013, 18, 407–414. [Google Scholar] [CrossRef]
- Purba, E.; Saita, E.; Maruyama, I. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.L.; Vellanki, P.J.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Shen, Y.L.; Xia, H.; Li, Y.; Liu, J.; Zirkelbach, J.F.; et al. FDA Approval Summary: Osimertinib for Adjuvant Treatment of Surgically Resected Non–Small Cell Lung Cancer, a Collaborative Project Orbis Review. Clin. Cancer Res. 2021, 27, 6638–6643. [Google Scholar] [CrossRef]
- Fan, W.-L.; Shiao, M.-S.; Hui, R.C.-Y.; Su, S.-C.; Wang, C.-W.; Chang, Y.-C.; Chung, W.-H. HLA Association with Drug-Induced Adverse Reactions. J. Immunol. Res. 2017, 2017, 3186328. [Google Scholar] [CrossRef]
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; McGuinn, W.D.; Morse, D.; Abraham, S.; Rahman, A.; Liang, C.; Lostritto, R.; et al. United States Food and Drug Administration Drug Approval Summary. Clin. Cancer Res. 2004, 10, 1212–1218. [Google Scholar] [CrossRef] [Green Version]
- Barker, A.J.; Gibson, K.H.; Grundy, W.; Godfrey, A.A.; Barlow, J.J.; Healy, M.P.; Woodburn, J.R.; Ashton, S.E.; Curry, B.J.; Scarlett, L.; et al. Studies Leading to the Identification of ZD1839 (IressaTM): An Orally Active, Selective Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Targeted to the Treatment of Cancer. Bioorg. Med. Chem. Lett. 2001, 11, 1911–1914. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. A Novel Approach in the Treatment of Cancer: Targeting the Epidermal Growth Factor Receptor. Clin. Cancer Res. 2001, 7, 2958–2970. [Google Scholar] [PubMed]
- Fukuoka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J.-Y.; Nishiwaki, Y.; Vansteenkiste, J.; Kudoh, S.; Rischin, D.; et al. Multi-Institutional Randomized Phase II Trial of Gefitinib for Previously Treated Patients with Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Classification of Small Molecule Protein Kinase Inhibitors Based upon the Structures of Their Drug-Enzyme Complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Schiller, J.; Fukuoka, M.; Natale, R.; Lynch, T.; Averbuch, S.; Kay, A. Results from Two Phase II Trials (IDEAL 1 and IDEAL 2) of ZD1839 in Patients with Locally Advanced or Matastatic Non-Small Cell Lung Cancer. (Thoracic Oncology: 12:00 p.m.–1:45 p.m.). Chest 2002, 122, 168S. [Google Scholar]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch, J.T.J.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A.; et al. Efficacy of Gefitinib, an Inhibitor of the Epidermal Growth Factor Receptor Tyrosine Kinase, in Symptomatic Patients with Non–Small Cell Lung Cancer. JAMA 2003, 290, 2149–2158. [Google Scholar] [CrossRef] [Green Version]
- Cella, D.; Herbst, R.S.; Lynch, T.J.; Prager, D.; Belani, C.P.; Schiller, J.H.; Heyes, A.; Ochs, J.S.; Wolf, M.K.; Kay, A.C.; et al. Clinically Meaningful Improvement in Symptoms and Quality of Life for Patients With Non-Small-Cell Lung Cancer Receiving Gefitinib in a Randomized Controlled Trial. J. Clin. Oncol. 2005, 23, 2946–2954. [Google Scholar] [CrossRef]
- Nishiwaki, Y.; Yano, S.; Tamura, T.; Nakagawa, K.; Kudoh, S.; Horai, T.; Noda, K.; Takata, I.; Watanabe, K.; Saka, H.; et al. Subset Analysis of Data in the Japanese Patients with NSCLC from IDEAL 1 Study on Gefitinib. Gan To Kagaku Ryoho 2004, 31, 567–573. [Google Scholar]
- Thatcher, N.; Chang, A.; Parikh, P.; Rodrigues Pereira, J.; Ciuleanu, T.; von Pawel, J.; Thongprasert, S.; Tan, E.H.; Pemberton, K.; Archer, V.; et al. Gefitinib plus Best Supportive Care in Previously Treated Patients with Refractory Advanced Non-Small-Cell Lung Cancer: Results from a Randomised, Placebo-Controlled, Multicentre Study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005, 366, 1527–1537. [Google Scholar] [CrossRef]
- Chang, A.; Parikh, P.; Thongprasert, S.; Huat Tan, E.; Perng, R.P.; Ganzon, D.; Yang, C.-H.; Tsao, C.-J.; Watkins, C.; Botwood, N.; et al. Gefitinib (IRESSA) in Patients of Asian Origin with Refractory Advanced Non-Small Cell Lung Cancer: Subset Analysis from the ISEL Study. J. Thorac. Oncol. 2006, 1, 847–855. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Varella-Garcia, M.; Bunn, P.A.; Franklin, W.A.; Dziadziuszko, R.; Thatcher, N.; Chang, A.; Parikh, P.; Pereira, J.R.; Ciuleanu, T.; et al. Molecular Predictors of Outcome with Gefitinib in a Phase III Placebo-Controlled Study in Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2006, 24, 5034–5042. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, F.R.; Dziadziuszko, R.; Thatcher, N.; Mann, H.; Watkins, C.; Parums, D.V.; Speake, G.; Holloway, B.; Bunn, P.A.; Franklin, W.A. Epidermal Growth Factor Receptor Immunohistochemistry. Cancer 2008, 112, 1114–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib Binds Both Inactive and Active Conformations of the EGFR Tyrosine Kinase Domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jänne, P.A.; Wang, X.; Socinski, M.A.; Crawford, J.; Stinchcombe, T.E.; Gu, L.; Capelletti, M.; Edelman, M.J.; Villalona-Calero, M.A.; Kratzke, R.; et al. Randomized Phase II Trial of Erlotinib Alone or With Carboplatin and Paclitaxel in Patients Who Were Never or Light Former Smokers with Advanced Lung Adenocarcinoma: CALGB 30406 Trial. J. Clin. Oncol. 2012, 30, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Ansari, R.; Bustin, F.; Flynn, P.; Hart, L.; Otterson, G.A.; Vlahovic, G.; Soh, C.-H.; O’Connor, P.; Hainsworth, J. Efficacy of Bevacizumab plus Erlotinib versus Erlotinib Alone in Advanced Non-Small-Cell Lung Cancer after Failure of Standard First-Line Chemotherapy (BeTa): A Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2011, 377, 1846–1854. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.; Stern, H.; Amler, L.; Otterson, G.; Lin, M.; O’Connor, P.; Hainsworth, J. Biomarker Evaluation in the Phase III, Placebo (P)-Controlled, Randomized BeTa Trial of Bevacizumab (B) and Erlotinib (E) for Patients (Pts) with Advanced Non-Small Cell Lung Cancer (NSCLC) after Failure of Standard 1st-Line Chemotherapy: Correlation with Treatment Outcomes. J. Thorac. Oncol. 2009, 4, 530. [Google Scholar]
- Stinchcombe, T.E.; Jänne, P.A.; Wang, X.; Bertino, E.M.; Weiss, J.; Bazhenova, L.; Gu, L.; Lau, C.; Paweletz, C.; Jaslowski, A.; et al. Effect of Erlotinib Plus Bevacizumab vs Erlotinib Alone on Progression-Free Survival in Patients with Advanced EGFR -Mutant Non–Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 1448–1455. [Google Scholar] [CrossRef]
- Blencke, S.; Zech, B.; Engkvist, O.; Greff, Z.; Őrfi, L.; Horváth, Z.; Kéri, G.; Ullrich, A.; Daub, H. Characterization of a Conserved Structural Determinant Controlling Protein Kinase Sensitivity to Selective Inhibitors. Chem. Biol. 2004, 11, 691–701. [Google Scholar] [CrossRef]
- Dungo, R.T.; Keating, G.M. Afatinib: First Global Approval. Drugs 2013, 73, 1503–1515. [Google Scholar] [CrossRef]
- Abdallah, S.M.; Hirsh, V. Irreversible Tyrosine Kinase Inhibition of Epidermal Growth Factor Receptor with Afatinib in Egfr Activating Mutation–Positive Advanced Non-Small-Cell Lung Cancer. Curr. Oncol. 2018, 25, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef]
- Langer, C.J. Epidermal Growth Factor Receptor Inhibition in Mutation-Positive Non–Small-Cell Lung Cancer: Is Afatinib Better or Simply Newer? J. Clin. Oncol. 2013, 31, 3303–3306. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M.; et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients with Metastatic Lung Adenocarcinoma with EGFR Mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.-L.; Zhou, C.; Hu, C.-P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus Cisplatin plus Gemcitabine for First-Line Treatment of Asian Patients with Advanced Non-Small-Cell Lung Cancer Harbouring EGFR Mutations (LUX-Lung 6): An Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Wu, Y.-L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.-P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus Cisplatin-Based Chemotherapy for EGFR Mutation-Positive Lung Adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of Overall Survival Data from Two Randomised, Phase 3 Trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Smaill, J.B.; Gonzales, A.J.; Spicer, J.A.; Lee, H.; Reed, J.E.; Sexton, K.; Althaus, I.W.; Zhu, T.; Black, S.L.; Blaser, A.; et al. Tyrosine Kinase Inhibitors. 20. Optimization of Substituted Quinazoline and Pyrido[3,4-d]Pyrimidine Derivatives as Orally Active, Irreversible Inhibitors of the Epidermal Growth Factor Receptor Family. J. Med. Chem. 2016, 59, 8103–8124. [Google Scholar] [CrossRef]
- Ou, S.-H.; Soo, R. Dacomitinib in Lung Cancer: A “Lost Generation” EGFR Tyrosine-Kinase Inhibitor from a Bygone Era? Drug Des. Dev. Ther. 2015, 9, 5641–5653. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Lee, M.; Linke, R.; Rosell, R.; Corral, J.; et al. Improvement in Overall Survival in a Randomized Study That Compared Dacomitinib With Gefitinib in Patients with Advanced Non–Small-Cell Lung Cancer and EGFR-Activating Mutations. J. Clin. Oncol. 2018, 36, 2244–2250. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.-M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. PF00299804, an Irreversible Pan-ERBB Inhibitor, Is Effective in Lung Cancer Models with EGFR and ERBB2 Mutations That Are Resistant to Gefitinib. Cancer Res. 2007, 67, 11924–11932. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Fujino, T.; Nishino, M.; Koga, T.; Chiba, M.; Sesumi, Y.; Ohara, S.; Shimoji, M.; Tomizawa, K.; Takemoto, T.; et al. EGFR T790M and C797S Mutations as Mechanisms of Acquired Resistance to Dacomitinib. J. Thorac. Oncol. 2018, 13, 727–731. [Google Scholar] [CrossRef] [Green Version]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S Mutation Mediates Resistance to AZD9291 in Non–Small Cell Lung Cancer Harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus Gefitinib as First-Line Treatment for Patients with EGFR-Mutation-Positive Non-Small-Cell Lung Cancer (ARCHER 1050): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Jänne, P.A.; Yang, J.C.-H.; Kim, D.-W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.-J.; Kim, S.-W.; Su, W.-C.; Horn, L.; et al. AZD9291 in EGFR Inhibitor–Resistant Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finlay, M.R.V.; Anderton, M.; Ashton, S.; Ballard, P.; Bethel, P.A.; Box, M.R.; Bradbury, R.H.; Brown, S.J.; Butterworth, S.; Campbell, A.; et al. Discovery of a Potent and Selective EGFR Inhibitor (AZD9291) of Both Sensitizing and T790M Resistance Mutations That Spares the Wild Type Form of the Receptor. J. Med. Chem. 2014, 57, 8249–8267. [Google Scholar] [CrossRef]
- Yan, X.-E.; Ayaz, P.; Zhu, S.-J.; Zhao, P.; Liang, L.; Zhang, C.H.; Wu, Y.-C.; Li, J.-L.; Choi, H.G.; Huang, X.; et al. Structural Basis of AZD9291 Selectivity for EGFR T790M. J. Med. Chem. 2020, 63, 8502–8511. [Google Scholar] [CrossRef]
- Yosaatmadja, Y.; Silva, S.; Dickson, J.M.; Patterson, A.V.; Smaill, J.B.; Flanagan, J.U.; McKeage, M.J.; Squire, C.J. Binding Mode of the Breakthrough Inhibitor AZD9291 to Epidermal Growth Factor Receptor Revealed. J. Struct. Biol. 2015, 192, 539–544. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus Standard Chemotherapy as First-Line Treatment for European Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (EURTAC): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.-H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.-H.; Lee, K.H.; Lu, S.; et al. Afatinib versus Gefitinib as First-Line Treatment of Patients with EGFR Mutation-Positive Non-Small-Cell Lung Cancer (LUX-Lung 7): A Phase 2B, Open-Label, Randomised Controlled Trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Walter, A.O.; Sjin, R.T.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a Mutant-Selective Covalent Inhibitor of EGFR That Overcomes T790M-Mediated Resistance in NSCLC. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequist, L.V.; Soria, J.-C.; Goldman, J.W.; Wakelee, H.A.; Gadgeel, S.M.; Varga, A.; Papadimitrakopoulou, V.; Solomon, B.J.; Oxnard, G.R.; Dziadziuszko, R.; et al. Rociletinib in EGFR -Mutated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-E.; Zhu, S.-J.; Liang, L.; Zhao, P.; Choi, H.G.; Yun, C.-H. Structural Basis of Mutant-Selectivity and Drug-Resistance Related to CO-1686. Oncotarget 2017, 8, 53508–53517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.C.-H.; Reckamp, K.L.; Kim, Y.-C.; Novello, S.; Smit, E.F.; Lee, J.-S.; Su, W.-C.; Akerley, W.L.; Blakely, C.M.; Groen, H.J.M.; et al. Efficacy and Safety of Rociletinib Versus Chemotherapy in Patients With EGFR-Mutated NSCLC: The Results of TIGER-3, a Phase 3 Randomized Study. JTO Clin. Res. Rep. 2021, 2, 100114. [Google Scholar] [CrossRef]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [Green Version]
- Ercan, D.; Choi, H.G.; Yun, C.-H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Jänne, P.A. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Yao, M.-Y.; Zhu, S.-J.; Chen, J.-Y.; Yun, C.-H. Crystal Structure of EGFR T790M/C797S/V948R in Complex with EAI045. Biochem. Biophys. Res. Commun. 2018, 502, 332–337. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Liu, D. EAI045: The Fourth-Generation EGFR Inhibitor Overcoming T790M and C797S Resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef]
- Caporuscio, F.; Tinivella, A.; Restelli, V.; Semrau, M.S.; Pinzi, L.; Storici, P.; Broggini, M.; Rastelli, G. Identification of Small-Molecule EGFR Allosteric Inhibitors by High-Throughput Docking. Future Med. Chem. 2018, 10, 1545–1553. [Google Scholar] [CrossRef]
- Sickmier, E.A.; Kurzeja, R.J.M.; Michelsen, K.; Vazir, M.; Yang, E.; Tasker, A.S. The Panitumumab EGFR Complex Reveals a Binding Mechanism That Overcomes Cetuximab Induced Resistance. PLoS ONE 2016, 11, e0163366. [Google Scholar] [CrossRef]
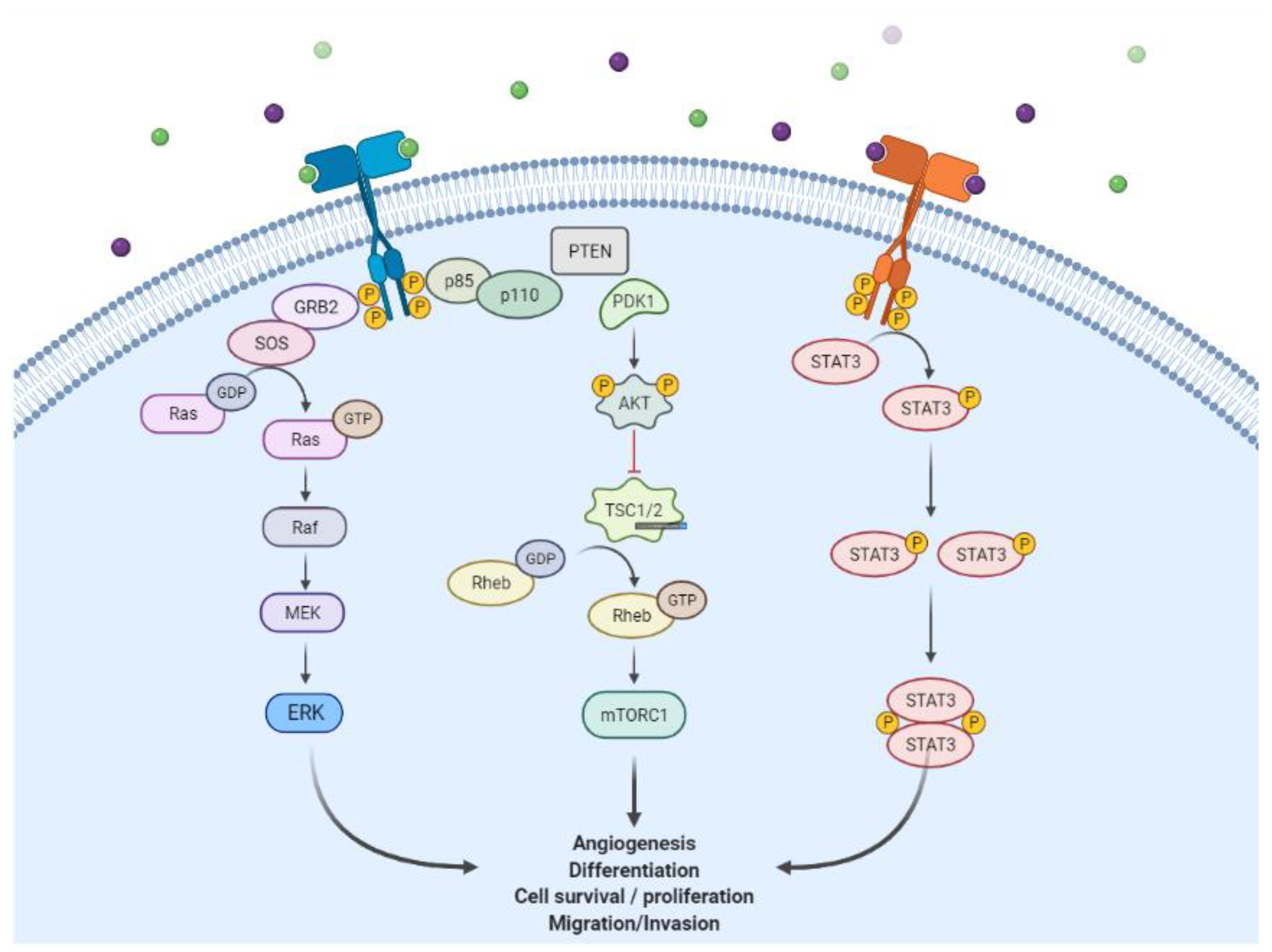
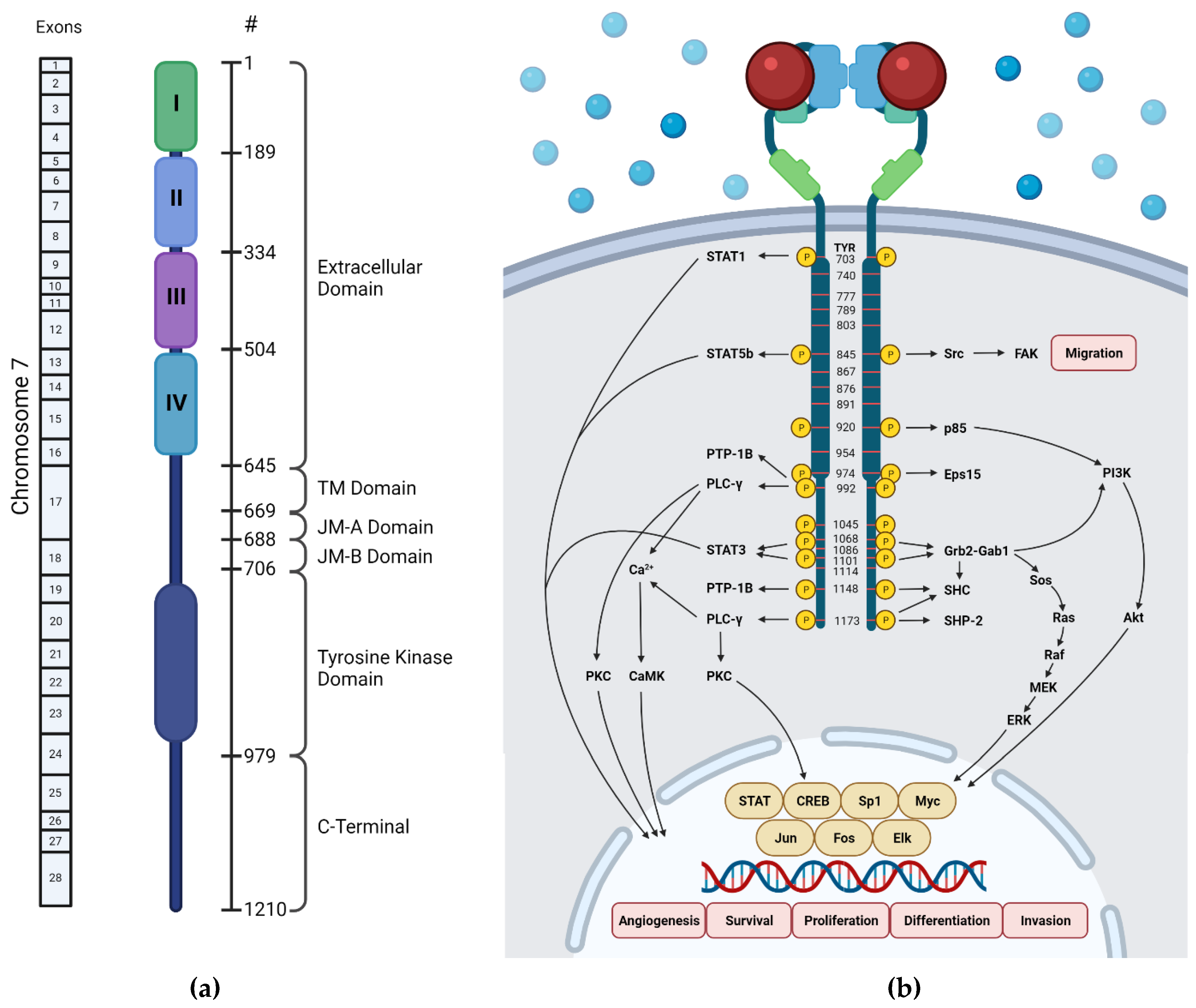
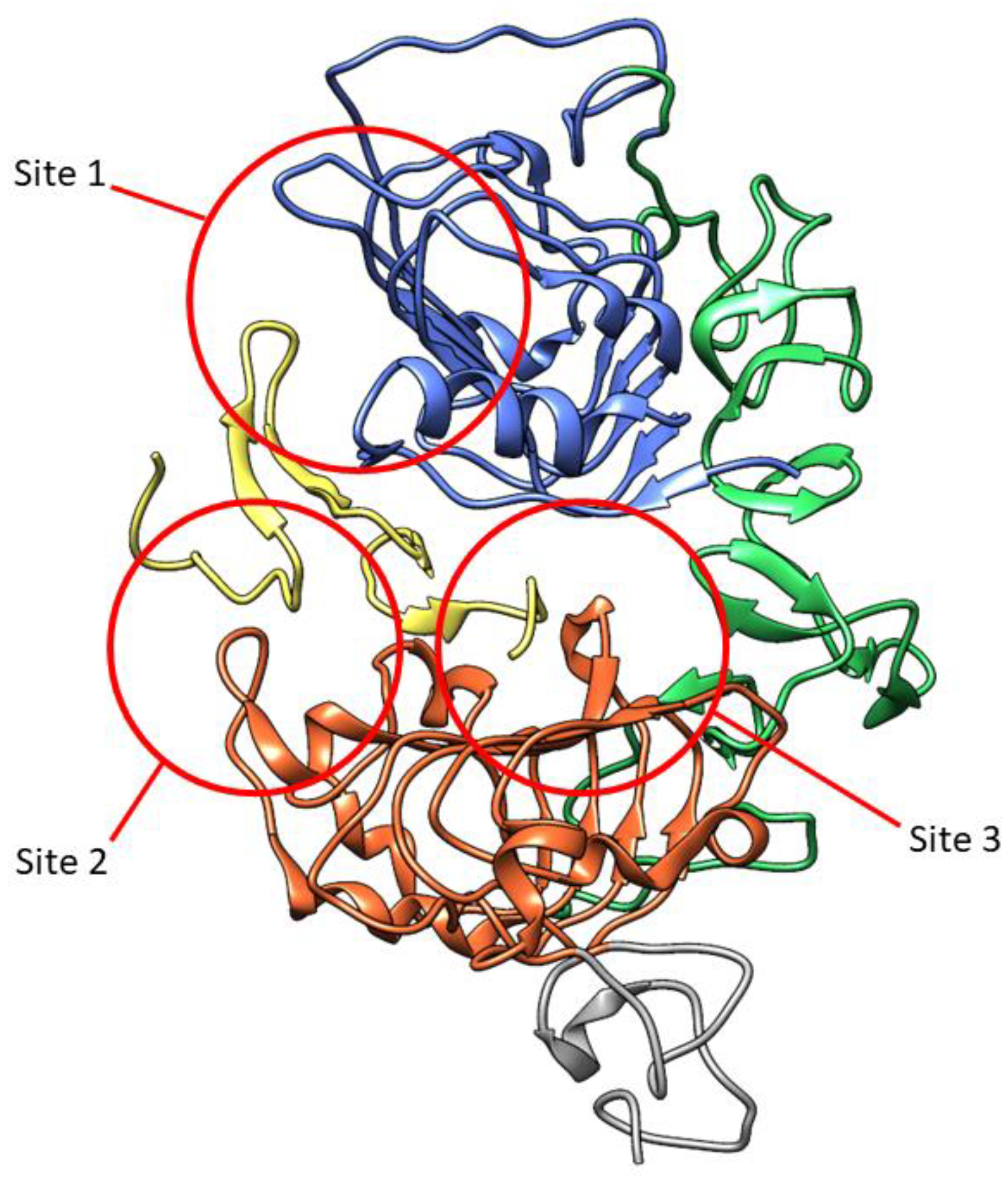
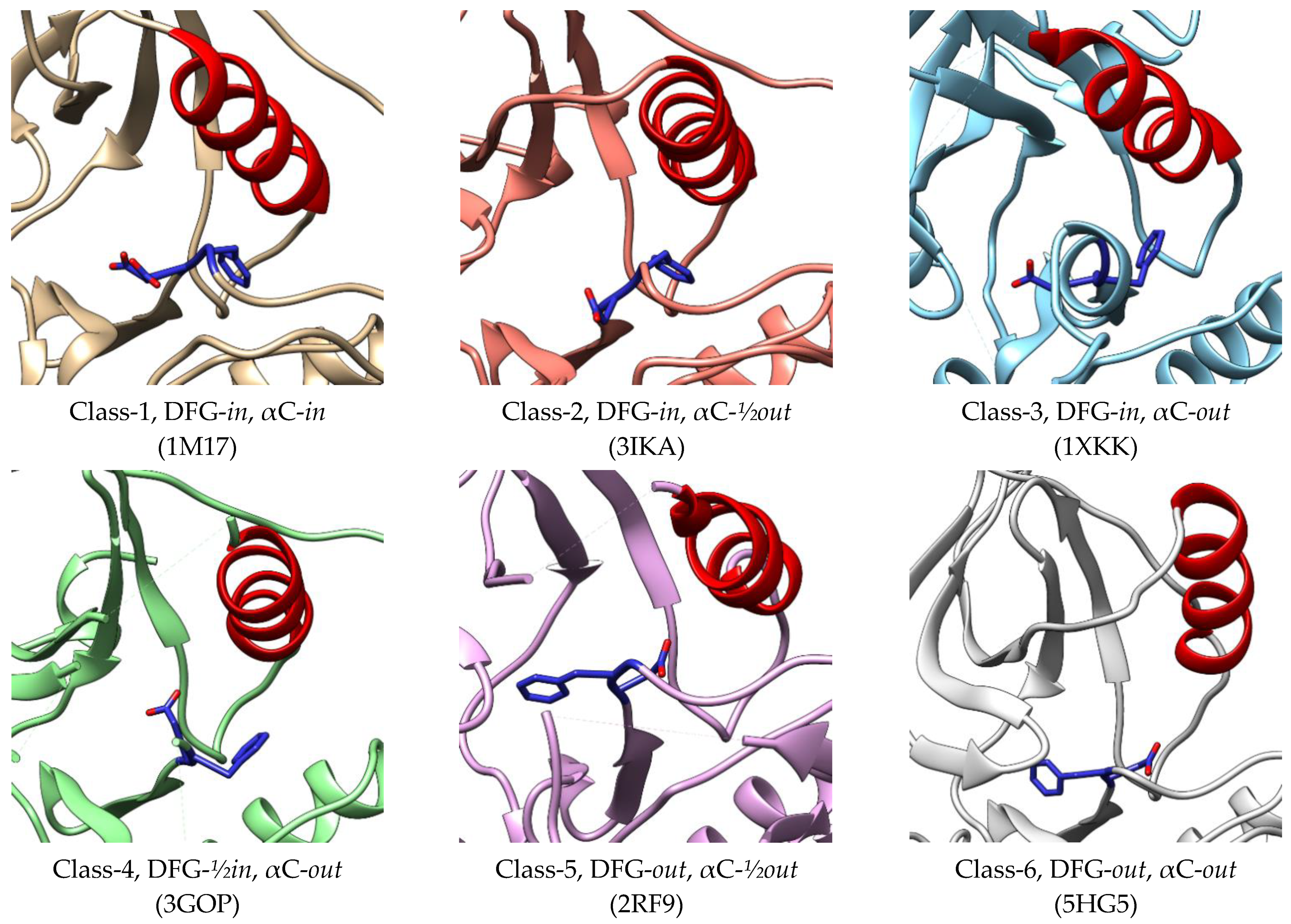
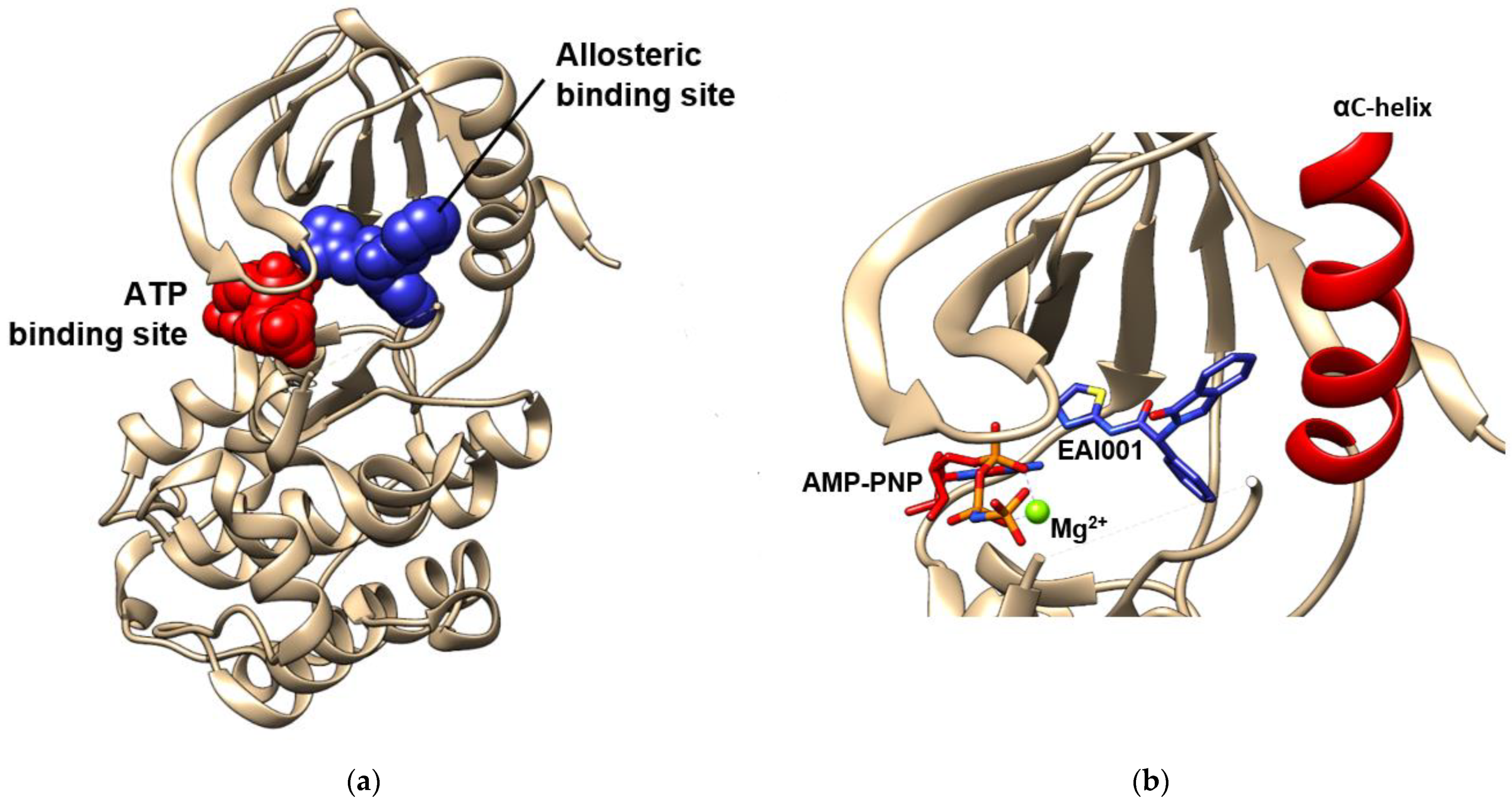
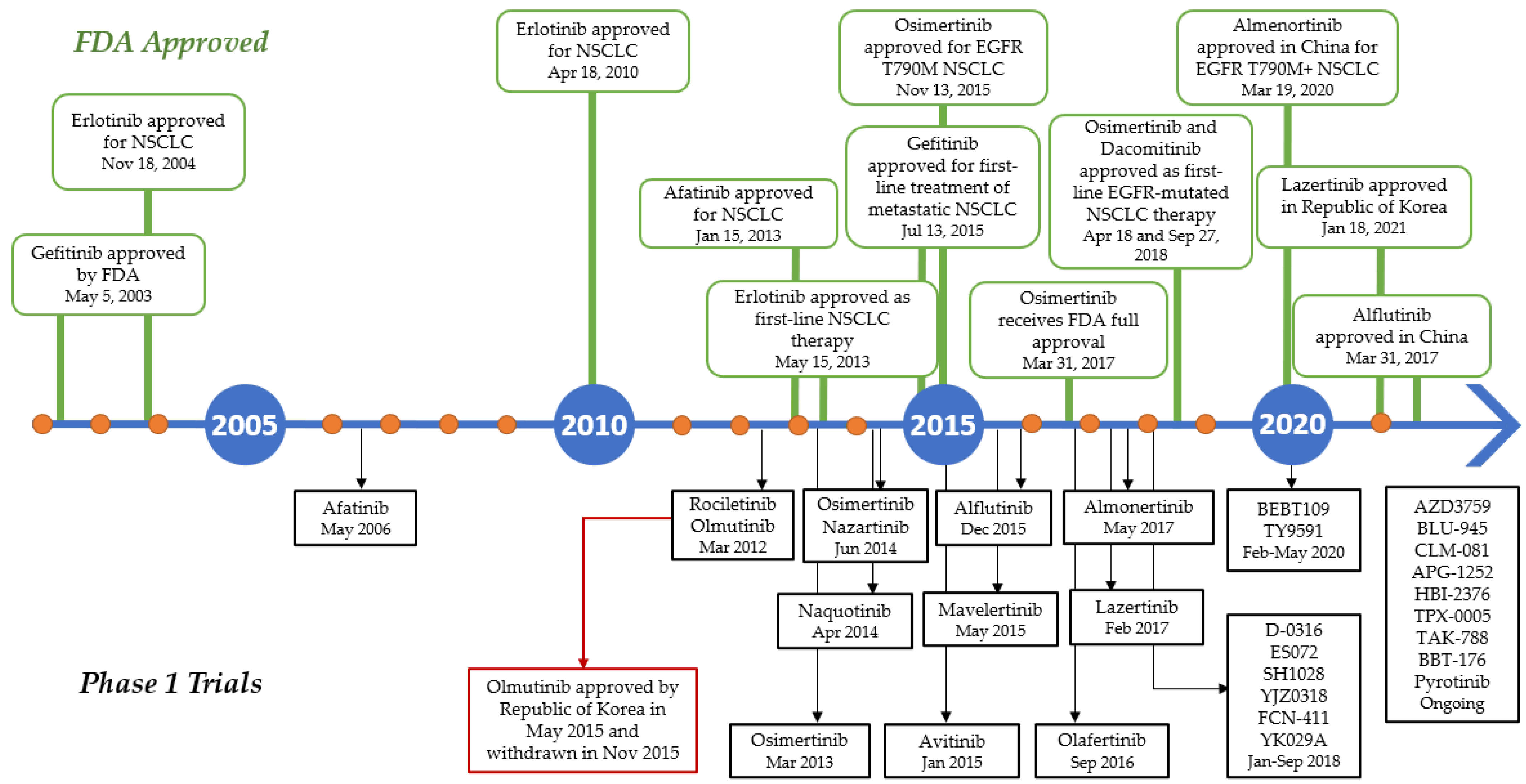
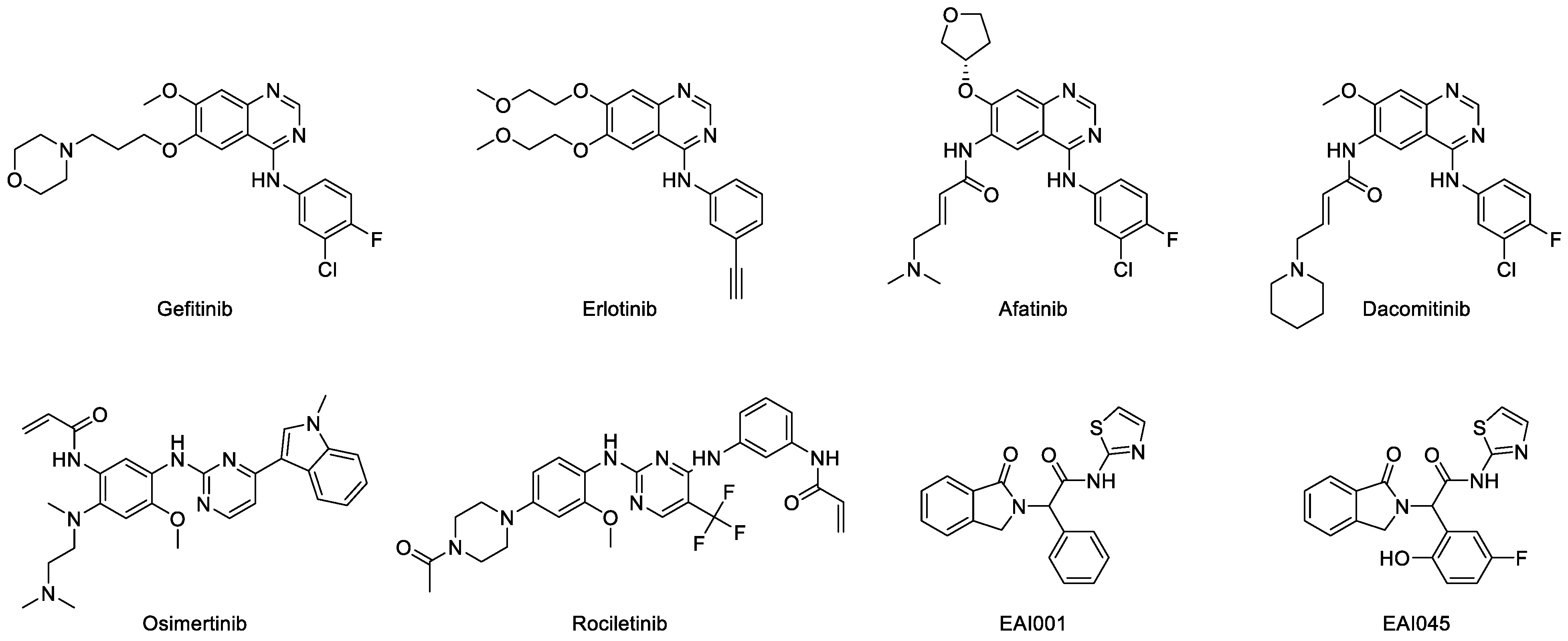
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).