Abstract
We report a new method for a tandem Pd-catalyzed intramolecular addition of active methylene compounds to internal alkynes followed by coupling with aryl and heteroaryl bromides. Highly substituted vinylidenecyclopentanes were obtained with good yields, complete selectivity, and excellent functional group tolerance. A plausible mechanism, supported by DFT calculations, involves the oxidative addition of bromoarene to Pd(0), followed by cyclization and reductive elimination. The excellent regio- and stereoselectivity arises from the 5-exo-dig intramolecular addition of the enol form of the substrate to alkyne activated by the π-acidic Pd(II) center, postulated as the rate-determining step.
1. Introduction
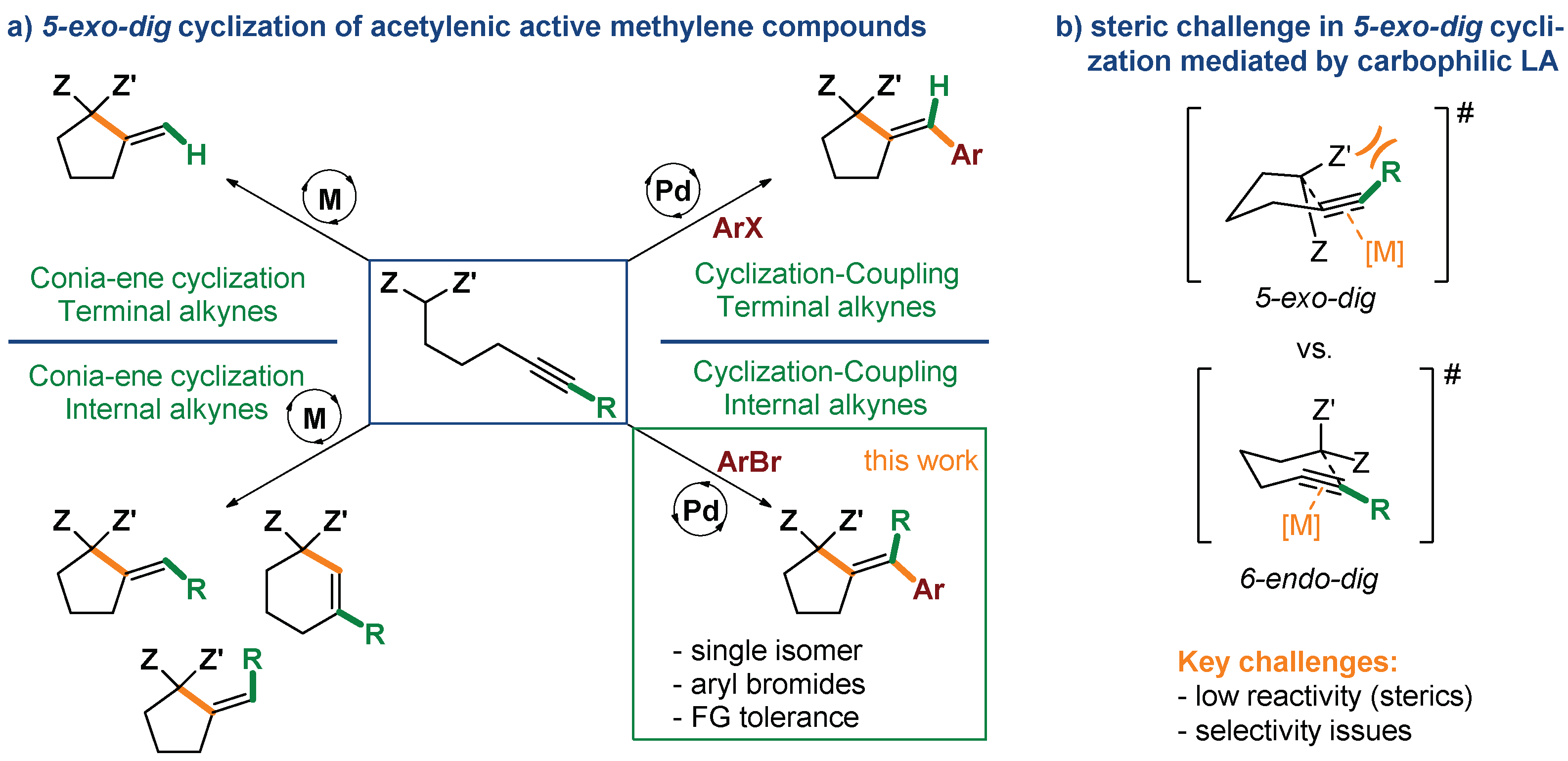
Cyclization of alkynes and alkenes bearing a tethered nucleophilic group constitutes a direct and effective strategy for the construction of a range of carbo- and heterocyclic scaffolds [1,2,3,4,5,6,7,8,9,10]. Since the seminal work by Conia [11,12] on thermal cyclization of unsaturated carbonyl compounds, a range of catalytic methods towards carbocyclic motifs, featuring excellent atom economy and high efficiency under mild conditions were developed (Scheme 1a) [13]. One of the most effective of such strategies relies on the use of transition metal complexes exhibiting carbophilic Lewis acidic character (e.g., Au, Pd, Pt, Cu, Ag), capable of coordination to the C-C multiple bond, and in consequence, its activation for nucleophilic attack [14,15,16,17,18,19,20,21,22]. This approach offers another opportunity for harnessing the reactivity of the metal–vinyl intermediate for further functionalization via cross-coupling, generating higher molecular complexity in a single step [23,24,25,26,27,28]. In this regard, palladium complexes are catalysts of choice due to the combination of sufficient π-acidity with redox activity for driving both cyclization and cross-coupling [1,4,29].
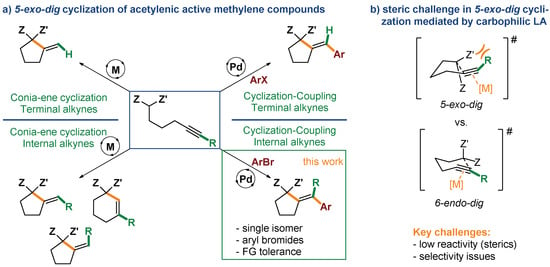
Scheme 1.
Strategies and challenges in methodologies involving 5-exo-dig cyclization of acetylenic active methylene compounds.
This strategy was first realized by Balme in the early 1990s for acetylenic keto esters and malonates with simple aryl iodides [30,31,32]. More recently, employing a catalyst based on a bulky, electron-rich monophosphine, we have solved one of the major limitations of the reaction, disclosing mild and functional group-tolerant conditions suitable for considerably less reactive (hetero)aryl bromides and chlorides [33,34]. However, one of the remaining challenges of both 5-exo-dig cycloisomerization and cyclization/coupling processes catalyzed by carbophilic Lewis acids is the low reactivity of substrates derived from internal alkynes. As the challenge arises from steric interactions of metal catalyst with a substituent at the alkyne terminus, shorter homologues cyclizing through a 5-endo-dig manifold, and thus lacking such constrains, undergo facile cyclization [16,26,28,35]. Therefore, only few protocols for Conia-ene 5-exo-dig cyclization of nonterminal acetylenes were reported to date [16,36,37,38]. Moreover, as observed for Au-catalyzed cycloisomerization of nonterminal acetylenic β-keto esters, competition between 5-exo-dig and 6-endo-dig cyclization manifolds could be expected, which ultimately could compromise the selectivity of the transformation (Scheme 1b) [16,37].
Herein, we present a protocol for effective Pd-catalyzed tandem 5-exo-dig carbocyclization of nonterminal acetylenic active methylene compounds with subsequent coupling with aryl and heteroaryl bromides. The present method exhibits high efficiency, excellent functional group tolerance and selectivity towards a single isomer of the cyclization/coupling product.
2. Results and Discussion
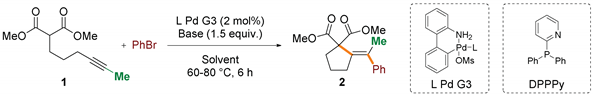
First, in the quest for a method suitable for challenging substrates bearing an internal alkyene motif, a benchmark reaction of dimethyl hex-4-yn-1-ylmalonate 1 with bromobenzene was evaluated over a range of Pd-catalysts and reaction conditions (Table 1) [39]. Third generation Buchwald-type palladacyclic precatalysts were chosen as a platform for testing of phosphine ligands due to availability, moisture and oxygen insensitivity, and rapid and clean activation under mildly basic conditions [40]. Initial screening of palladium complexes based on various mono- and bi-dentate phosphines (See Table S1 in the Supporting Information for details) revealed that bulky, electron-rich monophosphines, catalysts of choice for analogous terminal acetylenic active methylene compounds, exhibit poor or at best moderate efficiency in the model reaction involving internal alkyne (Table 1, entries 1–3). For instance, employment of 2 mol% of Pd-complexes with XPhos or RuPhos at 60 °C provided only 31% and 0% of expected cyclization/coupling product 2, respectively. It sharply contrasts the full conversion and ~90% yields observed for the reaction of dimethyl pent-4-yn-1-ylmalonate, a terminal analogue of 1. Despite the moderate yield of the transformation, the product was isolated as a single isomer, constituting a promising starting point for further optimization. Increase of the temperature to 80 °C only slightly improved the efficiency of the transformation catalyzed by the Pd/XPhos system (Table 1, entries 1 and 2). Generally, bidentate phosphines exhibited even lower efficiency, except for diphenyl-2-pyridylphosphine (DPPPY), which performed best of all tested ligands. Further evaluation of the experimental variables delivered satisfactory conditions (See Tables S2–S6 in the Supporting Information for details) involving the use of potassium phosphate in DMF. Employment of less polar solvents, as well as strong organic bases, resulted in considerably worse results (entries 7–11).

Table 1.
Evaluation of the reaction conditions 1.
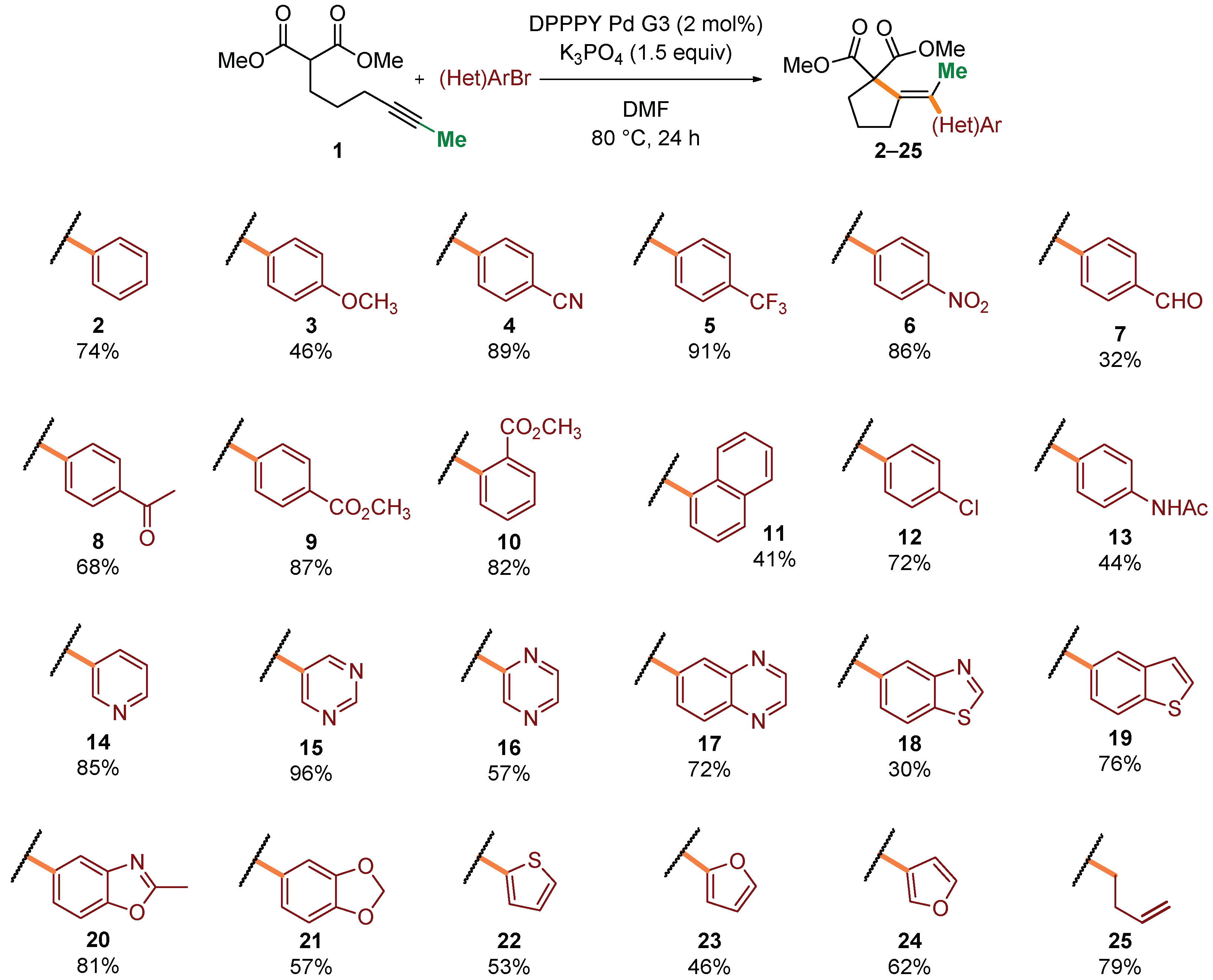
Having established satisfactory conditions for a benchmark reaction, we proceeded to the investigation of the scope of the process. A range of structurally and electronically diverse aryl and heteroaryl bromides were initially tested in the reaction with malonate 1 (Scheme 2). Generally, electron-deficient coupling partners provided higher yields of desired products 4–10 (up to 91%). Steric hindrance caused by shift of a substituent to the ortho position seemed to have little influence on the overall efficiency of the process (9 and 10). Various functional groups, including amides, ketones, and aldehydes were well tolerated (7–10, 14). Moreover, a range of heteroaryl bromides, including medicinally relevant N-heterocycles, were competent reaction partners, generally delivering products 14–24 in very good yields. Finally, vinyl bromide was also compatible with the reaction conditions, giving rise to diene 25 in 79% yield.
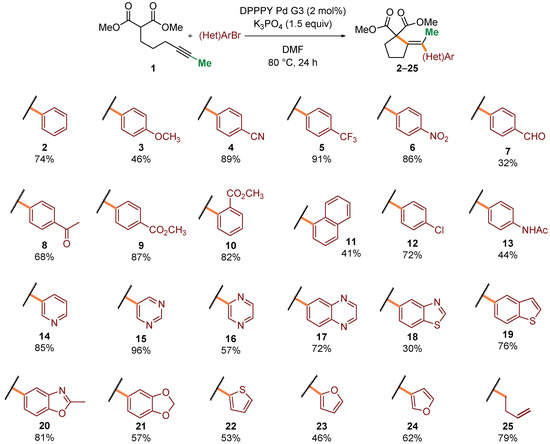
Scheme 2.
Scope in respect to aryl bromides. Standard conditions: Dimethyl 2-(hex-4-yn-1-yl)malonate (0.4 mmol), (hetero)aryl bromide (0.5 mmol, 1.25 equiv), DPPPY Pd G3 (5.1 mg, 8.0 μmol, 2 mol%), K3PO4 (127 mg, 0.6 mmol, 1.5 equiv), DMF (1.0 mL), 80 °C, 24 h. Isolated yields of pure products (after column chromatography) were reported.
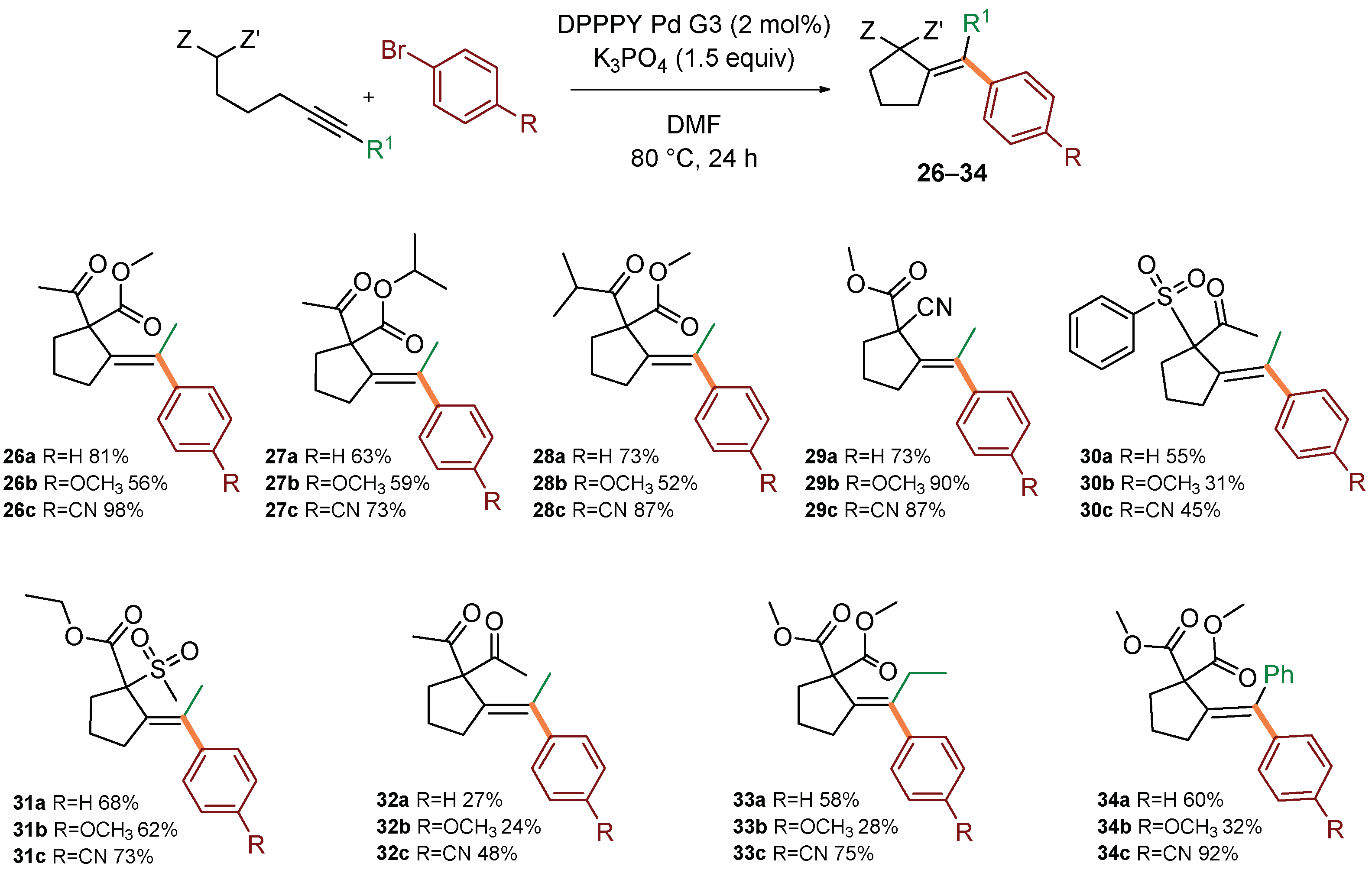
Next, the scope in respect to various actetylenic active methylenes was investigated in reactions with three prototypical aryl bromides of various electronic properties: bromobenzene, p-methoxybromobenzene, and p-cyanobromobenzene (Scheme 3). Generally, β-keto esters (leading to 26–28), cyanomalonates (leading to 29), sulfones (leading to 30–31) reacted with similar efficiency to model malonate 1, contrary to β-diketones which reacted in moderate yields (leading to 32). It could be speculated that low reactivity of dikentones arises from lower nucleophilicity of the enol form. In most cases, better yields were observed for the reaction with electron-deficient aryl bromides. Change of the substituent at the alkyne terminus to ethyl or phenyl was also tolerated, although slightly lower yields were observed (leading to 33–34).
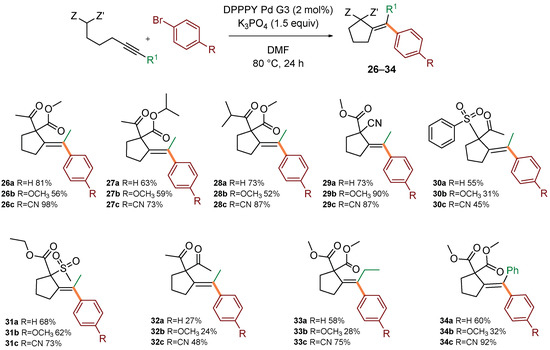
Scheme 3.
Scope in respect to acetylenic active methylenes. Standard conditions: acetylenic active methylene compound (0.4 mmol), (hetero)aryl bromide (0.5 mmol, 1.25 equiv), DPPPY Pd G3 (5.1 mg, 8.0 μmol, 2 mol%), K3PO4 (127 mg, 0.6 mmol, 1.5 equiv), DMF (1.0 mL), 80 °C, 24 h. Isolated yields of pure products (after column chromatography) were reported.
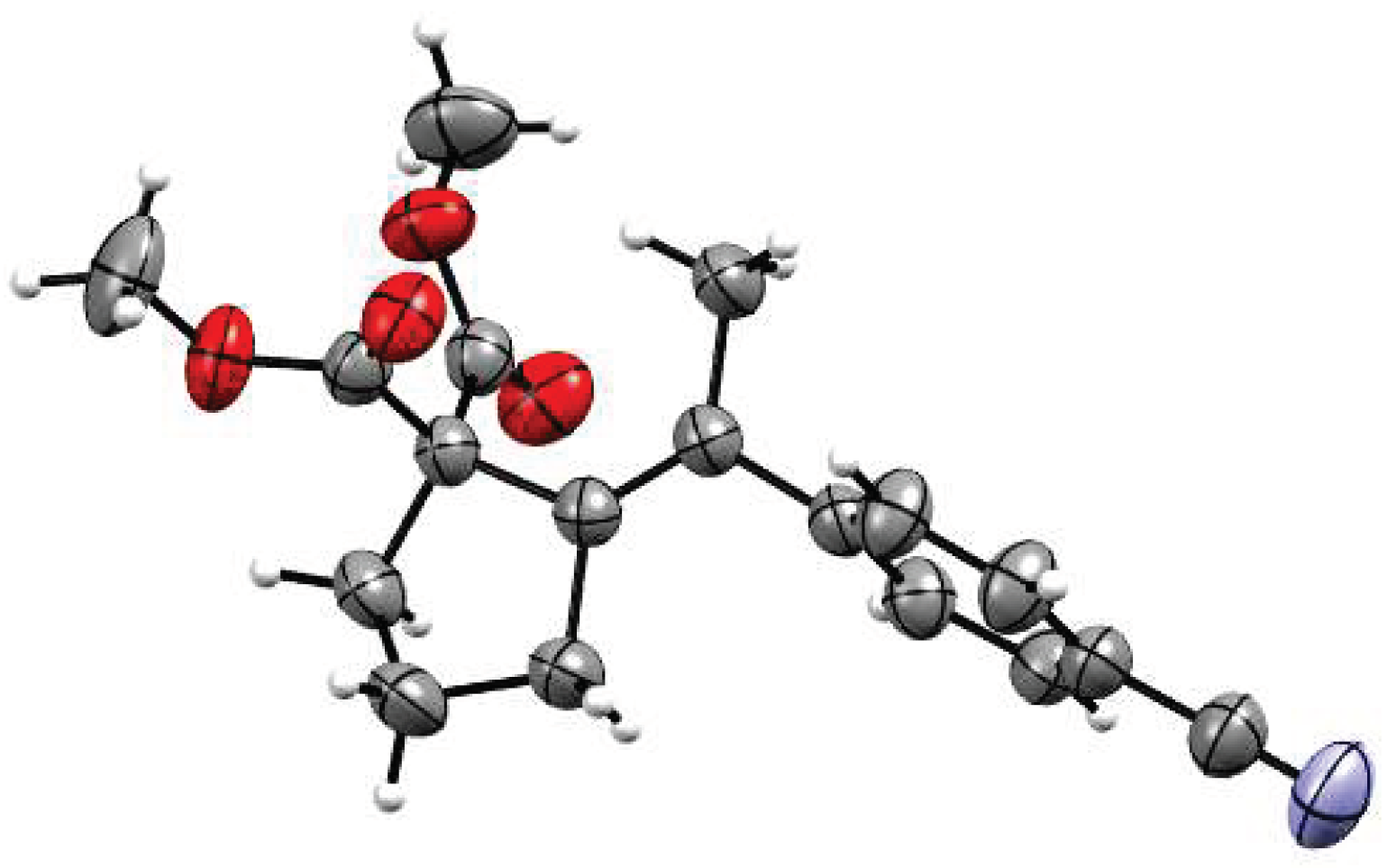
As mentioned, all reactions proceeded with complete regio- and stereoselectivity, delivering products as single isomers. The structure was unambiguously confirmed by X-ray crystallographic analysis of a representative compound 4 (Figure 1). Mechanistically, the stereoselectivity of the transformation implies involvement of anti carbometalation of the alkyne. Such selectivity could be achieved only if the C-nucleophilic fragment of the molecule (e.g., enolate) attacks the alkyne motif activated through coordination of the carbophilic Pd(II) centre. Other scenarios involving either simultaneous coordination of enolate and alkyne moieties, or initial insertion of alkyne to aryl-Pd species would preferentially lead to syn dicarbofunctionalization products.
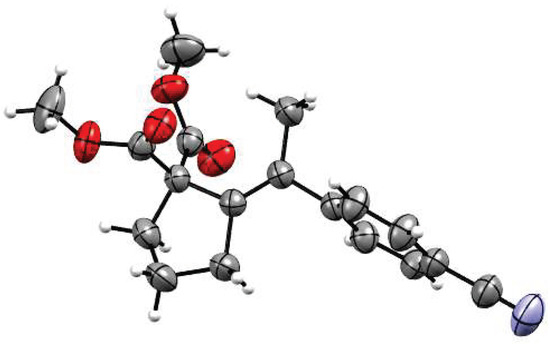
Figure 1.
X-ray molecular structure of 4.
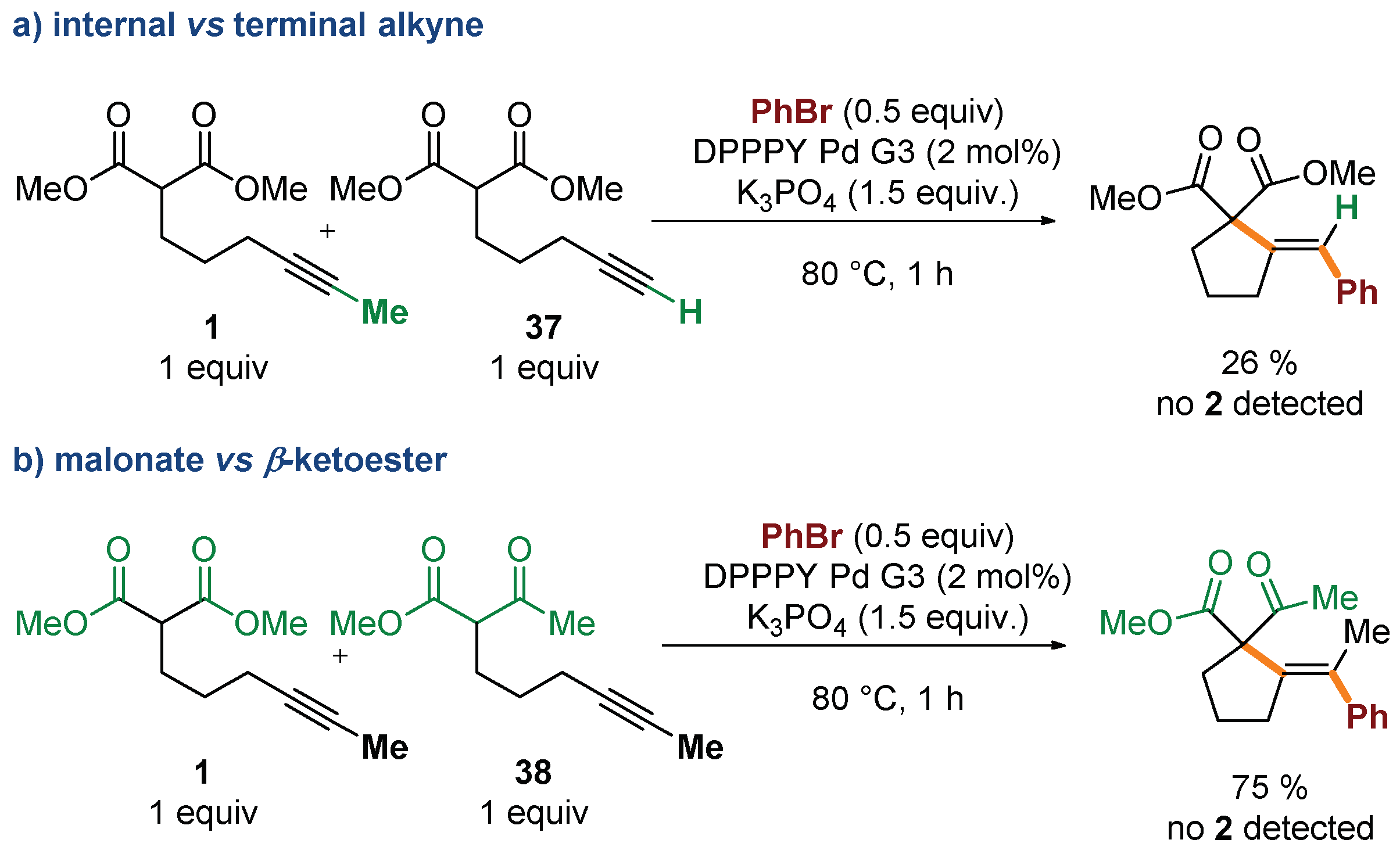
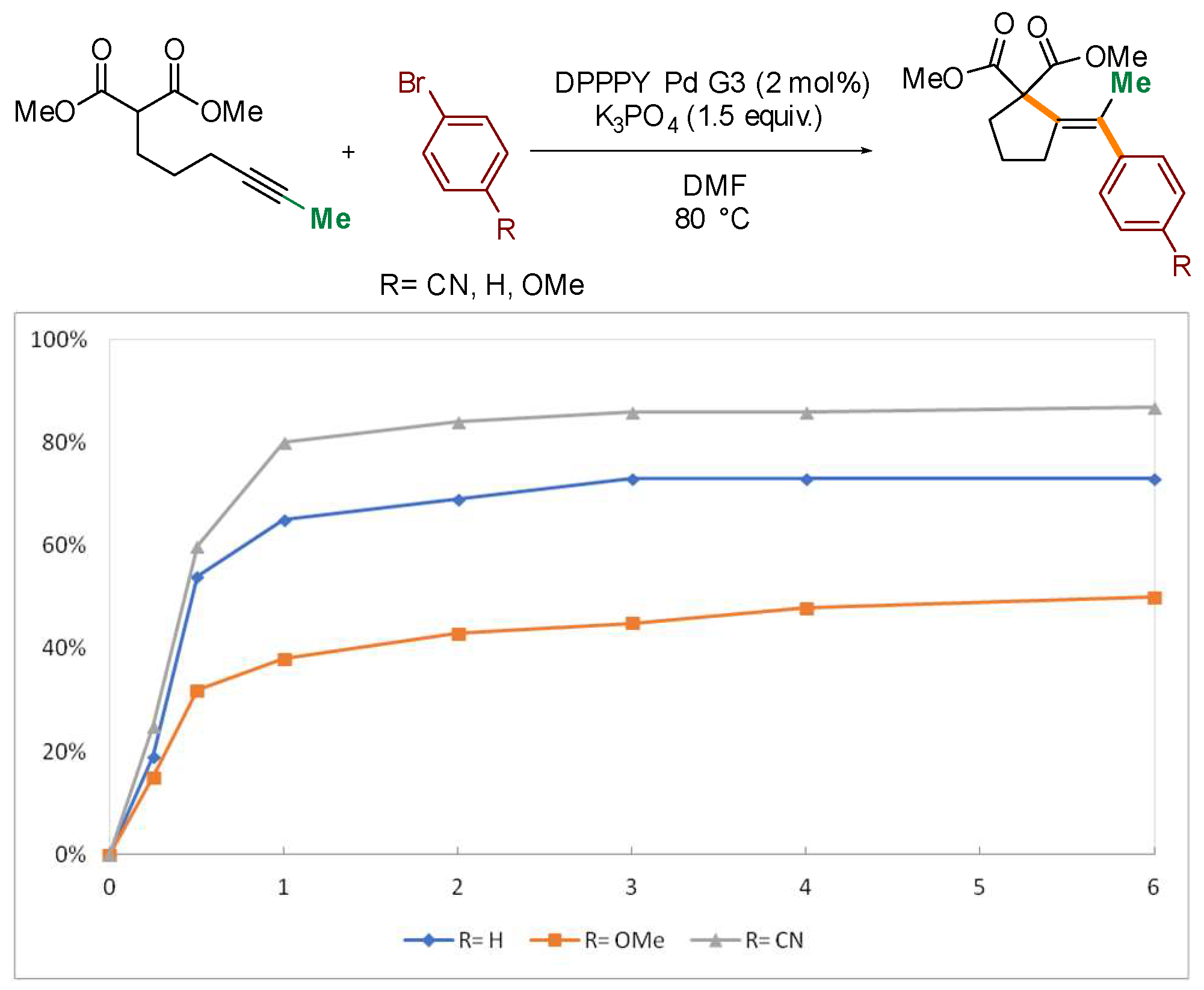
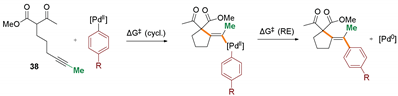
Several control experiments were conducted to gain additional insight into the mechanism of the process. Competition experiments confirmed dramatic difference in reactivity of substrates based on internal and terminal alkynes (Scheme 4a). Reaction of bromobenzene with an equimolar mixture of 1 and its terminal analogue 37 delivered exclusively the product of cyclization/coupling of the latter, without even traces of 2 detected by GC. Similarly, no consumption of 1 was observed in competition experiments with the structurally related β-keto ester 38 (Scheme 4b). Such a huge difference in the reactivity of malonates and β-keto esters, differing in acidity by only ca. 1 pKa unit, ref. [41] could suggest that deprotonation of the starting material is not involved in the rate-determining step (vide infra). The impact of the electronic nature of the electrophilic partner on the reaction progress was also investigated through a comparison of the reactivity of 1 with three electronically distinct bromoarenes–bromobenzene, p-bromoanisole, and p-bromobenzonitrile. Higher rates were observed for more electron-deficient partners (Figure 2).
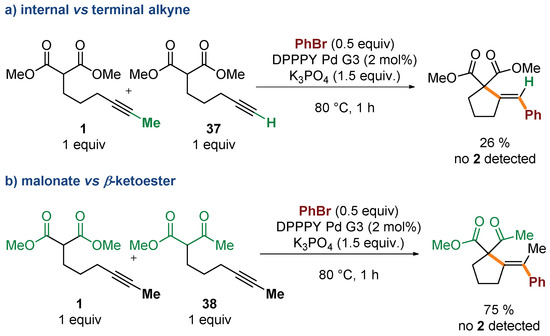
Scheme 4.
Competition experiments.
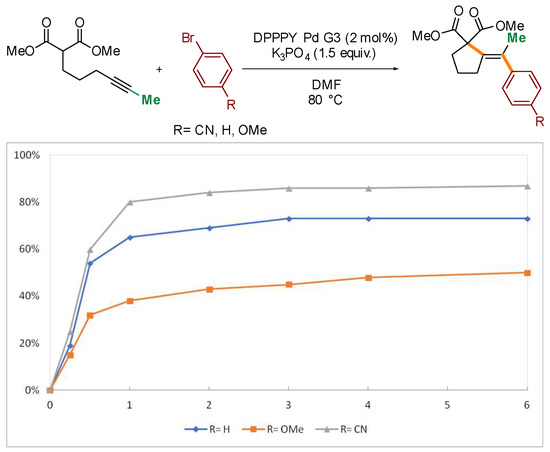
Figure 2.
Kinetic profiles for the reaction of 1 with electronically varied bromoarenes.
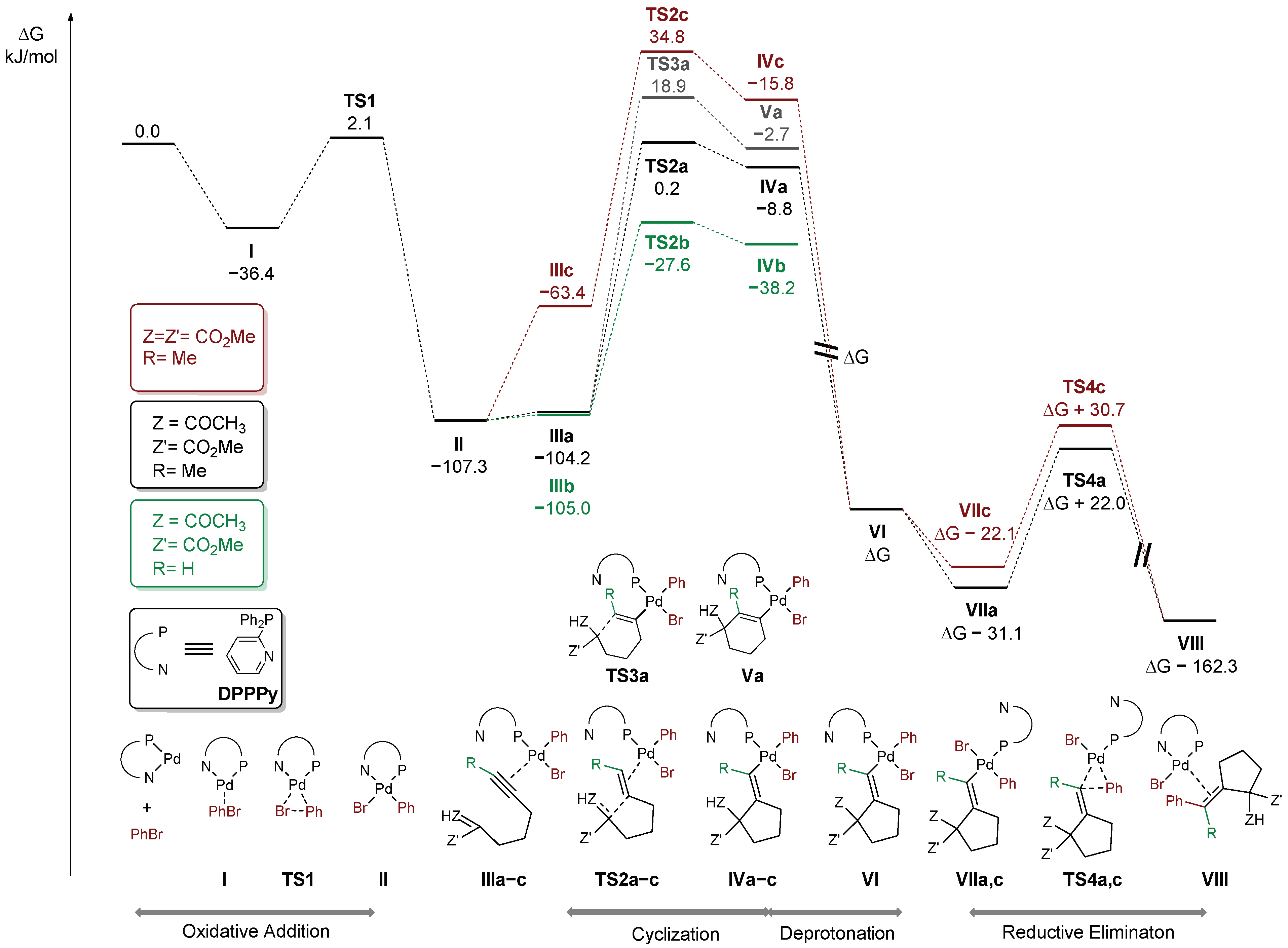
Density functional theory (DFT) calculations were conducted to elaborate the detailed reaction mechanism. All calculations were performed using Gaussian 16 package [42]. Structures of minima and transition states were optimized employing B3LYP and LANL2DZ basis sets for Pd, 6-31G(d) basis set for the other atoms, D3 version of Grimme’s empirical dispersion correction [43] and solvation (DMF) with SMD model [44]. Frequency analysis was performed at the same level to provide correction to thermodynamic functions and confirm the nature of optimized structures (minima and transition states featured zero or one imaginary frequency, respectively). Single point energies were calculated at M06 level of theory employing SDD basis set for Pd, 6-311++g(d,p) basis set for the other atoms and solvation (DMF) with SMD model [44]. Various conformers of intermediates and transition states were investigated and only the lowest energy conformers are shown in the work.
The free energy profile of the postulated pathway for model reaction of β-ketoester 38 with bromobenzene (black) is shown in Figure 3. Initially, bromobenzene undergoes facile oxidative addition to palladium(0) species (ΔG‡ = 38.5 kJ/mol), leading to Pd(II) intermediate II. Then, alkyne coordinates to a Pd(II) centre ultimately leading to a complex III, with enol form of the substrate coordinated in the conformation pre-organized for the cyclization, featuring ΔG‡ = 107.5 kJ/mol. Due to attack of the C-nucleophilic center of the enol from the opposite side of the alkyne in respect to the coordinated metal centre, the carbopalladation can proceed only in the anti fashion. The cyclization of the terminal analogue proceeds through a considerably lower barrier (ΔG‡ = 79.7 kJ/mol, green path). It is fully consistent with previously discussed competition experiments (Scheme 1a) and known examples of Conia-ene reactions catalyzed by carbophilic Lewis acids [37]. Interestingly, steric hindrance caused by the substituent at the alkyne moiety seems to have little if any effect on the coordination of the C-C triple bond to the Pd(II) species (cf. intermediates IIIa and IIIb). On the contrary, cyclization of the related malonate 1 (red path) is considerably more difficult than β-ketoester 38 (ΔG‡ = 142.1 vs. 107.5 kJ/mol) which is also in line with the competition experiment depicted in Scheme 1b. A significant fraction of the barrier arises from unfavorable enolization of the malonate, which is reflected in endergonic formation of intermediate IIIc. An alternative path involving deprotonation prior to the cyclization was also considered. However, the calculated barriers for the cyclization involving deprotonated malonate and keto ester fragments exhibited similar magnitudes (ΔG‡ = 41.5 and 40.4 kJ/mol, respectively). Taking into account the small difference in acidity (ca. 1 pKa unit at ambient temperature), formation of at least some amount of both products should be observed in the competition experiment (see Scheme 1b). Therefore, this path is less probable, at least if the reaction is carried out with a relatively weak, insoluble base (K3PO4). The other important factor associated with the cyclization step is the observed preference of 5-exo-dig over 6-endo-dig manifold which determines the excellent regioselectivity of the overall process. In fact, the calculated barrier for 6-endo-dig carbocyclization (grey path) is higher by 18.7 kJ/mol than the observed 5-exo-dig (black path). Furthermore, the vinyl–palladium intermediate IVa, bearing a protonated ketone moiety undergoes facile deprotonation to VIa. It then undergoes isomerization to VIIa, with vinyl and phenyl ligands in the cis position required for reductive elimination, which proceeds with ΔG‡ = 53.1 kJ/mol (through a transition state TS4a). Reductive elimination from the malonate analogue (red path) is similarly easy (ΔG‡ = 52.8 kJ/mol).
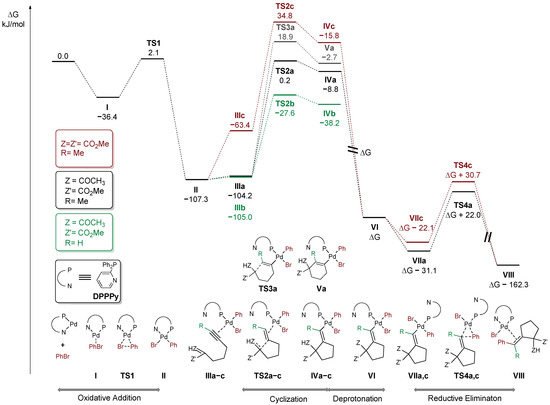
Figure 3.
Gibbs free energy profile.
As previously mentioned, the reaction with electron-deficient bromoarenes proceeds with slightly higher rates (see Figure 2). Presumably, substituent at the arene moiety perturbs the Lewis acidity of the Pd(II) centre and thus exerts impact on the facility of the cyclization step, considered rate-determining. Free energies of activation for the cyclization step were calculated for a series of three electronically distinct arylpalladium species bearing CN, H, and OMe at the para position of the phenyl moiety (Table 1). The lowest barrier was found for the complex with the most electron-deficient arene. To complete the mechanistic picture of the overall transformation, barriers for reductive elimination were also calculated and are summarized in Table 2.
Table 2.
Free energy of activation for cyclization and reductive elimination steps in the reaction of 38 with aryl bromides.
3. Conclusions
In conclusion, we developed a broadly applicable method for the carbocyclization-coupling of active methylene compounds bearing internal alkyne motif with aryl and heteroaryl bromides. The tandem Pd-catalyzed process exhibits excellent regio- and stereoselectivity, functional group tolerance, and high efficiency of the challenging 5-exo-dig intramolecular addition to nonterminal alkynes activated by a carbophilic Lewis acid. The mechanistic investigations, involving computational studies, support a mechanism involving oxidative addition, cyclization, and reductive elimination. The 5-exo-dig intramolecular nucleophilic addition of the enol form of the substrate to alkyne activated through coordination to the Pd(II) centre was identified as the rate- and configuration-determining step.
4. Experimental Section
General procedure for Pd-catalyzed carbocyclization-coupling of aryl bromides with acetylenic active methylene compounds: In a drybox, a 4 mL screw-cap vial was charged with DPPPY Pd G3 (5.10 mg, 8 mmol), aryl halide (0.5 mmol), K3PO4 (127.2 mg, 0.6 mmol), DMF (1 mL), and a magnetic stirring bar. Then, acetylenic active methylene compound (e.g., dimethyl 2-(hex-4-yn-1-yl)malonate) was added (0.4 mmol), the vial was tightly sealed and removed from drybox. The reaction mixture was stirred for 24 h at 80 °C in a heating block, then cooled to room temperature, quenched with 20 mL of a saturated NH4Cl solution, added to 10 mL of water, and extracted with MTBE (3 × 10 mL). The combined organic phases were dried with Na2SO4, filtered, and concentrated. The crude product was purified by column chromatography on silica gel.
Dimethyl (E)-2-(1-phenylethylidene)cyclopentane-1,1-dicarboxylate was prepared in a reaction of dimethyl 2-(hex-4-yn-1-yl)malonate and bromobenzene following a general procedure (98 mg, yield 85%). Product was isolated as an oil after column chromatography on silica gel (15g, hex/AcOEt 9:1). 1H NMR (400 MHz, CDCl3) δ 7.34–7.28 (m, 2H), 7.23–7.16 (m, 3H), 3.79 (s, 6H), 2.42 (t, J = 6.8 Hz, 2H), 2.29–2.22 (m, 2H), 1.96 (t, J = 2.0 Hz, 3H), 1.64 (p, J = 7.0 Hz, 2H);13C NMR (101 MHz, CDCl3) δ 172.0, 145.2, 135.8, 135.5, 128.1, 127.3, 126.3, 63.4, 52.5, 39.2, 33.5, 24.8, 22.3; IR(CH2Cl2): 2952, 2879, 2850, 1732, 1599, 1436, 1252, 1157, 1074, 988, 919, 764, 700, 429 cm−1; MS (EI): m/z (%) = 289(9), 288(43) [M+], 256(27), 230(24), 229(97), 228(59), 197(23), 170(24), 169(100), 168(27), 141(27), 128(16), 115(17), 105(7), 91(27), 77(10), 59(11); HRMS (EI): m/z calcd for C17H20O4 288.1362; found 288.1364.
Supplementary Materials
The following supporting information can be downloaded. Details of optimization and control experiments, experimental procedures and characterization of all compounds, crystallographic data, details of DFT calculations (atom coordinates, energies and corrections to thermodynamic functions for calculated structures).
Author Contributions
A.B.—Performed experiments, analyzed data and prepared Supporting Information; W.C.—conceived and supervised the project, acquired funding, performer DFT calculations, and wrote a manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support from the Polish National Science Centre (Grant 2016/22/E/ST5/00537) is gratefully acknowledged. Calculations have been carried out using resources provided by Wroclaw Centre for Networking and Supercomputing (grant No. 518).
Acknowledgments
The authors thank Maja Morawiak (Institute of Organic Chemistry PAS) for the X-ray crystal structure determination of 4.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Balme, G.; Bossharth, E.; Monteiro, N. Pd-Assisted Multicomponent Synthesis of Heterocycles. Eur. J. Org. Chem. 2003, 2003, 4101–4111. [Google Scholar] [CrossRef]
- Balme, G.; Bouyssi, D.; Lomberget, T.; Monteiro, N. Cyclisations Involving Attack of Carbo- and Heteronucleophiles on Carbon-Carbon π-Bonds Activated by Organopalladium Complexes. Synthesis 2003, 2003, 2115–2134. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-Metal-Catalyzed Addition of Heteroatom−Hydrogen Bonds to Alkynes. Chem. Rev. 2004, 104, 3079–3160. [Google Scholar] [CrossRef]
- Balme, G.; Bouyssi, D.; Monteiro, N. Palladium-Mediated Cascade or Multicomponent Reactions: A New Route to Carbo- and Heterocyclic Compounds. Pure Appl. Chem. 2006, 78, 231–239. [Google Scholar] [CrossRef]
- Zeni, G.; Larock, R.C. Synthesis of Heterocycles via Palladium-Catalyzed Oxidative Addition. Chem. Rev. 2006, 106, 4644–4680. [Google Scholar] [CrossRef] [PubMed]
- Dénès, F.; Pérez-Luna, A.; Chemla, F. Addition of Metal Enolate Derivatives to Unactivated Carbon−Carbon Multiple Bonds. Chem. Rev. 2010, 110, 2366–2447. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. Chemicals from Alkynes with Palladium Catalysts. Chem. Rev. 2014, 114, 1783–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, H. Recent Advances in the Construction of Polycyclic Compounds by Palladium-Catalyzed Atom-Economical Cascade Reactions. Asian J. Org. Chem. 2013, 2, 18–28. [Google Scholar] [CrossRef]
- Chen, L.; Chen, K.; Zhu, S. Transition-Metal-Catalyzed Intramolecular Nucleophilic Addition of Carbonyl Groups to Alkynes. Chem 2018, 4, 1208–1262. [Google Scholar] [CrossRef] [Green Version]
- Albano, G.; Aronica, L.A. From Alkynes to Heterocycles through Metal-Promoted Silylformylation and Silylcarbocyclization Reactions. Catalysts 2020, 10, 1012. [Google Scholar] [CrossRef]
- Rouessac, F.; Conia, J.-M. La cyclisation thermique des cetones εζ-ethyleniques. Tetrahedron Lett. 1965, 6, 3313–3318. [Google Scholar] [CrossRef]
- Conia, J.M.; Perchec, P.L. The Thermal Cyclisation of Unsaturated Carbonyl Compounds. Synthesis 1975, 1975, 1–19. [Google Scholar] [CrossRef]
- Hack, D.; Blümel, M.; Chauhan, P.; Philipps, A.R.; Enders, D. Catalytic Conia-Ene and Related Reactions. Chem. Soc. Rev. 2015, 44, 6059–6093. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A.; Davies, P.W. Catalytic Carbophilic Activation: Catalysis by Platinum and Gold π Acids. Angew. Chem. Int. Ed. 2007, 46, 3410–3449. [Google Scholar] [CrossRef] [PubMed]
- Kennedy-Smith, J.J.; Staben, S.T.; Toste, F.D. Gold(I)-Catalyzed Conia-Ene Reaction of β-Ketoesters with Alkynes. J. Am. Chem. Soc. 2004, 126, 4526–4527. [Google Scholar] [CrossRef]
- Staben, S.T.; Kennedy-Smith, J.J.; Toste, F.D. Gold(I)-Catalyzed 5-Endo-Dig Carbocyclization of Acetylenic Dicarbonyl Compounds. Angew. Chem. Int. Ed. 2004, 43, 5350–5352. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ma, X.; Zheng, Z.; Ma, S. Controllable Cyclization Reactions of 2-(2′,3′-Allenyl)Acetylacetates Catalyzed by Gold and Palladium Affording Substituted Cyclopentene and 4,5-Dihydrofuran Derivatives with Distinct Selectivity. Chem. Eur. J. 2008, 14, 8572–8578. [Google Scholar] [CrossRef] [PubMed]
- Gou, F.-R.; Bi, H.-P.; Guo, L.-N.; Guan, Z.-H.; Liu, X.-Y.; Liang, Y.-M. New Insight into Ni(II)-Catalyzed Cyclization Reactions of Propargylic Compounds with Soft Nucleophiles: Novel Indenes Formation. J. Org. Chem. 2008, 73, 3837–3841. [Google Scholar] [CrossRef] [PubMed]
- Montaignac, B.; Vitale, M.R.; Ratovelomanana-Vidal, V.; Michelet, V. Cooperative Copper(I) and Primary Amine Catalyzed Room-Temperature Carbocyclization of Formyl Alkynes. Eur. J. Org. Chem. 2011, 2011, 3723–3727. [Google Scholar] [CrossRef]
- Pei, T.; Wang, X.; Widenhoefer, R.A. Palladium-Catalyzed Intramolecular Oxidative Alkylation of Unactivated Olefins. J. Am. Chem. Soc. 2003, 125, 648–649. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Widenhoefer, R.A. Mechanism of the Palladium-Catalyzed Intramolecular Hydroalkylation of 7-Octene-2,4-Dione. J. Am. Chem. Soc. 2003, 125, 2056–2057. [Google Scholar] [CrossRef]
- Reeves, R.D.; Phelps, A.M.; Raimbach, W.A.T.; Schomaker, J.M. Diastereoselective Au-Catalyzed Allene Cycloisomerizations to Highly Substituted Cyclopentenes. Org. Lett. 2017, 19, 3394–3397. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Lu, X. Palladium(II)-Catalyzed Coupling Reactions of Alkynes and Allylic Compounds Initiated by Intramolecular Carbopalladation of Alkynes. Tetrahedron Lett. 2002, 43, 6791–6794. [Google Scholar] [CrossRef]
- Fujino, D.; Yorimitsu, H.; Oshima, K. Palladium-Catalyzed Intramolecular Carboacetoxylation of 4-Pentenyl-Substituted Malonate Esters with Iodobenzene Diacetate. Chem. Asian J. 2010, 5, 1758–1760. [Google Scholar] [CrossRef] [PubMed]
- Fujino, D.; Yorimitsu, H.; Oshima, K. Synthesis of Alkylidenecyclopropanes by Palladium-Catalyzed Reaction of Propargyl-Substituted Malonate Esters with Aryl Halides by Anti-Carbopalladation Pathway. J. Am. Chem. Soc. 2011, 133, 9682–9685. [Google Scholar] [CrossRef]
- Fujino, D.; Yorimitsu, H.; Osuka, A. Synthesis of 1,2-Disubstituted Cyclopentenes by Palladium-Catalyzed Reaction of Homopropargyl-Substituted Dicarbonyl Compounds with Organic Halides via 5-Endo-Dig Cyclization. Org. Lett. 2012, 14, 2914–2917. [Google Scholar] [CrossRef]
- Hess, W.; Burton, J.W. Palladium-Catalysed Cyclisation of N-Alkynyl Aminomalonates. Chem. Eur. J. 2010, 16, 12303–12306. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.-N.; Duan, X.-H.; Bi, H.-P.; Liu, X.-Y.; Liang, Y.-M. Synthesis of Indenes via Palladium-Catalyzed Carboannulation of Diethyl 2-(2-(1-Alkynyl)Phenyl)Malonate and Organic Halides. J. Org. Chem. 2006, 71, 3325–3327. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Ballav, T.; Biswas, K.; Ghosh, S.; Ganesh, V. Exploiting the Versatility of Palladium Catalysis: A Modern Toolbox for Cascade Reactions. Eur. J. Org. Chem. 2021, 2021, 4566–4602. [Google Scholar] [CrossRef]
- Fournet, G.; Balme, G.; Van Hemelryck, B.; Gore, J. Stereospecific Synthesis of Arylidene and Allylidene Cyclopentanes by a Palladium-Catalyzed Cylisation. Tetrahedron Lett. 1990, 31, 5147–5150. [Google Scholar] [CrossRef]
- Fournet, G.; Balme, G.; Gore, J. Synthese de (e)-Arylidene et Allylidene Cyclopentanes Par Une Annelation Catalysee Par Un Complexe de Palladium(0). Tetrahedron 1991, 47, 6293–6304. [Google Scholar] [CrossRef]
- Fournet, G.; Balme, G.; Gore, J. New Palladium Mediated Cyclopentanation of Alkenes Bearing a δ Nucleophilic Substituent. Tetrahedron Lett. 1989, 30, 69–70. [Google Scholar] [CrossRef]
- Chaładaj, W.; Domański, S. Mild and Functional Group Tolerant Method for Tandem Palladium-Catalyzed Carbocyclization–Coupling of ε-Acetylenic β-Ketoesters with Aryl Bromides and Chlorides. Adv. Synth. Catal. 2016, 358, 1820–1825. [Google Scholar] [CrossRef]
- Błocka, A.; Woźnicki, P.; Stankevič, M.; Chaładaj, W. Pd-Catalyzed Intramolecular Addition of Active Methylene Compounds to Alkynes with Subsequent Cross-Coupling with (Hetero)Aryl Halides. RSC Adv. 2019, 9, 40152–40167. [Google Scholar] [CrossRef] [Green Version]
- Kołodziejczyk, A.; Domański, S.; Chaładaj, W. Tandem Palladium-Catalyzed 6-Exo-Dig Oxocyclization Coupling of δ-Acetylenic β-Ketoesters with Aryl Bromides and Chlorides: Route to Substituted Dihydropyrans. J. Org. Chem. 2018, 83, 12887–12896. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Makida, Y.; Ochida, A.; Ohmiya, H.; Sawamura, M. Cyclization of Nonterminal Alkynic β-Keto Esters Catalyzed by Gold(I) Complex with a Semihollow, End-Capped Triethynylphosphine Ligand. Org. Lett. 2008, 10, 5051–5054. [Google Scholar] [CrossRef]
- Chaładaj, W.; Kołodziejczyk, A.; Domański, S. Gold(I)-Catalyzed Conia-Ene Cyclization of Internal ϵ-Acetylenic β-Ketoesters under High Pressure. ChemCatChem 2017, 9, 4334–4339. [Google Scholar] [CrossRef]
- Montel, S.; Bouyssi, D.; Balme, G. An Efficient and General Microwave-Assisted Copper-Catalyzed Conia-Ene Reaction of Terminal and Internal Alkynes Tethered to a Wide Variety of Carbonucleophiles. Adv. Synth. Catal. 2010, 352, 2315–2320. [Google Scholar] [CrossRef]
- See Supporting Information for Details.
- Bruno, N.C.; Tudge, M.T.; Buchwald, S.L. Design and Preparation of New Palladium Precatalysts for C–C and C–N Cross-Coupling Reactions. Chem. Sci. 2013, 4, 916–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordwell PKa Table. Available online: https://organicchemistrydata.org/hansreich/resources/pka/ (accessed on 9 November 2021).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).