
Part I: NiMoO4 Nanostructures Synthesized by the Solution Combustion Method: A Parametric Study on the Influence of Synthesis Parameters on the Materials’ Physicochemical, Structural, and Morphological Properties



Abstract
:
1. Introduction
2. Results and Discussion
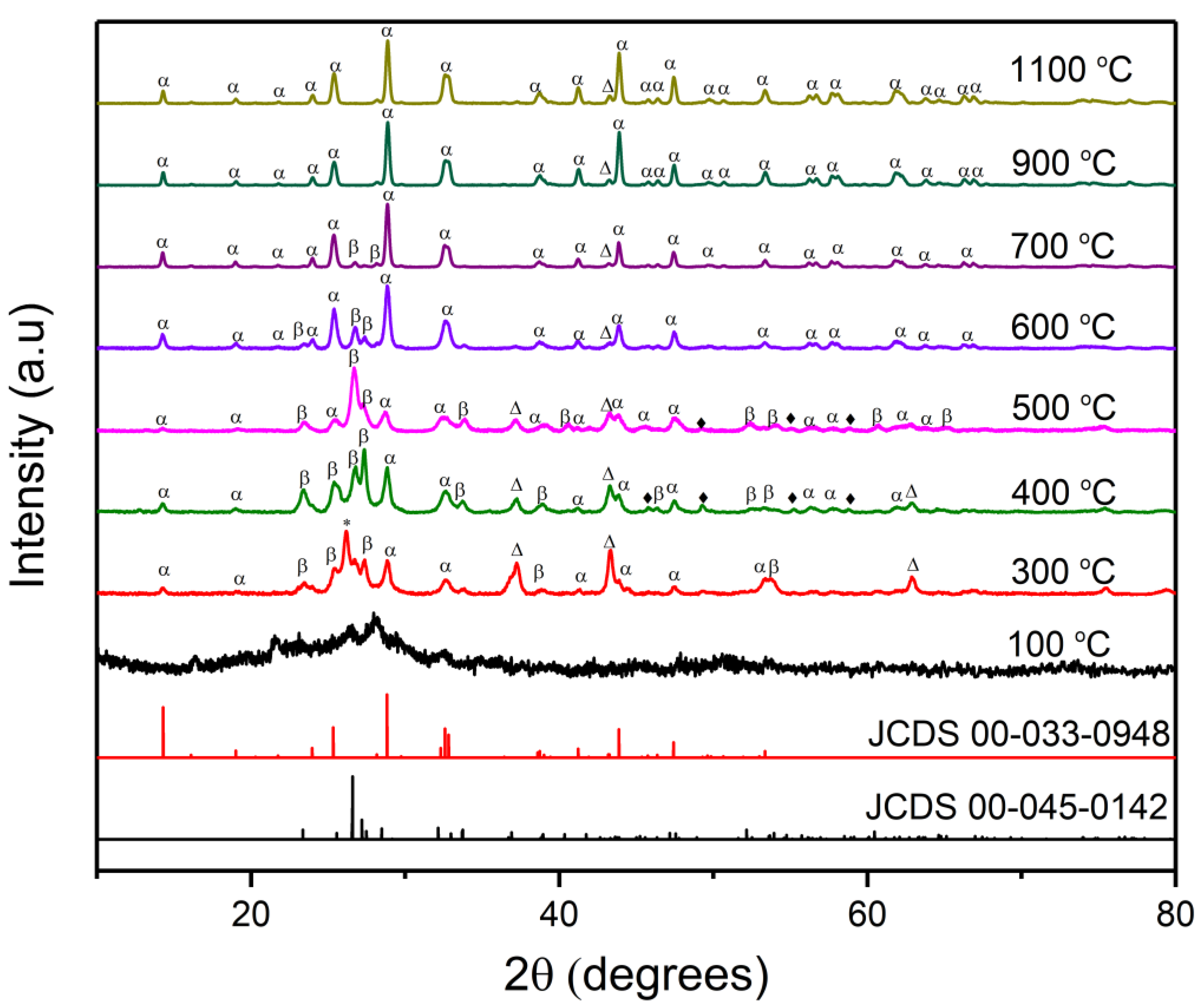
2.1. Effect of Calcination Temperature
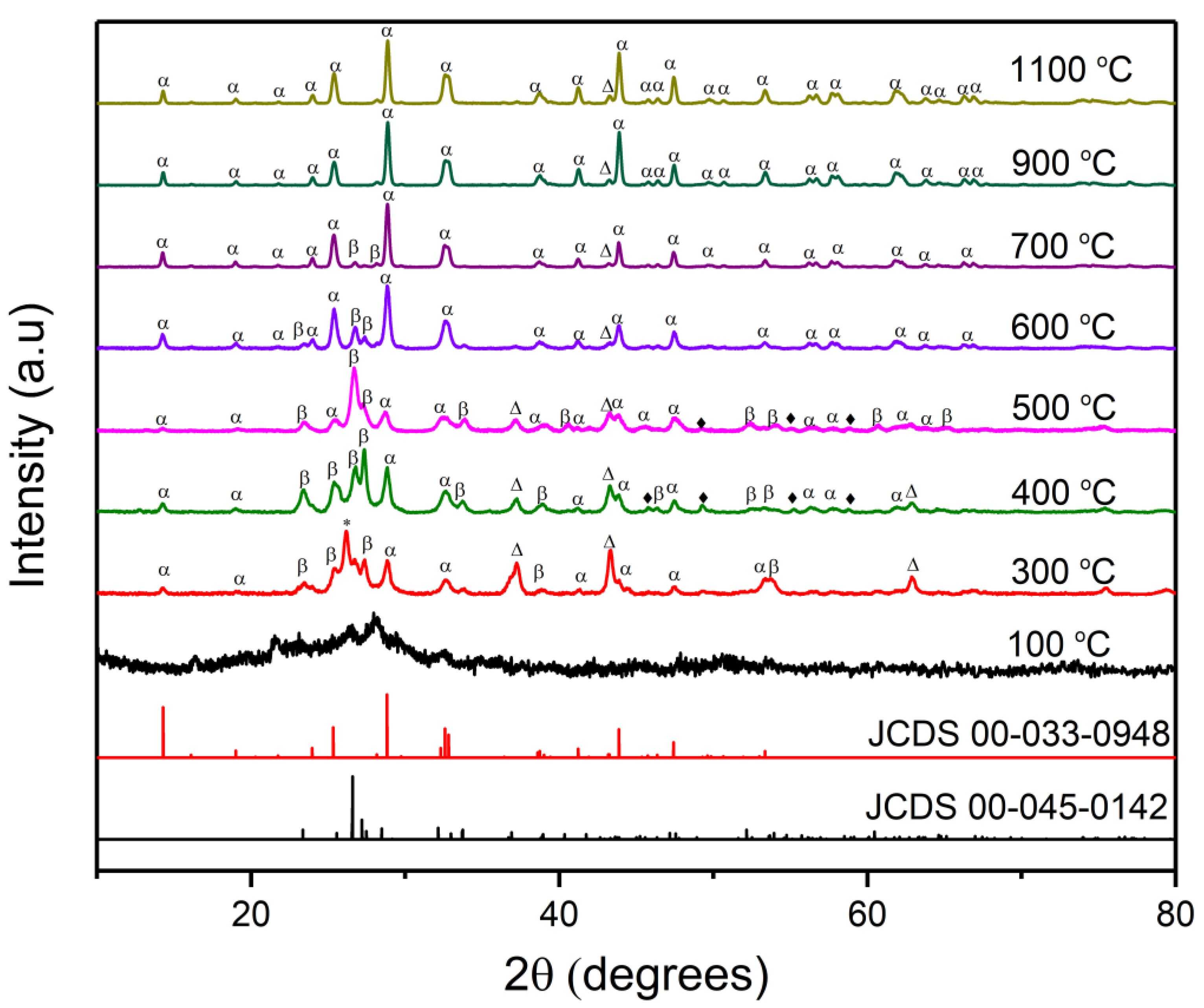
2.1.1. XRD Measurements
2.1.2. FTIR Characterization
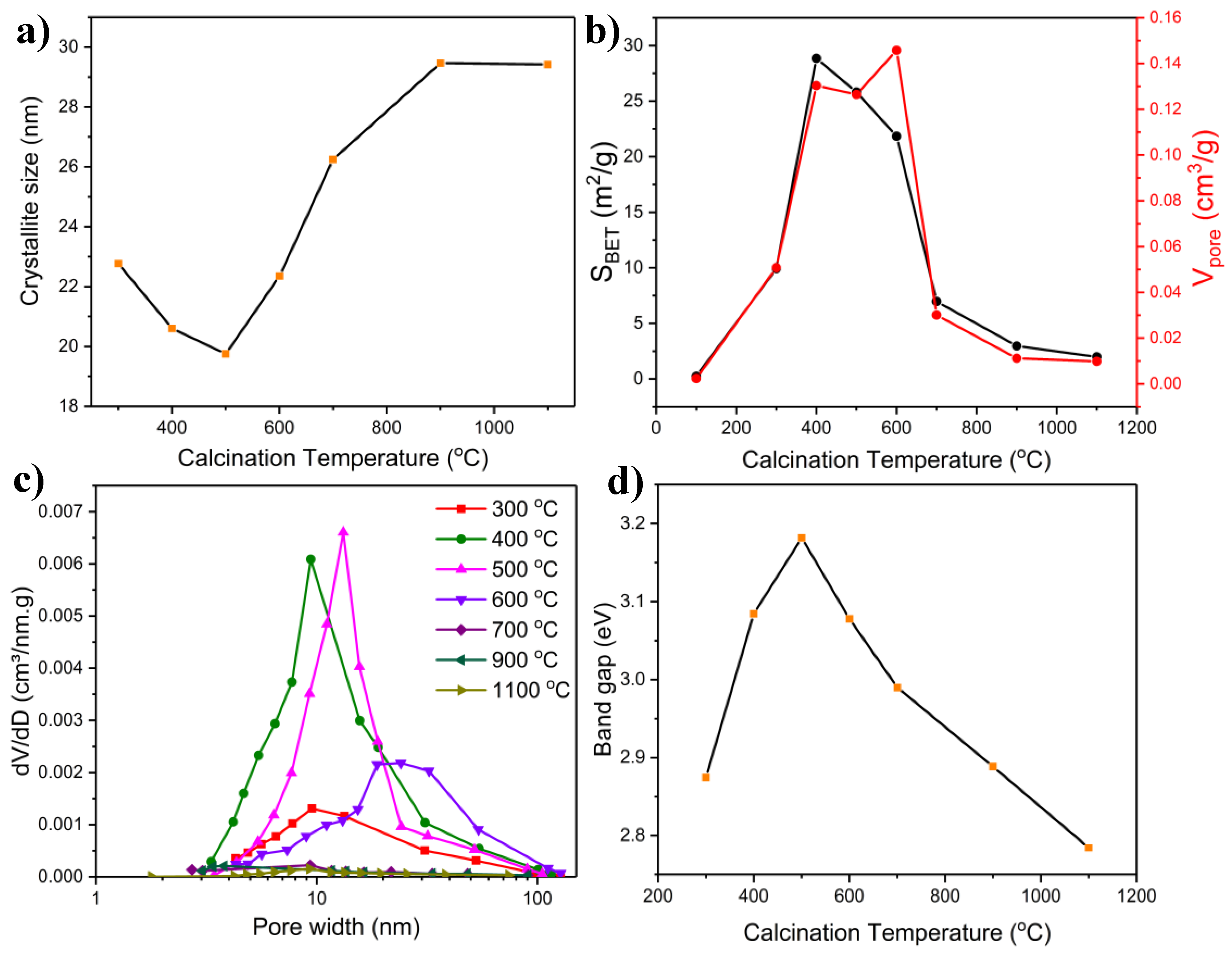
2.1.3. BET Measurements
2.1.4. Microscopy Characterization
2.1.5. Band Gap Measurements
2.2. Effect of Calcination Time
2.2.1. XRD Measurements
2.2.2. FTIR Characterization
2.2.3. BET Measurements
2.2.4. Microscopy Characterization
2.2.5. Band Gap Measurements
2.3. Effect of Composition
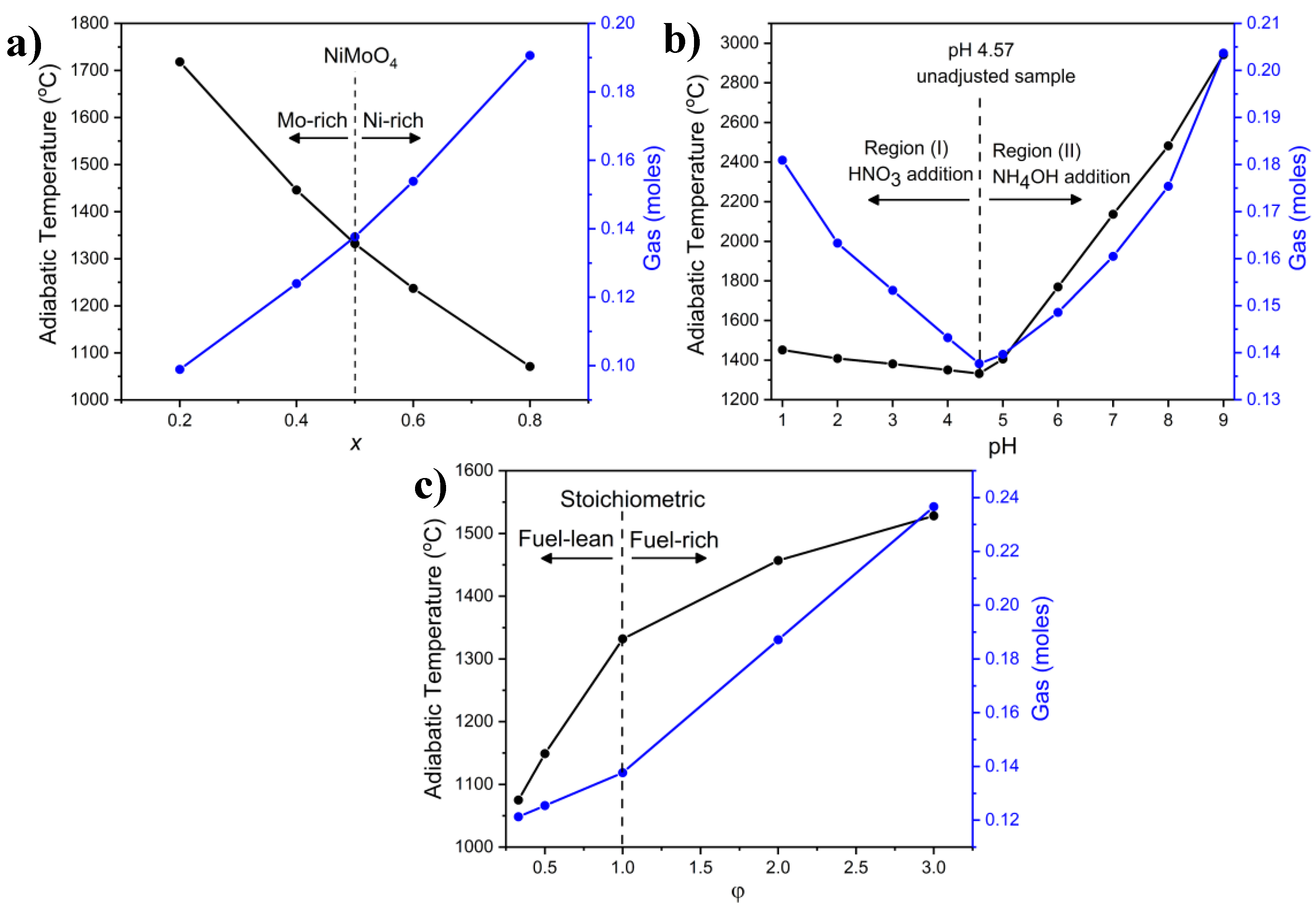
2.3.1. Combustion Parameters
2.3.2. XRD Measurements
2.3.3. FTIR Characterization
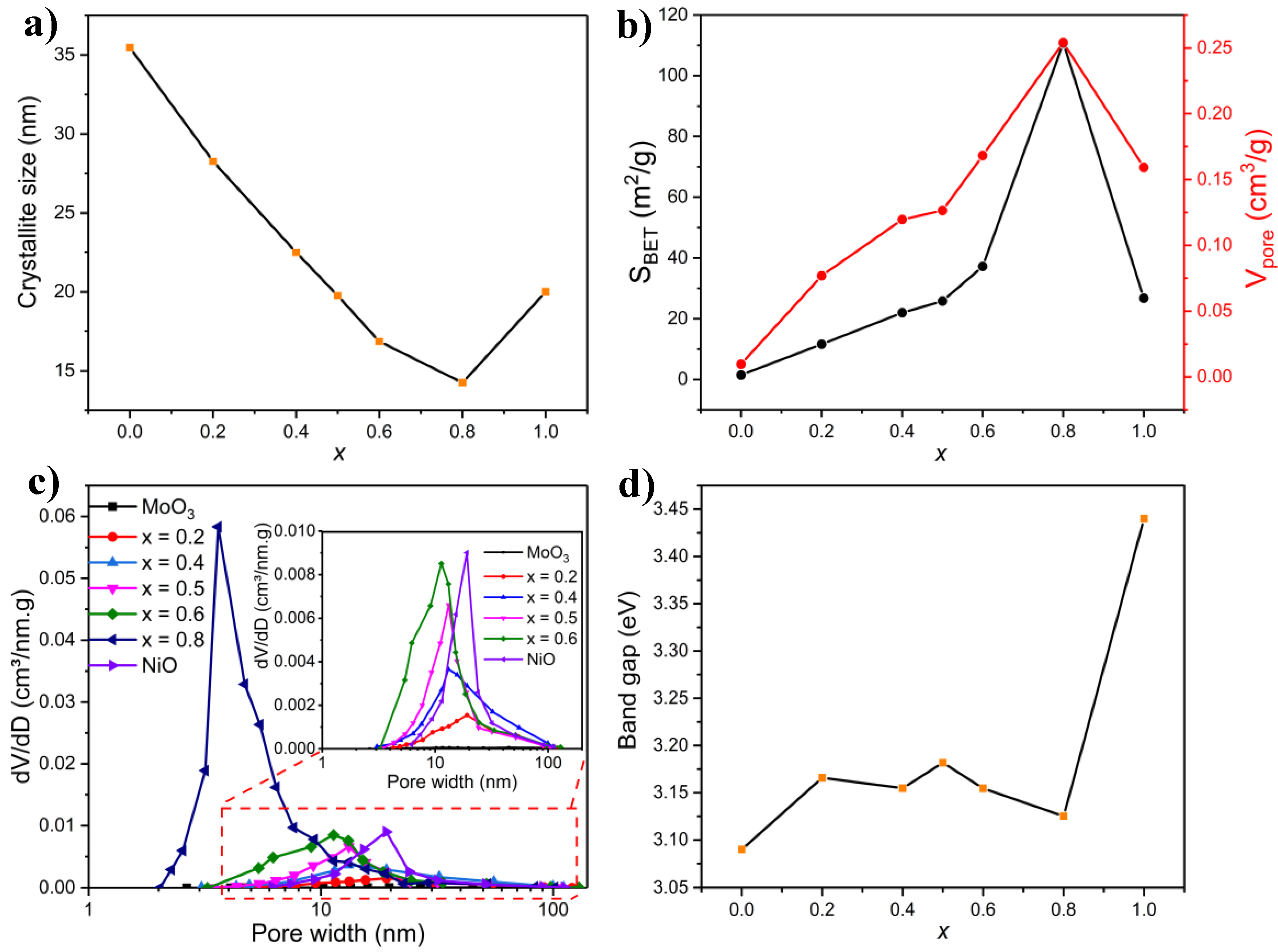
2.3.4. BET Measurements
2.3.5. Microscopy Characterization
2.3.6. Band Gap Measurements
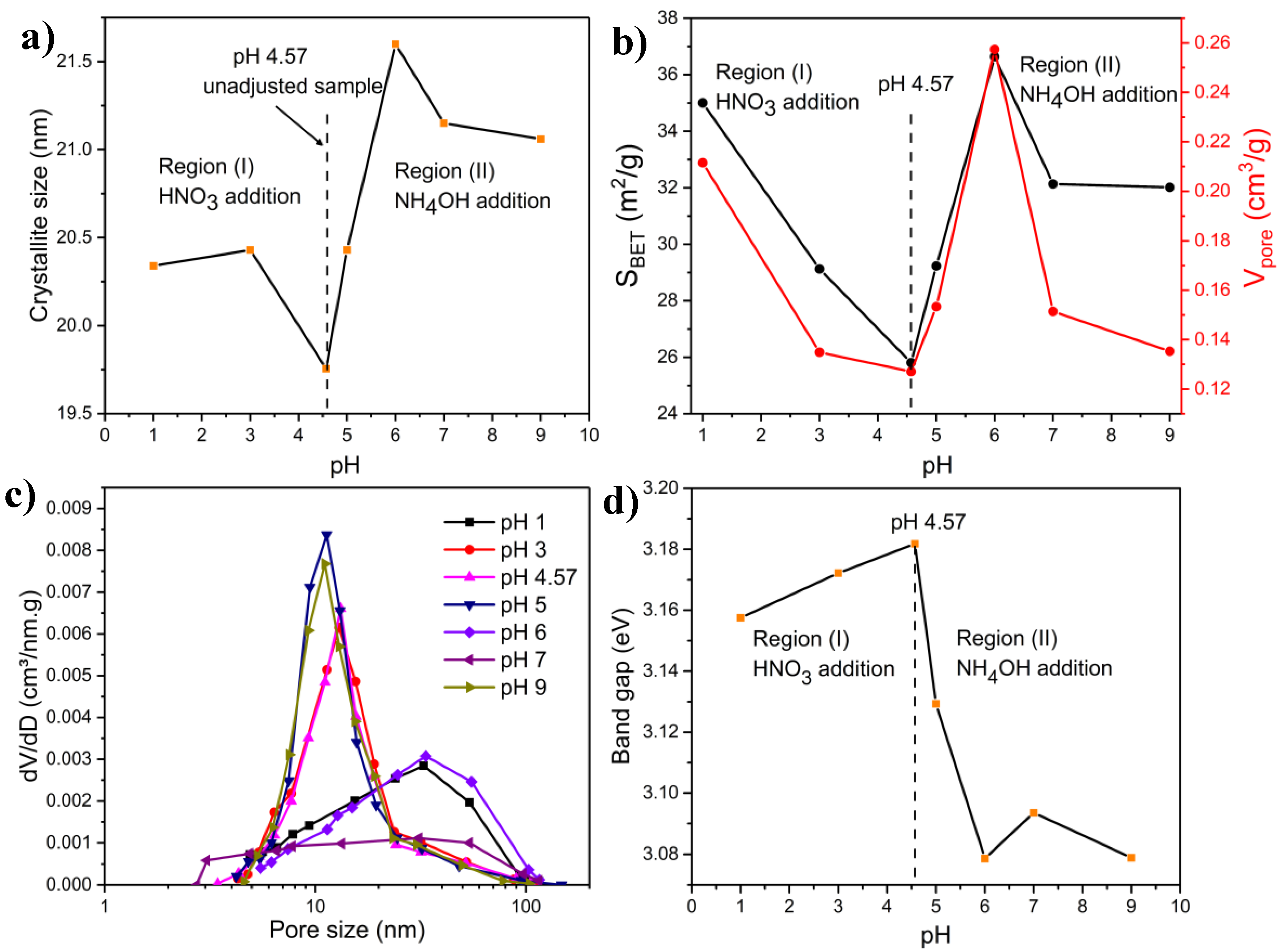
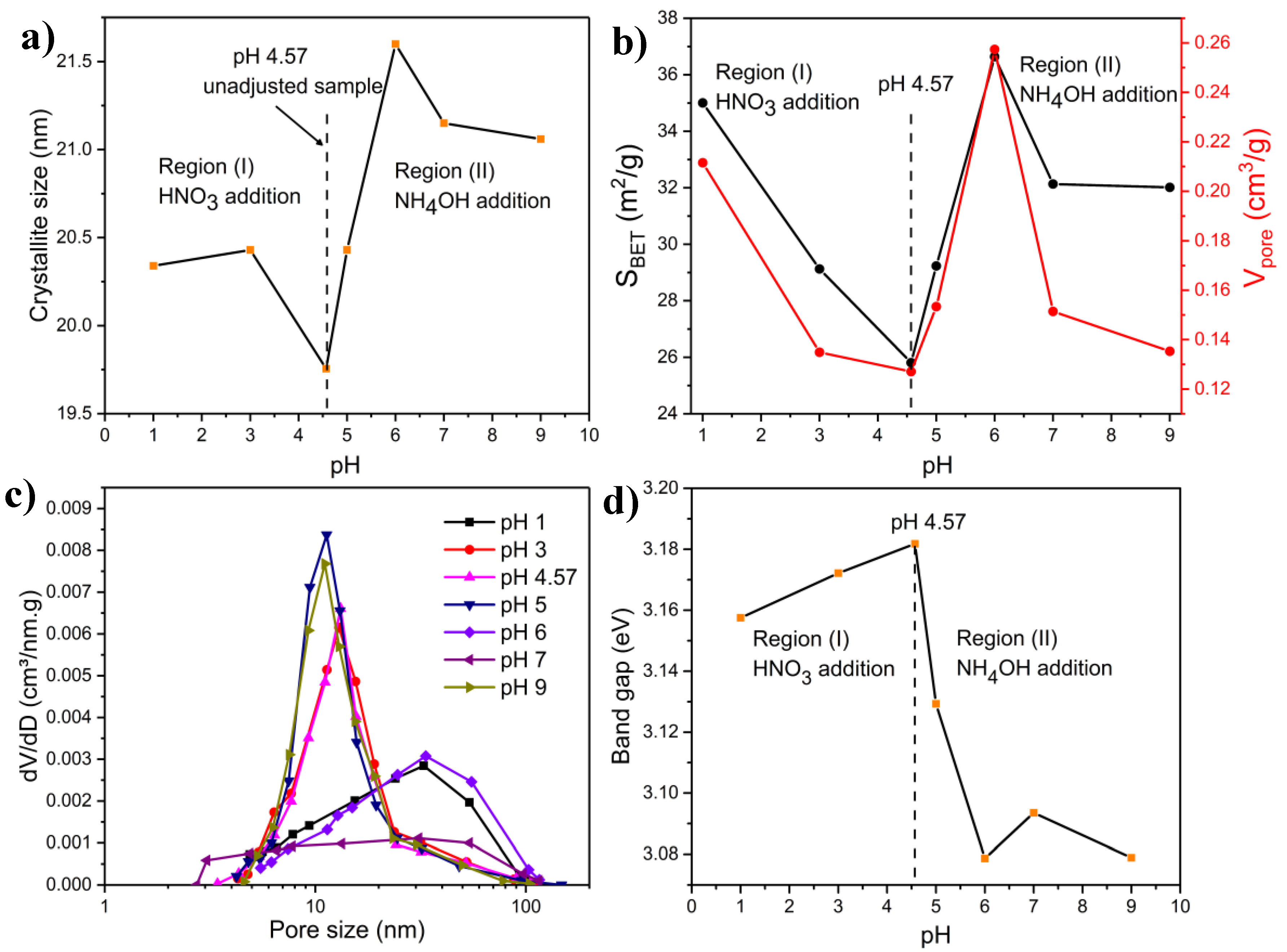
2.4. Effect of the pH of the Precursor Solution
2.4.1. Combustion Parameters
2.4.2. XRD Measurements
2.4.3. FTIR Characterization
2.4.4. BET Measurements
2.4.5. Microscopy Characterization
2.4.6. Band Gap Measurements
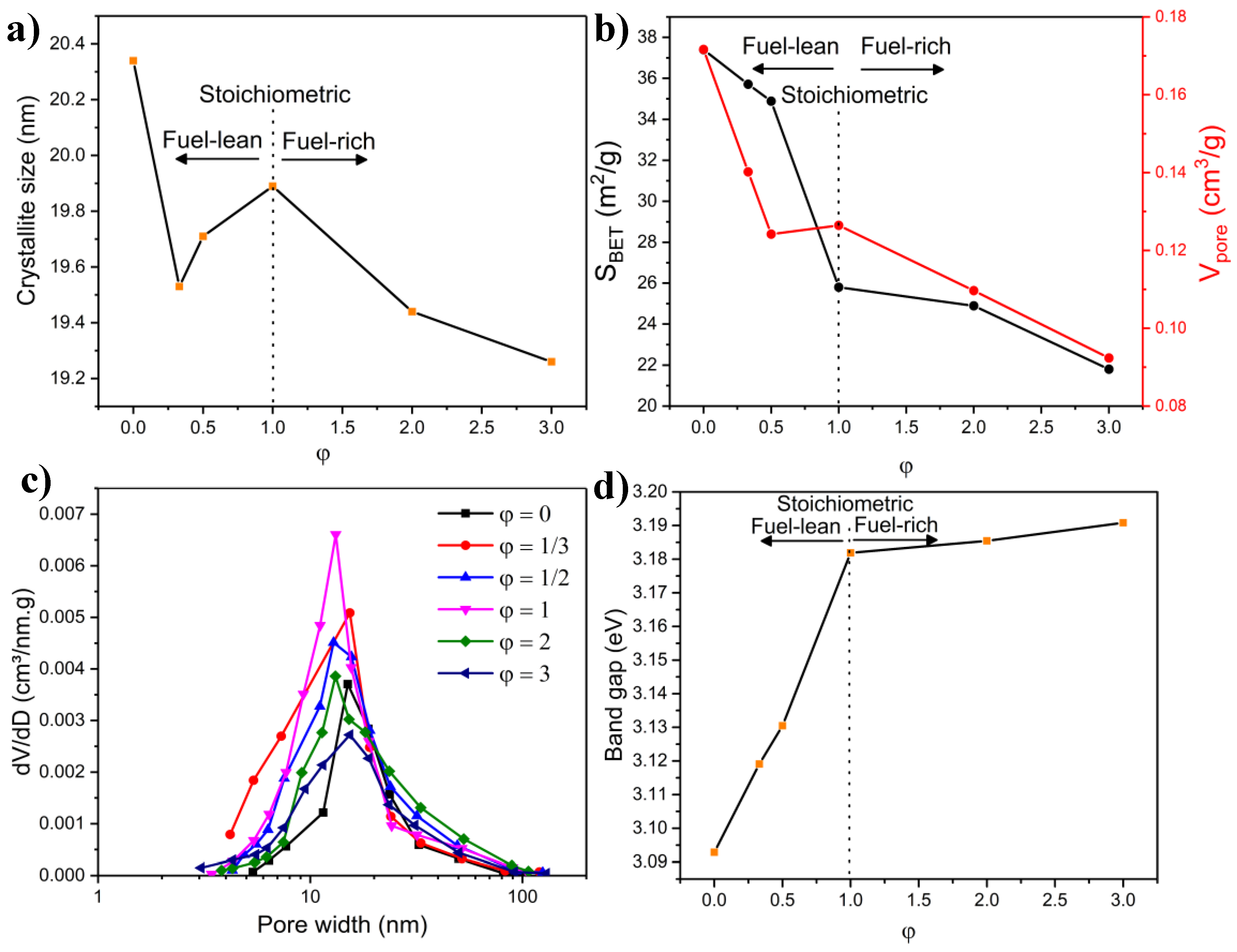
2.5. Effect of Fuel Content
2.5.1. Combustion Parameters
2.5.2. XRD Measurements
2.5.3. FTIR Characterization
2.5.4. BET Measurements
2.5.5. Microscopy Characterization
2.5.6. Band Gap Measurements
3. Materials and methods
3.1. Material Synthesis
3.2. Characterization Techniques
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gawande, M.B.; Pandey, R.K.; Jayaram, R.V. Role of mixed metal oxides in catalysis science—Versatile applications in organic synthesis. Catal. Sci. 2012, 2, 1113–1125. [Google Scholar] [CrossRef]
- Madeira, L.; Portela, M.; Mazzocchia, C. Nickel molybdate catalysts and their use in the selective oxidation of hydrocarbons. Catal. Rev. 2004, 46, 53–110. [Google Scholar] [CrossRef]
- Jothi, P.R.; Shanthi, K.; Salunkhe, R.R.; Pramanik, M.; Malgras, V.; Alshehri, S.M.; Yamauchi, Y. Synthesis and Characterization of α-NiMoO4 Nanorods for Supercapacitor Application. Eur. J. Inorg. Chem. 2015, 2015, 3694–3699. [Google Scholar] [CrossRef]
- Adhikary, M.C.; Priyadarsini, M.; Rath, S.K.; Das, C.K. 3D porous NiMoO4 nanoflakes arrays for advanced supercapacitor electrodes. J. Nanopart. Res. 2017, 19, 314. [Google Scholar] [CrossRef]
- Yesuraj, J.; Padmaraj, O.; Suthanthiraraj, S.A. Synthesis, Characterization, and Improvement of Supercapacitor Properties of NiMoO4 Nanocrystals with Polyaniline. J. Inorg. Organomet. Polym. 2020, 30, 310–321. [Google Scholar] [CrossRef]
- Klissurski, D.; Mancheva, M.; Iordanova, R.; Tyuliev, G.; Kunev, B. Mechanochemical synthesis of nanocrystalline nickel molybdates. J. Alloys Compd. 2006, 422, 53–57. [Google Scholar] [CrossRef]
- Tadjarodi, A.; Pradehkhorram, R.; Imani, M.; Ebrahimi, S.; Salehi, M. A simplified microwave-assisted synthesis of NiMoO4 nanoparticles by using organic driving agent and study of photocatalytic activity. In Proceedings of the 18th International Electronic Conference on Synthetic Organic Chemistry (ECSOC-18), Basel, Switzerland, 10 October 2014. [Google Scholar]
- Ahn, J.H.; Park, G.D.; Kang, Y.C.; Lee, J.-H. Phase-pure β-NiMoO4 yolk-shell spheres for high-performance anode materials in lithium-ion batteries. Electrochim. Acta 2015, 174, 102–110. [Google Scholar] [CrossRef]
- Moreno, B.; Chinarro, E.; Colomer, M.; Jurado, J. Combustion synthesis and electrical behavior of nanometric β-NiMoO4. J. Phys. Chem. 2010, 114, 4251–4257. [Google Scholar] [CrossRef]
- Lezla, O.; Bordes, E.; Courtine, P.; Hecquet, G. Synergetic effects in the Ni-Mo-O system: Influence of preparation on catalytic performance in the oxidative dehydrogenation of propane. J. Catal. 1997, 170, 346–356. [Google Scholar] [CrossRef]
- Kakihana, M. Invited review “sol-gel” preparation of high temperature superconducting oxides. J. Sol-Gel Sci. Technol. 1996, 6, 7–55. [Google Scholar] [CrossRef]
- Varma, A.; Mukasyan, A.S.; Rogachev, A.S.; Manukyan, K.V. Solution combustion synthesis of nanoscale materials. Chem. Rev. 2016, 116, 14493–14586. [Google Scholar] [CrossRef]
- Carlos, E.; Martins, R.; Fortunato, E.; Branquinho, R. Frontispiece: Solution Combustion Synthesis: Towards a Sustainable Approach for Metal Oxides. Chem. A Eur. J. 2020, 26. [Google Scholar] [CrossRef]
- González-Cortés, S.L.; Imbert, F.E. Fundamentals, properties and applications of solid catalysts prepared by solution combustion synthesis (SCS). Appl. Catal. A Gen. 2013, 452, 117–131. [Google Scholar] [CrossRef]
- Kingsley, J.; Patil, K. A novel combustion process for the synthesis of fine particle α-alumina and related oxide materials. Mater. Lett. 1988, 6, 427–432. [Google Scholar] [CrossRef]
- Senthilkumar, B.; Sankar, K.V.; Selvan, R.K.; Danielle, M.; Manickam, M. Nano α-NiMoO4 as a new electrode for electrochemical supercapacitors. RSC Adv. 2013, 3, 352–357. [Google Scholar] [CrossRef]
- Mentus, S.; Tomić-Tucaković, B.; Majstorović, D.; Dimitrijević, R. Gel-combustion synthesis of NiO–MoO3 mixtures and their reduction to Ni–Mo alloys. Mater. Chem. Phys. 2008, 112, 254–261. [Google Scholar] [CrossRef]
- Neeraj, N.S.; Mordina, B.; Srivastava, A.K.; Mukhopadhyay, K.; Prasad, N.E. Impact of process conditions on the electrochemical performances of NiMoO4 nanorods and activated carbon based asymmetric supercapacitor. Appl. Surf. Sci. 2019, 473, 807–819. [Google Scholar] [CrossRef]
- Alibe, I.M.; Matori, K.A.; Sidek, H.A.A.; Yaakob, Y.; Rashid, U.; Alibe, A.M.; Zaid, M.; Hafiz, M.; Khiri, A.; Zulhasif, M. Effects of calcination holding time on properties of wide band gap willemite semiconductor nanoparticles by the polymer thermal treatment method. Molecules 2018, 23, 873. [Google Scholar] [CrossRef] [Green Version]
- Lwin, N.; Othman, R.; Noor, A.F.M.; Sreekantan, S.; Yong, T.C.; Singh, R.; Tin, C.-C. Influence of pH on the physical and electromagnetic properties of Mg–Mn ferrite synthesized by a solution combustion method. Mater. Charact. 2015, 110, 109–115. [Google Scholar] [CrossRef]
- Wen, W.; Wu, J.-M. Nanomaterials via solution combustion synthesis: A step nearer to controllability. RSC Adv. 2014, 4, 58090–58100. [Google Scholar] [CrossRef]
- Rammal, M.B.; Omanovic, S. Synthesis and characterization of NiO, MoO3, and NiMoO4 nanostructures through a green, facile method and their potential use as electrocatalysts for water splitting. Mater. Chem. Phys. 2020, 255, 123570. [Google Scholar] [CrossRef]
- Nethravathi, P.; Kumar, M.P.; Suresh, D.; Lingaraju, K.; Rajanaika, H.; Nagabhushana, H.; Sharma, S. Tinospora cordifolia mediated facile green synthesis of cupric oxide nanoparticles and their photocatalytic, antioxidant and antibacterial properties. Mater. Sci. Semicond. Process. 2015, 33, 81–88. [Google Scholar]
- Popa, M.; Crespo, D.; Calderon-Moreno, J.M.; Preda, S.; Fruth, V. Synthesis and structural characterization of single-phase BiFeO3 powders from a polymeric precursor. J. Am. Ceram. Soc. 2007, 90, 2723–2727. [Google Scholar] [CrossRef]
- Lee, D.W.; Won, J.H.; Shim, K.B. Low temperature synthesis of BaCeO3 nano powders by the citrate process. Mater. Lett. 2003, 57, 3346–3351. [Google Scholar] [CrossRef]
- Thomas, A.; Janáky, C.; Samu, G.F.; Huda, M.N.; Sarker, P.; Liu, J.P.; Van Nguyen, V.; Wang, E.H.; Schug, K.A.; Rajeshwar, K. Time-and energy-efficient solution combustion synthesis of binary metal tungstate nanoparticles with enhanced photocatalytic activity. ChemSusChem 2015, 8, 1652–1663. [Google Scholar] [CrossRef]
- Zãvoianu, R.; Dias, C.R.; Portela, M.F. Stabilisation of β-NiMoO4 in TiO2-supported catalysts. Catal. Commun. 2001, 2, 37–42. [Google Scholar] [CrossRef]
- Senthilkumar, B.; Meyrick, D.; Lee, Y.-S.; Selvan, R.K. Synthesis and improved electrochemical performances of nano β-NiMoO4–CoMoO4 xH2O composites for asymmetric supercapacitors. RSC Adv. 2013, 3, 16542–16548. [Google Scholar] [CrossRef]
- Brito, J.L.; Barbosa, A.L.; Albornoz, A.; Severino, F.; Laine, J. Nickel molybdate as precursor of HDS catalysts: Effect of phase composition. Catal. Lett. 1994, 26, 329–337. [Google Scholar] [CrossRef]
- Brito, J.L.; Laine, J.; Pratt, K.C. Temperature-programmed reduction of Ni-Mo oxides. J. Mater. Sci. 1989, 24, 425–431. [Google Scholar] [CrossRef]
- De Melo, O.; González, Y.; Climent-Font, A.; Galán, P.; Ruediger, A.; Sánchez, M.; Calvo-Mola, C.; Santana, G.; Torres-Costa, V. Optical and electrical properties of MoO2 and MoO3 thin films prepared from the chemically driven isothermal close space vapor transport technique. J. Phys. Condens. Matter 2019, 31, 295703. [Google Scholar] [CrossRef]
- Plyasova, L.M.; Andrushkevich, M.M. Study of Phase State of Molybdates Under Reaction Conditions of Partial Oxidation of Hydrocarbons. Kinet. Catal. 1974, 15, 1207–1210. [Google Scholar]
- Rodriguez, J.A.; Chaturvedi, S.; Hanson, J.C.; Albornoz, A.; Brito, J.L. Electronic properties and phase transformations in CoMoO4 and NiMoO4: XANES and time-resolved synchrotron XRD studies. J. Phys. Chem. B 1998, 102, 1347–1355. [Google Scholar] [CrossRef]
- Sekar, C.; Selvan, R.K.; Senthilkumar, S.; Senthilkumar, B.; Sanjeeviraja, C. Combustion synthesis and characterization of spherical α-MnMoO4 nanoparticles. Powder Technol. 2012, 215, 98–103. [Google Scholar] [CrossRef]
- Mazzocchia, C.; Aboumrad, C.; Diagne, C.; Tempesti, E.; Herrmann, J.; Thomas, G. On the NiMoO4 oxidative dehydrogenation of propane to propene: Some physical correlations with the catalytic activity. Catal. Lett. 1991, 10, 181–191. [Google Scholar] [CrossRef]
- Pillay, B.; Mathebula, M.R.; Friedrich, H.B. The oxidative dehydrogenation of n-hexane over Ni–Mo–O catalysts. Appl. Catal. A Gen. 2009, 361, 57–64. [Google Scholar] [CrossRef]
- Pourgolmohammad, B.; Masoudpanah, S.; Aboutalebi, M. Effects of the fuel type and fuel content on the specific surface area and magnetic properties of solution combusted CoFe2O4 nanoparticles. Ceram. Int. 2017, 43, 8262–8268. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Gaber, A.; Abdel-Rahim, M.; Abdel-Latief, A.; Abdel-Salam, M.N. Influence of calcination temperature on the structure and porosity of nanocrystalline SnO2 synthesized by a conventional precipitation method. Int. J. Electrochem. Sci. 2014, 9, 81–95. [Google Scholar]
- Sun, Z.-X.; Zheng, T.-T.; Bo, Q.-B.; Du, M.; Forsling, W. Effects of calcination temperature on the pore size and wall crystalline structure of mesoporous alumina. J. Colloid Interface Sci. 2008, 319, 247–251. [Google Scholar] [CrossRef]
- Sathiskumar, P.; Thomas, C.; Madras, G. Solution combustion synthesis of nanosized copper chromite and its use as a burn rate modifier in solid propellants. Ind. Eng. Chem. Res. 2012, 51, 10108–10116. [Google Scholar] [CrossRef]
- Takht Ravanchi, M.; Rahimi Fard, M.; Fadaeerayeni, S.; Yaripour, F. Effect of calcination conditions on crystalline structure and pore size distribution for a mesoporous alumina. Chem. Eng. Commun. 2015, 202, 493–499. [Google Scholar] [CrossRef]
- Yang, L.; Wang, J.; Wan, Y.; Li, Y.; Xie, H.; Cheng, H.; Seo, H.J. Structure and effective visible-light-driven photocatalytic activity of α-NiMoO4 for degradation of methylene blue dye. J. Alloys Compd. 2016, 664, 756–763. [Google Scholar] [CrossRef]
- Dhanasekar, M.; Ratha, S.; Rout, C.S.; Bhat, S.V. Efficient sono-photocatalytic degradation of methylene blue using nickel molybdate nanosheets under diffused sunlight. J. Environ. Chem. Eng. 2017, 5, 2997–3004. [Google Scholar] [CrossRef]
- Pavithra, N.; Lingaraju, K.; Raghu, G.; Nagaraju, G. Citrus maxima (Pomelo) juice mediated eco-friendly synthesis of ZnO nanoparticles: Applications to photocatalytic, electrochemical sensor and antibacterial activities. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 185, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Teoh, L.G.; Li, K.-D. Synthesis and characterization of NiO nanoparticles by sol–gel method. Mater. Trans. 2012, 53, 2135–2140. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Xie, X.; Zhu, Q.; Lin, L.; Jiang, Y.-F.; Yang, Z.-K.; Zhou, X.; Xu, A.-W. Metallic MoO2 cocatalyst significantly enhances visible-light photocatalytic hydrogen production over MoO2/Zn0.5Cd0.5S heterojunction. Nanoscale 2015, 7, 5752–5759. [Google Scholar] [CrossRef]
- Li, M. Finite size and length effects on bandgap of CdSe nanorods. Phys. Chem. Chem. Phys. 2017, 19, 5513–5518. [Google Scholar] [CrossRef]
- Ratha, S.; Samantara, A.K.; Singha, K.K.; Gangan, A.S.; Chakraborty, B.; Jena, B.K.; Rout, C.S. Urea-assisted room temperature stabilized metastable β-NiMoO4: Experimental and theoretical insights into its unique bifunctional activity toward oxygen evolution and supercapacitor. ACS Appl. Mater. Interfaces 2017, 9, 9640–9653. [Google Scholar] [CrossRef]
- Shaat, S.; Musleh, H.; Zayed, H.; Tamous, H.; Issa, A.; Shurrab, N.; Asad, J.; Mousa, A.; Abu-Shawish, M.; AlDahoudi, N. Solution combustion-derived ZnO nanoparticles for photoanode of solar cells. Mater. Sci. Eng. B 2019, 241, 75–81. [Google Scholar] [CrossRef]
- Raphael, E.; Avellaneda, C.O.; Manzolli, B.; Pawlicka, A. Agar-based films for application as polymer electrolytes. Electrochim. Acta 2010, 55, 1455–1459. [Google Scholar] [CrossRef]
- Zhang, W.; Li, R.; Yang, B. Porous TiO2-Al2O3 composite photocatalyst prepared by sol-gel method: The role of calcination temperature. Nanosci. Nanotechnol.-Asia 2012, 2, 59–65. [Google Scholar]
- Porter, J.F.; Li, Y.-G.; Chan, C.K. The effect of calcination on the microstructural characteristics and photoreactivity of Degussa P-25 TiO2. J. Mater. Sci. 1999, 34, 1523–1531. [Google Scholar] [CrossRef]
- Venci, X.; George, A.; Raj, A.D.; Irudayaraj, A.A.; Raj, D.M.A.; Jayakumar, G.; Sundaram, S.J. Tuning the morphology and band gap of CdSe nanoparticles via solvothermal method. Mater. Today Proc. 2020, 36, 459–463. [Google Scholar] [CrossRef]
- Wu, Y.; He, Y.; Wu, T.; Chen, T.; Weng, W.; Wan, H. Influence of some parameters on the synthesis of nanosized NiO material by modified sol–gel method. Mater. Lett. 2007, 61, 3174–3178. [Google Scholar] [CrossRef]
- Ozkan, U.; Schrader, G.L. NiMoO4 selective oxidation catalysts containing excess MoO3 for the conversion of C4 hydrocarbons to maleic anhydride: I. Preparation and characterization. J. Catal. 1985, 95, 120–136. [Google Scholar] [CrossRef]
- Barrault, J.; Batiot, C.; Magaud, L.; Ganne, M. Selective oxidation of propane into oxygenated compounds over promoted nickel-molybdenum catalysts. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1997; Volume 110, pp. 375–382. [Google Scholar]
- Pilipenko, F.; Tsailingold, A.; Stepanov, G. Catalytic Activity of Nickel-Molybdenum Catalysts during Oxidative Dehydrogenation of N-Butane to 1, 3-Butadiene. Kinet. Catal. 1976, 17, 842–846. [Google Scholar]
- Thomas, G.; Sautel, M.; Kaddouri, A.; Mazzocchia, C. Comparison between electrical conductivity properties and catalytic activity of nickel molybdate. Solid State Ion. 1997, 101, 775–780. [Google Scholar] [CrossRef]
- Itenberg, I.S.; Andrushkevich, M.; Buyanov, R.; Khramova, G.; Sitnikov, V. Catalytic activity of an NiO-MoO3 system in a single-stage oxidative dehydrogenation of n-butane into divinyl. Kinet. Catal. 1976, 17, 867–870. [Google Scholar]
- Deganello, F.; Tyagi, A.K. Solution combustion synthesis, energy and environment: Best parameters for better materials. Prog. Cryst. Growth Charact. Mater. 2018, 64, 23–61. [Google Scholar] [CrossRef]
- Plyasova, L.; Ivanchenko, I.Y.; Andrushkevich, M.; Buyanov, R.; Itenberg, I.S.; Khramova, G.; Karakchiev, L.; Kustova, G.; Stepanov, G.; Tsailingol’d, A. Study of the phase composition of nickel-molybdenum catalysts. Kinet. Catal. 1973, 14, 882–886. [Google Scholar]
- Guo, X.; Mao, D.; Lu, G.; Wang, S.; Wu, G. Glycine–nitrate combustion synthesis of CuO–ZnO–ZrO2 catalysts for methanol synthesis from CO2 hydrogenation. J. Catal. 2010, 271, 178–185. [Google Scholar] [CrossRef]
- Saha, B.; Sarkar, K.; Bera, A.; Deb, K.; Thapa, R. Schottky diode behaviour with excellent photoresponse in NiO/FTO heterostructure. Appl. Surf. Sci. 2017, 418, 328–334. [Google Scholar] [CrossRef]
- Jaimeewong, P.; Promsawat, M.; Jiansirisomboon, S.; Watcharapasorn, A. Influence of pH values on the surface and properties of BCZT nanopowders synthesized via sol-gel auto-combustion method. Surf. Coat. Technol. 2016, 306, 16–20. [Google Scholar] [CrossRef]
- Komova, O.; Simagina, V.; Mukha, S.; Netskina, O.; Odegova, G.; Bulavchenko, O.; Ishchenko, A.; Pochtar, A. A modified glycine–nitrate combustion method for one-step synthesis of LaFeO3. Adv. Powder Technol. 2016, 27, 496–503. [Google Scholar] [CrossRef]
- Peng, T.; Liu, X.; Dai, K.; Xiao, J.; Song, H. Effect of acidity on the glycine–nitrate combustion synthesis of nanocrystalline alumina powder. Mater. Res. Bull. 2006, 41, 1638–1645. [Google Scholar] [CrossRef]
- Pathak, L.; Singh, T.; Das, S.; Verma, A.; Ramachandrarao, P. Effect of pH on the combustion synthesis of nano-crystalline alumina powder. Mater. Lett. 2002, 57, 380–385. [Google Scholar] [CrossRef]
- Junliang, L.; Wei, Z.; Cuijing, G.; Yanwei, Z. Synthesis and magnetic properties of quasi-single domain M-type barium hexaferrite powders via sol–gel auto-combustion: Effects of pH and the ratio of citric acid to metal ions (CA/M). J. Alloys Compd. 2009, 479, 863–869. [Google Scholar] [CrossRef]
- Deraz, N. Glycine-assisted fabrication of nanocrystalline cobalt ferrite system. J. Anal. Appl. Pyrolysis 2010, 88, 103–109. [Google Scholar] [CrossRef]
- Yue, Z.; Guo, W.; Zhou, J.; Gui, Z.; Li, L. Synthesis of nanocrystilline ferrites by sol–gel combustion process: The influence of pH value of solution. J. Magn. Magn. Mater. 2004, 270, 216–223. [Google Scholar] [CrossRef]
- Kaneti, Y.V.; Zakaria, Q.M.; Zhang, Z.; Chen, C.; Yue, J.; Liu, M.; Jiang, X.; Yu, A. Solvothermal synthesis of ZnO-decorated α-Fe2O3 nanorods with highly enhanced gas-sensing performance toward n-butanol. J. Mater. Chem. A 2014, 2, 13283–13292. [Google Scholar] [CrossRef]
- de Moura, A.P.; de Oliveira, L.H.; Rosa, I.L.; Xavier, C.S.; Lisboa-Filho, P.N.; Li, M.S.; La Porta, F.A.; Longo, E.; Varela, J.A. Structural, optical, and magnetic properties of NiMoO4 nanorods prepared by microwave sintering. Sci. World J. 2015, 2015, 315084. [Google Scholar] [CrossRef] [Green Version]
- Mak, W.C.; Olesen, K.; Sivlér, P.; Lee, C.-J.; Moreno-Jimenez, I.; Edin, J.; Courtman, D.; Skog, M.; Griffith, M. Controlled delivery of human cells by temperature responsive microcapsules. J. Funct. Biomater. 2015, 6, 439–453. [Google Scholar] [CrossRef]
- Adedirin, O.; Adamu, U.; Nnabuk, E.O. Removal of Cd (II) from aqueous solution using Bacillus subtilis and Escherichia coli immobilized in agarose gel: Equilibrium, kinetics and thermodynamic study. Arch. Appl. Sci. Res 2011, 3, 59–76. [Google Scholar]
- Liu, J.; Xue, Z.; Zhang, W.; Yan, M.; Xia, Y. Preparation and properties of wet-spun agar fibers. Carbohydr. Polym. 2018, 181, 760–767. [Google Scholar] [CrossRef]
- Danks, A.; Hall, S.; Schnepp, Z. The evolution of ‘sol–gel’chemistry as a technique for materials synthesis. Mater. Horiz. 2016, 3, 91–112. [Google Scholar] [CrossRef] [Green Version]
- Pourgolmohammad, B.; Masoudpanah, S.; Aboutalebi, M. Effect of starting solution acidity on the characteristics of CoFe2O4 powders prepared by solution combustion synthesis. J. Magn. Magn. Mater. 2017, 424, 352–358. [Google Scholar] [CrossRef]
- Musa, I.; Qamhieh, N. Study of optical energy gap and quantum confinment effects in Zinc Oxide nanoparticles and nanorods. Dig. J. Nanomater. Biostruct. 2019, 14, 119–125. [Google Scholar]
- Qu, R.; Liu, N.; Chen, Y.; Zhang, W.; Zhu, G.; Zhang, Q.; Feng, L. Morphology-Induced TiO2 Bandgap Change for Super Rapid Treatment of Dye Wastewater under Visible Light. Adv. Mater. Technol. 2017, 2, 1700125. [Google Scholar] [CrossRef]
- Deganello, F.; Marcì, G.; Deganello, G. Citrate–nitrate auto-combustion synthesis of perovskite-type nanopowders: A systematic approach. J. Eur. Ceram. Soc. 2009, 29, 439–450. [Google Scholar] [CrossRef]
- Nagabhushana, B.; Chakradhar, R.; Ramesh, K.; Prasad, V.; Shivakumara, C.; Chandrappa, G. Effect of fuel on the formation structure, transport and magnetic properties of LaMnO3+ δ nanopowders. Philos. Mag. 2010, 90, 2009–2025. [Google Scholar] [CrossRef]
- Schnepp, Z. Biopolymers as a flexible resource for nanochemistry. Angew. Chem. Int. Ed. 2013, 52, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, J.C.; Takimi, A.; Bergmann, C.P. Nanostructured cobalt oxides (Co3O4 and CoO) and metallic Co powders synthesized by the solution combustion method. Mater. Res. Bull. 2010, 45, 672–676. [Google Scholar] [CrossRef]
- Ribeiro, N.F.; Neto, R.C.; Moya, S.F.; Souza, M.M.; Schmal, M. Synthesis of NiAl2O4 with high surface area as precursor of Ni nanoparticles for hydrogen production. Int. J. Hydrogen Energy 2010, 35, 11725–11732. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (°C) | Phase Composition (wt.%) | ||||
|---|---|---|---|---|---|
| α-NiMoO4 | β-NiMoO4 | MoO2 | MoO3 | NiO | |
| 300 | 22.4 | 22.7 | 46 | 1.8 | 7.1 |
| 400 | 41.9 | 40.7 | 0 | 12.3 | 5.1 |
| 500 | 19.6 | 72.8 | 0 | 4.1 | 3.5 |
| 600 | 73.7 | 23 | 0 | 2.2 | 1 |
| 700 | 90.9 | 7.4 | 0 | 1.4 | 0.3 |
| 900 | 100 | 0 | 0 | 0 | 0 |
| 1100 | 100 | 0 | 0 | 0 | 0 |
| Time (min) | Phase Composition (wt.%) | ||||
|---|---|---|---|---|---|
| α-NiMoO4 | β-NiMoO4 | MoO2 | MoO3 | NiO | |
| As-combusted | 22.1 | 21.9 | 44.4 | 4.4 | 7.1 |
| 5 | 37.9 | 44.8 | 0 | 10.4 | 6.9 |
| 20 | 24.7 | 55.5 | 0 | 7 | 3.4 |
| 60 | 26.9 | 62.5 | 0 | 5.8 | 4.8 |
| 360 | 19.6 | 72.8 | 0 | 4.1 | 3.5 |
| 720 | 26.3 | 67.7 | 0 | 2.8 | 3.1 |
| 1440 | 41.4 | 51 | 0 | 2.5 | 5.1 |
| Nominal | Observed |
|---|---|
| MoO3 | MoO3 |
| Ni0.20Mo0.80Ox | Ni0.20Mo0.80Ox |
| Ni0.40Mo0.60Ox | Ni0.40Mo0.60Ox |
| Ni0.50Mo0.50Ox | Ni0.51Mo0.49Ox |
| Ni0.60Mo0.40Ox | Ni0.59Mo0.39Ox |
| Ni0.80Mo0.20Ox | Ni0.80Mo0.20Ox |
| NiO | NiO |
| Composition (x in NixMo1-x-oxide) | Phase Composition (wt.%) | ||||
|---|---|---|---|---|---|
| α-NiMoO4 | β-NiMoO4 | MoO2 | MoO3 | NiO | |
| 0 | 0 | 0 | 0 | 100 | 0 |
| 0.2 | 25.9 | 4.5 | 0 | 69.6 | 0 |
| 0.4 | 44.4 | 26.8 | 0 | 27.7 | 1.1 |
| 0.5 | 19.6 | 72.8 | 0 | 4.1 | 3.5 |
| 0.6 | 19.2 | 70.3 | 0 | 2.7 | 7.8 |
| 0.8 | – | – | – | – | – |
| 1 | 0 | 0 | 0 | 0 | 100 |
| pH | Phase Composition (wt.%) | ||||
|---|---|---|---|---|---|
| α-NiMoO4 | β-NiMoO4 | MoO2 | MoO3 | NiO | |
| 1 | 22.4 | 73.3 | 0 | 3.2 | 1.1 |
| 3 | 21.3 | 74.2 | 0 | 2.7 | 1.8 |
| 4.57 | 19.6 | 72.8 | 0 | 4.1 | 3.5 |
| 5 | 27.8 | 58.7 | 0 | 3.9 | 9.6 |
| 6 | 79.6 | 15.6 | 0 | 3.2 | 1.6 |
| 7 | 75 | 20.8 | 0 | 1.5 | 2.7 |
| 9 | 69.1 | 28.5 | 0 | 1.1 | 1.4 |
| Fuel-to-Oxidant Ratio φ | Phase Composition (wt.%) | ||||
|---|---|---|---|---|---|
| α-NiMoO4 | β-NiMoO4 | MoO2 | MoO3 | NiO | |
| 0 | 85.9 | 13.1 | 0 | 0.7 | 0.3 |
| 1/3 | 39.8 | 56 | 0 | 2.7 | 1.8 |
| 1/2 | 30.3 | 64.7 | 0 | 3.5 | 1.4 |
| 1 | 19.6 | 72.8 | 0 | 4.1 | 3.5 |
| 2 | 24.8 | 71.3 | 0 | 2.7 | 1.2 |
| 3 | 26.9 | 69.6 | 0 | 2.5 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rammal, M.B.; Omanovic, S. Part I: NiMoO4 Nanostructures Synthesized by the Solution Combustion Method: A Parametric Study on the Influence of Synthesis Parameters on the Materials’ Physicochemical, Structural, and Morphological Properties. Molecules 2022, 27, 776. https://doi.org/10.3390/molecules27030776
Rammal MB, Omanovic S. Part I: NiMoO4 Nanostructures Synthesized by the Solution Combustion Method: A Parametric Study on the Influence of Synthesis Parameters on the Materials’ Physicochemical, Structural, and Morphological Properties. Molecules. 2022; 27(3):776. https://doi.org/10.3390/molecules27030776
Chicago/Turabian StyleRammal, Mahmoud Bassam, and Sasha Omanovic. 2022. "Part I: NiMoO4 Nanostructures Synthesized by the Solution Combustion Method: A Parametric Study on the Influence of Synthesis Parameters on the Materials’ Physicochemical, Structural, and Morphological Properties" Molecules 27, no. 3: 776. https://doi.org/10.3390/molecules27030776
APA StyleRammal, M. B., & Omanovic, S. (2022). Part I: NiMoO4 Nanostructures Synthesized by the Solution Combustion Method: A Parametric Study on the Influence of Synthesis Parameters on the Materials’ Physicochemical, Structural, and Morphological Properties. Molecules, 27(3), 776. https://doi.org/10.3390/molecules27030776